

Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) may serve as a useful target for drug development in non-diabetic diseases. However, some colorectal cancer cells are resistant to PPARγ agonists by mechanisms that are poorly understood. Here we provide the first evidence that elevated PPARδ expression and/or activation of PPARδ antagonize the ability of PPARγ to induce colorectal carcinoma cell death. More importantly, the opposing effects of PPARδ and PPARγ in regulating programmed cell death are mediated by survivin and caspase-3. We found that activation of PPARγ results in decreased survivin expression and increased caspase-3 activity, whereas activation of PPARδ counteracts these effects. Our findings suggest that PPARδ and PPARγ coordinately regulate cancer cell fate by controlling the balance between the cell death and survival and demonstrate that inhibition of PPARδ can reprogram PPARγ ligand-resistant cells to respond to PPARγ agonists.
Keywords: Peroxisome proliferator-activated receptors, colorectal cancer, surviving, apoptosis
Introduction
The peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily and are also ligand-dependent transcription factors. To date, three mammalian PPARs have been identified and are referred to as PPARα (NR1C1), PPARδ/β (NR1C2) and PPARγ (NR1C3), respectively. It is well established that modulation of PPAR activity maintains cellular and whole-body glucose and lipid homeostasis. Hence, great efforts have been made to develop drugs targeting these receptors. For example, PPARγ synthetic agonists, rosiglitazone and pioglitazone, are anti-diabetic agents which suppress insulin resistance in adipose tissue, while a PPARδ agonist are currently being evaluated for treatment of dyslipidemias, obesity, and/or Type-2 diabetes. Recent studies suggest that PPARγ and PPARδ may play an important role in modulating colorectal carcinogenesis as well as other types of cancer (Cellai et al., 2006; Gupta et al., 2004; Panigrahy et al., 2005; Takayama et al., 2006; Wang et al., 2004; Yin et al., 2005).
Cellular transformation and tumor progression involve cooperative interactions between signaling pathways that affect both tumor cell proliferation and death. Dysregulation of apoptosis with increased resistance to cell death is a common feature of malignant cells and represents a significant obstacle to successful cancer therapy. A growing body of evidence has shown that PPARγ agonists exhibit antitumor and apoptosis-inducing effects in a broad range of human malignancies. In cell culture studies, activation of PPARγ results in tumor cell growth arrest through induction of apoptosis and/or differentiation in many cell types, including colorectal carcinoma cells. In animal models, activation of PPARγ inhibits colorectal tumor growth in xenograft studies and in azoxymethane (AOM)-treated mice (Osawa et al., 2003; Sarraf et al., 1998). However, PPARγ agonists have been reported to have tumor-promoting effects in the ApcMin/+ mice (Lefebvre et al., 1998; Pino et al., 2004; Saez et al., 1998). These divergent effects of PPARγ might be related to drug doses and bioavailability and/or animal models employed. These paradoxic observations appear to have been resolved by genetic studies showing that the heterozygous disruption of PPARγ is sufficient to increase tumor number(s) in AOM-treated mice and that ligand-activation of PPARγ inhibits tumor growth only in the presence of functional APC but not in cells with loss of APC function (Girnun et al., 2002). The ApcMin/+ mouse contains an inherited mutation in one allele of Apc gene and eventually develop intestinal adenomas (Williams et al., 1996). These results suggest that loss of APC may alter the normal response of intestinal epithelial tumor cells to PPARγ agonists. This is consistent with the negative outcome of clinical trials examining the efficacy of PPARγ agonists in humans with advanced colorectal cancer (CRC) (Burstein et al., 2003; Kulke et al., 2002) since an APC mutation occurs in about 85% of human CRCs. Loss of APC function results in upregulation of PPARδ through β-catenin/Tcf cascade (He et al., 1999). Therefore, one potential mechanism by which colorectal carcinoma cells become resistant to PPARγ agonists could include antagonism by PPARδ.
The role of PPARδ in cancer biology remains unclear since results generated by different groups do not agree. One study showed that deletion of PPARδ exon 8 enhances polyp growth in ApcMin/+ and AOM-treated mice (Harman et al., 2004). On the other hand, the studies from our group and others revealed that loss of PPARδ by deletion of its exons 4-5 or 4 attenuated both small and large intestinal adenoma growth in ApcMin/+ mice and AOM-treated mice (Wang et al., 2006b; Zuo et al., 2009). Moreover, our results revealed that activation of PPARδ accelerates intestinal tumor growth by promoting cell survival in vitro and in vivo (Gupta et al., 2004; Wang et al., 2004). These studies indicate that PPARγ and PPARδ have opposing effects on CRC progression. Hence it is critical to evaluate the relationship between PPARγ and PPARδ in CRC in order to develop strategies for cancer prevention and treatment and further establish their respective roles in cancer biology.
Survivin is a unique member of the IAP family since it is overexpressed in almost every human tumor that has been studied, but is barely detectable in most normal adult tissues (Altieri, 2003). Overexpression of survivin is associated with poor clinical outcome with reduced tumor cell apoptosis in patients with CRC (Kawasaki et al., 1998; Sarela et al., 2001). Moreover, high expression of survivin correlates with resistance to certain anticancer agents and radiation therapy (Asanuma et al., 2000; Zaffaroni and Daidone, 2002). In contrast, inhibition of survivin expression or interference with survivin function inhibits tumor cell growth, induces apoptosis, and sensitizes tumor cells to radiation or chemotherapy (Kuo et al., 2004; Olie et al., 2000).
This study was designed to test the hypothesis that PPARδ causes colorectal carcinoma cells to become resistant to PPARγ–mediated apoptosis and to explore how PPARδ and PPARγ regulate tumor cell death. Here we report that overexpression of PPARδ or activation of PPARδ by its agonists attenuates the ability of PPARγ agonists to induce apoptosis. One intriguing finding is that treatment with a PPARγ agonist reduces survivin expression, which in turn induces apoptosis via increased caspase-3 activity. In contrast, treatment of PPARδ ligands protects cancer cells from PPARγ-induced apoptosis by inhibiting induction of the survivin-caspase-3 apoptotic pathway. Consistent with these findings, we also observed that PPARδ expression correlates well with survivin levels in human CRC specimens. Collectively, our results identify PPARδ as an anti-apoptotic gene that contributes to the resistance of CRC cells to the PPARγ agonist-induced apoptosis.
Results
PPARδ contributes to resistance of PPARγ ligand-induced apoptosis
To investigate whether PPARδ confers resistance to cell death induced by PPARγ, we first measured the relative levels of PPARγ and PPARδ in a panel of eight colorectal carcinoma cell lines. As shown in Figure 1a, all cell lines expressed fairly equivalent levels of PPARγ protein. In contrast, HCT-116 cells exhibited higher PPARδ expression than the other lines tested (Figure 1a). Since LS-174T and HCT-116 cells have been carefully evaluated to understand the anti-apoptotic effects of PPARδ (Gupta et al., 2004; Wang et al., 2004), we examined the ability of PPARγ agonists to induce apoptosis in these two cell lines by annexin V-FITC assay. Treatment of LS-174T cells with a selective synthetic PPARγ agonist GW7845 at 5 μM for 1, 2, and 3 days. The peak of apoptotic rate induced by GW7845 was observed in 2-day's treatment (Figure 1b, left panel). The treatment of GW7845 at 5 or 10 μM for 2 days resulted in a significant apoptosis (42% to 56%) compared to controls (DMSO) (Figure 1b, right panel). The pro-apoptotic effect of GW7845 in LS-174T cells was also confirmed by DNA fragment assays (Figure 1c). Furthermore, our results showed that blocking of PPARγ activation by treatment with its antagonist (GW9662) inhibited GW7845-induced apoptosis in LS-174T cells (Figure 1d), suggesting that PPARγ mediates the effect of GW7845 on induction of apoptosis. In contrast, HCT-116 cells with high PPARδ expression are more resistant to the PPARγ agonist GW7845 than LS-174T cells (Figure 1b and 1e). Similar results for the specificity of GW7845 in the HCT-116 cells were observed as well (data not shown). In addition, the results from cell viability assays with treatment of GW7845 at 10 μM revealed that viable LS-174T cells significantly decreased in a time-dependent manner, whereas the viable HCT-116 cells only reduced in day 3 but increased in day 5 (Supplementary Fig. 1a). The clonogenic cell survival assays further showed that all LS-174T cells were killed after 5-day's treatment, whereas HCT-116 cells formed colonies after 9-day's treatment (Supplementary Fig. 1b). These results demonstrate that HCT-116 cells are more resistant to the PPARγ agonist GW7845 than LS-174T cells. Since both cell lines express similar levels of the PPARγ receptor but have different amounts of PPARδ, these results suggest that PPARδ may be involved in protecting cancer cells from PPARγ-induced apoptosis.
Fig. 1. PPARδ expression inhibits PPARγ-induced apoptosis.
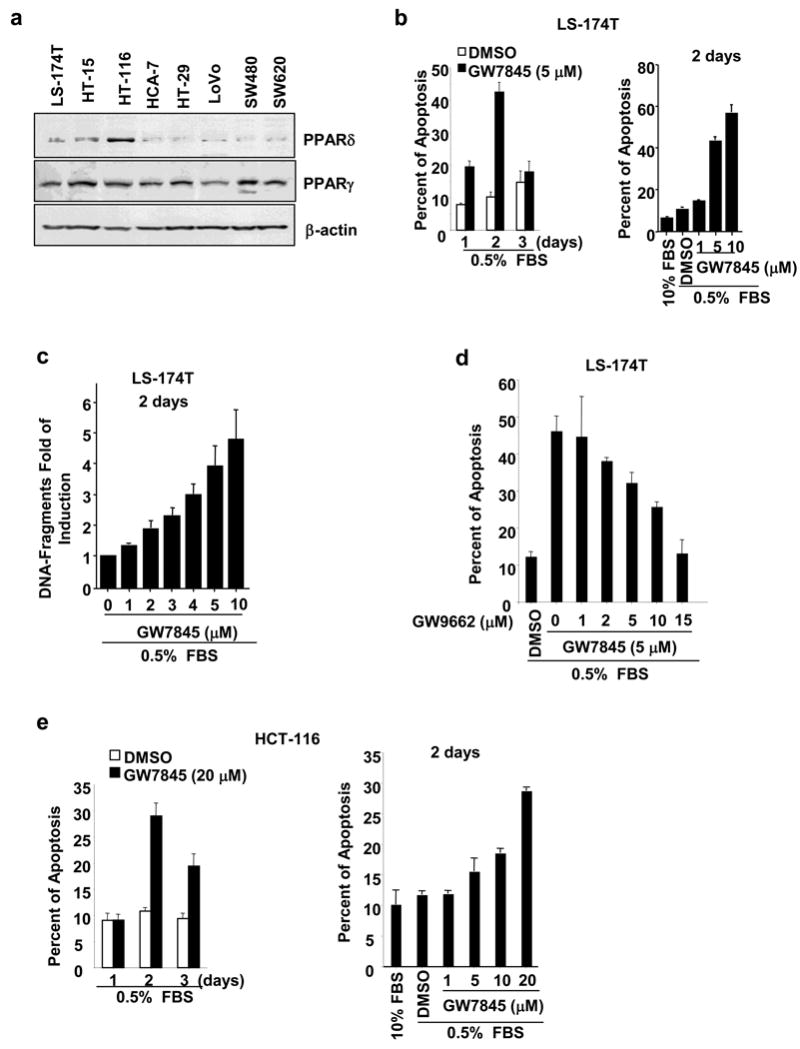
(a) Profile of PPARγ and PPARδ expression in CRC cell lines. The analysis of PPARδ and PPARγ protein in eight CRC cell lines were performed by Western blot analysis. The figure is a representative of three different experiments that showed similar results. (b-c) PPARγ agonist induces apoptosis in LS-174T cells. The cells were cultured in media with 0.5% fat-free FBS and treated with indicated dose of GW7845 for indicated times. The percent of apoptotic cells (b) and induction of DNA-fragments in cells (c) were determined by an annexin V-FITC kit and Cell Death Detection ELISA kit, respectively. Data are expressed as the mean + SE from three separate experiments. (d) PPARγ antagonist inhibits the pro-apoptotic effect of PPARγ agonist. The LS-174T cells were pretreated with GW9662 for 1 h and then were treated with GW7845 for 2 days. The apoptosis assays were conducted as described in Fig. 1b. (e) HCT-116 cells are resistant to PPARγ agonist. The cells were treated with indicated concentration of GW7845 for indicated times and apoptosis assays were carried out as described in Fig. 1b.
To directly study the function of PPARδ in PPARγ-induced apoptosis, we determined whether overexpression or deletion of PPARδ in cancer cells affects the ability of PPARγ agonist (GW7845) to induce apoptosis. Overexpression of PPARδ in LS-174T cells significantly reduces GW7845-induced apoptosis, as compared to control cells (Figure 2a, left panel). The level of PPARδ expression was confirmed by Western blot analysis (Figure 2a, right panel). In contrast, genetic disruption of both PPARδ alleles in HCT-116 cells (PPARδ-/-) by targeted homologous recombination restored the ability of PPARγ to induce apoptosis (Figure 2b). The HCT-116/PPARδ-/- cells don't express both PPARδ mRNA and protein (Figure 2b, right panel). These results demonstrate that PPARδ is responsible for colorectal carcinoma cells resistance to PPARγ ligand-induced apoptosis.
Fig. 2. Overexpression of PPARδ blocks the ability of PPARγ to induce apoptosis while disruption of PPARδ restores this ability.
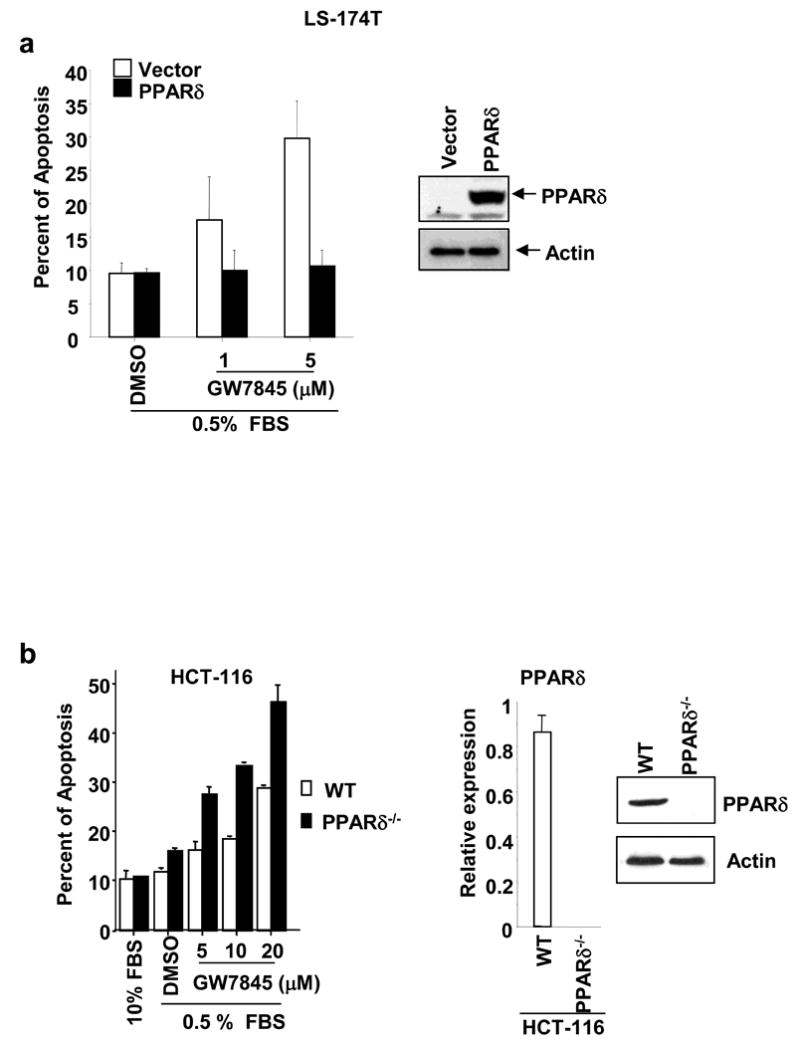
(a) The LS-174T/vector and LS-174T/PPARδ cells were treated with indicated concentration of GW7845 and apoptosis assays were carried out as described in Fig. 1b. PPARδ protein expression was determined by Western blot analysis (right panel). (b) The apoptosis assays were performed in parent and PPARδ-deficient HCT-116 cells treated with indicated concentration of GW7845 as described in Fig. 1b. The right panel represents the status of PPARδ at both mRNA and protein levels in HCT-116/WT and HCT-116/PPARδ-/-.
Activation of PPARδ inhibits PPARγ ligand-induced apoptosis
Amplification of PPARδ expression or deletion of PPARδ gene could have multiple biological effects independent of endogenous PPARδ activity. To overcome these limitations, we assessed whether PPARδ agonists inhibit the pro-apoptotic activity of PPARγ via the endogenous PPARδ receptor. LS-174T cells were treated with a selective PPARδ agonist GW501516 and/or PPARγ agonist GW7845. GW501516 attenuated GW7845-induced apoptosis to basal levels (DMSO) (Figure 3a). Similarly, an endogenous PPARγ ligand 15-PGJ2 induced apoptosis at a much lower concentration in LS-174T cells, while a PPARδ ligand cPGI2 inhibited 15-PGJ2-induced apoptosis (Figure 3b). To further confirm the specificity of PPARδ agonists, we examined the anti-apoptotic effects of PPARδ agonists in parental and PPARδ-deficient HCT-116 cells. Treatment of parental HCT-116 cells with GW501516 or cPGI2 significantly suppressed PPARγ-induced apoptosis in a dose-dependent manner. In contrast, the anti-apoptotic effect of PPARδ agonists was not seen in PPARδ-deficient HCT-116 cells, demonstrating that the effects of these PPARδ ligands are due to specific activation of PPARδ (Figure 3c-d). These results demonstrate that ligand-activated PPARδ inhibits PPARγ-induced apoptosis.
Fig. 3. Agonist-activated PPARδ counteracts the effect of PPARγ on inducing apoptosis.

(a) The LS-174T cells were pretreated with indicated concentration of GW501516 (a) or cPGI2 (b) for a half hour and then treated with GW7845 (a) or 15-PGJ2 (b) for 2 days. The apoptosis assays were carried out as described in Fig. 1b. (c-d) The parent and PPARδ-deficient HCT-116 cells were treated with agonists as described in panel a and b. The apoptosis assays were performed as same as described in Fig. 1b.
Survivin is a down-stream target of PPARγ
To further investigate PPARγ/δ-regulated intracellular events in apoptotic cascades, we first examined whether activation of PPARγ modulates genes involved in regulating cell apoptosis in CRC, such as Bcl-2, PTEN, and survivin. Treatment with the PPARγ agonist GW7845 decreased survivin expression but did not affect Bcl-2 and PTEN levels in LS-174T cells (Figure 4a). In addition, treatment of GW7845 did not affect the levels of phosphorylation of histone H3 (p-H3), indicating that PPARγ reduction of survivin is not due to a lack of G2 or/and M phase cells in GW7845-treated population. The PPARγ mediated downregulation of survivin was also seen in HCT-116 cells at a higher dose (Figure 4b). However, GW7845 failed to affect survivin expression at the mRNA level (Figure 4c), suggesting that PPARγ downregulates survivin protein expression via a post-translational modification mechanism. Since degradation of survivin protein is controlled by ubiquitylation and proteasome-dependent destruction in the cells, we examined whether treatment of LS-174T cells with a proteasome inhibitor (MG-132) blocks GW7845-induced downregulation of survivin. MG-132 inhibits the degradation of ubiquitin-conjugated proteins in cells. Indeed, treatment of MG-132 restored the survivin expression from 24 h to 48 h, suggesting that PPARγ downregulates survivin protein expression via enhancing its protein degradation (Figure 4d). Furthermore, forced-expression of survivin completely inhibits GW7845-induced apoptosis in LS-174T cells (Figure 4e). These results demonstrate that PPARγ induces cell death through downregulation of survivin.
Fig. 4. Survivin is a downstream target of PPARγ.
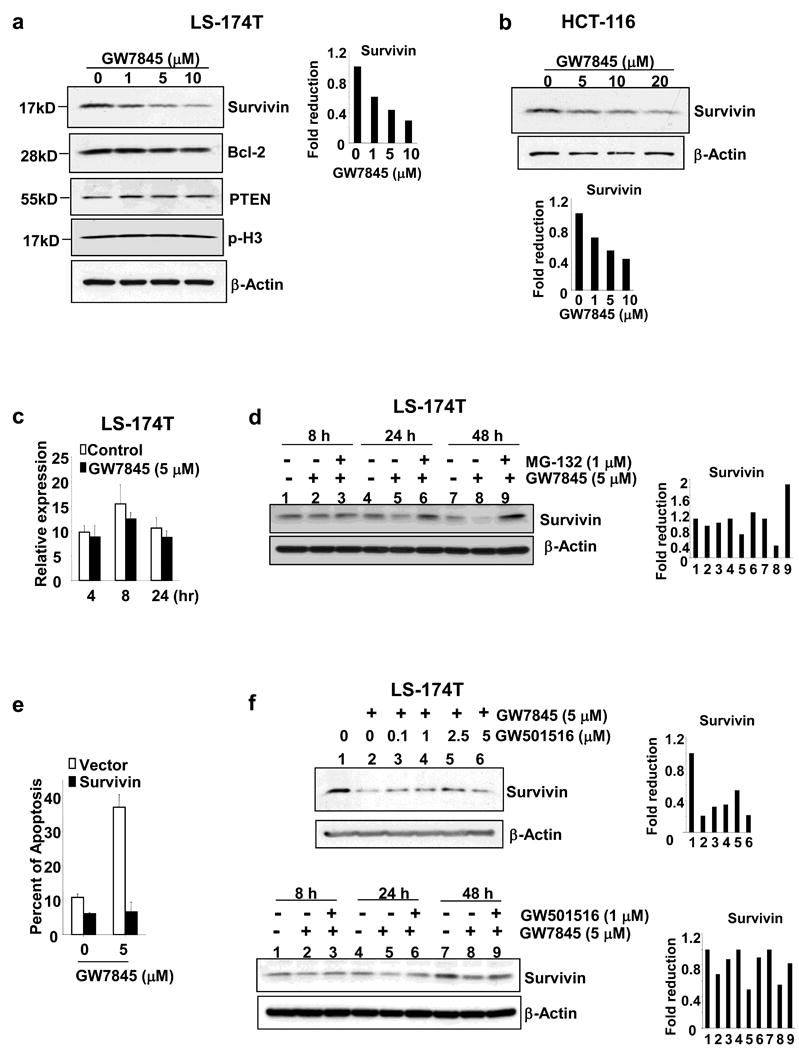
(a-b) The LS-174T cells (a) and HCT-116 (b) were cultured in media with 0.5% fat-free FBS and treated with indicated concentration of GW7845 for 1 day. Survivin, Bcl-2, PTEN, and phosphorylated histone H3 (p-H3) protein expression were analyzed by Western blotting. These figures are representative of three different experiments that showed similar results. The bar graph represents fold reduction of relative survivin band intensity. The relative survivin band intensity is survivin bend intensity normalized to β-actin bend intensity. (c) GW7845 does not affect survivin mRNA levels. The cells were treated as described in panel a and survivin mRNA was measured by quantitative real-time PCR as noted above. (d) Treatment of MG-132 inhibits GW7845 downregulation of survivin expression. The cells were pretreated with 1 μM of MG-132 and then treated with 5 μM of GW7845 for indicated time and survivin expression were determined by Western blot as described above. The bar graph represents fold reduction of relative survivin band intensity. (e) Overexpression of survivin inhibits PPARγ-induced apoptosis. The LS-174T/vector and LS-174T/survivin cells were treated with 5 μM of GW7845 and the percent of apoptotic cells was measured as described in Fig. 1b. (f) Activation of PPARδ rescues PPARγ downregulation of survivin expression at both dose- and time-dependent manner. The LS-174T cells were pretreated with indicated concentration of GW501516 and then treated with 5 μM of GW7845 for 1 day (top panel) as well as pretreated with 1 μM of GW501516 and then treated with 5 μM of GW7845 for indicated times (low panel). Survivin protein expression was determined by Western blot assay. The bar graph represents fold reduction of relative survivin band intensity.
Next, we investigated whether ligand-activated PPARδ attenuates the effect of PPARγ on downregulation of survivin. As shown in Figure 4f, treatment of PPARδ agonist GW501516 partially overcomes the effect of PPARγ ligand on survivin expression in both dose- and time-dependent manner. These results indicate that survivin is a downstream target for both PPARγ and PPARδ in regulating cell survival and death.
Caspase-3 mediates the effects of PPARγ and PPARδ in modulating apoptosis
We further examined whether PPARγ/δ affects caspase activity. Our results showed that a general caspase inhibitor (zVAD-fmk) reduced PPARγ agonist-induced apoptosis, suggesting that caspases are also downstream targets of PPARγ (Figure 5a). Furthermore, the PPARγ agonist GW7845 increased caspase-3 activity in both a dose- and time-dependent manner (Figure 5b). The activation of caspase-3 in turn resulted in cleavage of its target gene PARP in LS-174T cells (Figure 5b). Consistent with above results, PPARδ agonists (GW501516 and cPGI2) inhibited PPARγ-enhanced caspase-3 activity in LS-174T cells (Figure 5d-f). The ability of cPGI2 to inhibit 15-PGJ2-induced caspase-3 activity was observed in parental HCT116 cells, but not in PPARδ-deficient cells (Figure 5f), demonstrating that PPARδ mediates this effect of cPGI2. Taken together, these results support our hypothesis that activation of PPARδ inhibits the pro-apoptotic effects of PPARγ.
Fig. 5. Caspase-3 mediates the effects of PPARγ and PPARδ on regulating apoptosis.
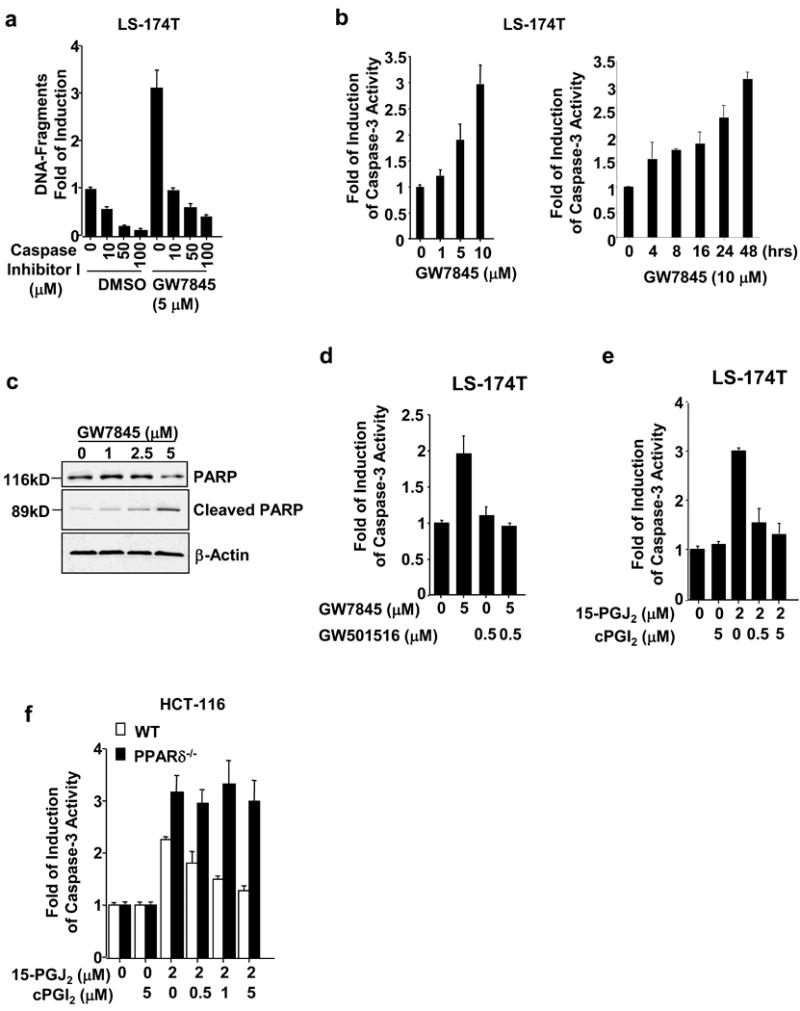
(a) Caspase inhibitor blocks PPARγ-induced apoptosis. The LS-174T cells were pretreated with indicated concentration of caspase inhibitor I and then treated with 5 μM of GW7845. The percent of apoptotic cells was measured as described in Fig. 1b. (b) Activation of PPARγ induces caspase-3 activity. The LS-174T cells were treated with indicated dose of GW7845 for indicated times. Caspase-3 activity was measured by a caspase-3 colorimetric assay. Data are represented as the mean + SE of fold-induction from three independent experiments. (c) PARP and cleaved PARP in the LS-174T cells treated with indicated concentration of GW7845 was measured by Western blot assays. (d-f) Activation of PPARδ inhibits PPARγ-induced caspase-3 activity in LS-174T cells. The cells were pretreated with indicated concentration of GW501516 (c) or cPGI2 (d) for a half hour and then treated with GW7845 (c) or 15-PGJ2 (d) for 1 day. The caspase-3 activity assays were carried out as described in panel b. Data are expressed as the mean + SE from three separate experiments. (e) The effect of PPARδ agonist is dependent on PPARδ. The caspase-3 activity assays were performed in parent and PPARδ-deficient HCT-116 cells treated with indicated concentration of GW7845 as described above.
Survivin expression correlates with PPARδ expression in human colorectal cancer
To determine the clinical relevance of our in vitro results, we examined whether survivin expression correlates with PPARδ in human colorectal carcinoma tissues. The analysis of Real-Time quantitative PCR revealed that the levels of survivin mRNA in all human colon carcinomas at grade II-III are elevated as compared to the matched normal tissues (Figure 6a). Similarly, PPARδ mRNA is also elevated in 8 of 12 (67%) cancer specimens (Figure 6b). The spearman rank coefficient was utilized to test if the correlation was significant in all 12-paired samples. A positive correlation of survivin and PPARδ is found in these samples (r=0.538, p=0.0066). Consistent with the above results, the positive correlation of survivin and PPARδ is also observed in eight CRC cell lines at both mRNA (Figure 6c, left panal) and protein levels (Figure 6c, right panal). Furthermore, our preliminary studies showed that ApcMin/+ mice treated with a PPARδ agonist (GW501516) exhibited higher survivin mRNA expression levels in the small intestine than control mice (Data not shown). These observations indicate that the anti-apoptotic effect of PPARδ correlate well with survivin expression in vivo.
Fig. 6. Survivin expression is associated with PPARδ expression in human colorectal tumors and CRC cell lines.
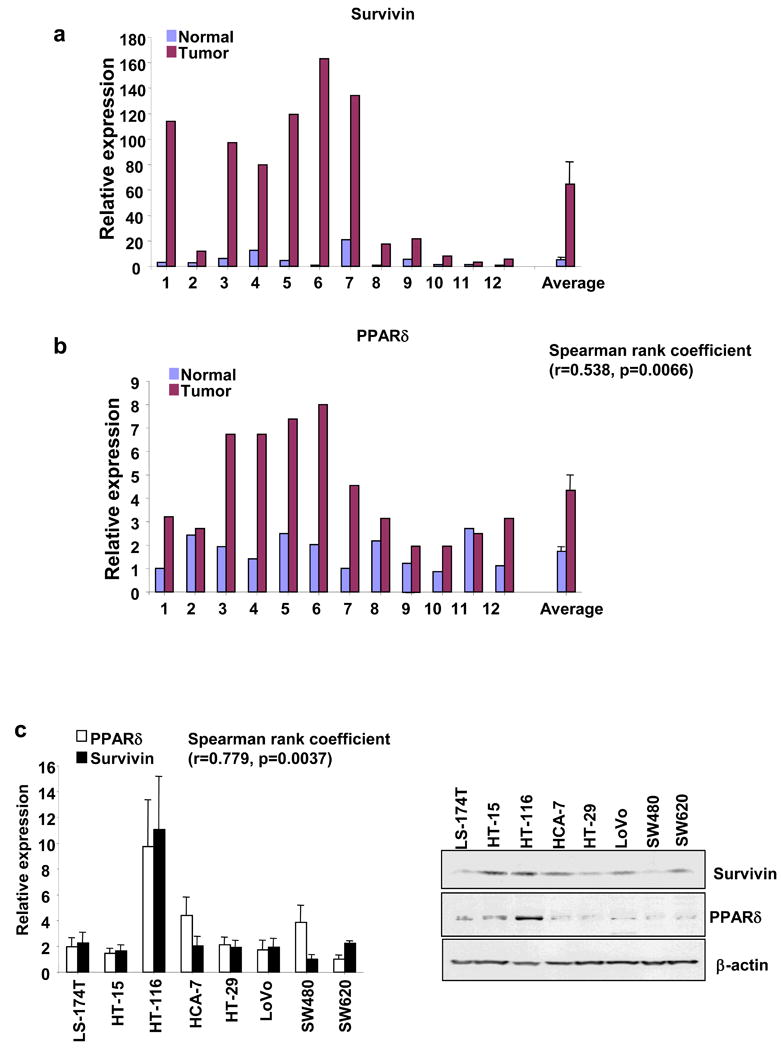
(a-b) The survivin (a) and PPARδ (b) expression in 12 pairs of tumors and the matched normal tissues is measured by Quantitative real-time PCR. (c) The survivin and PPARδ expression in eight CRC cell lines is measured by Quantitative real-time PCR (left panel) and Western blot (right panel).
Discussion
The conflicting results regarding the effect of PPARδ on intestinal tumorigenesis in ApcMin/+ and AOM-treated mice may be related to differences in the specific targeting strategy employed to delete PPARδ. Deletion of PPARδ exon 4 and/or 5, which encode an essential portion of the DNA binding domain, is thought to disrupt PPARδ function as a nuclear transcriptional factor and to inhibit tumorigenesis. The deletion of exon 8, the last PPARδ exon, is postulated to generate a hypomorphic allele, which retains some aporeceptor function. Indeed, the observation that the high rates of embryonic mortality, subsequent to abnormal trophoblastic giant cell differentiation and abnormal placental development occurred in deletion of PPARδ exon 4-5, but not in deletion of PPARδ exon 8 mice supports this hypothesis (Nadra et al., 2006; Peters et al., 2000).
In a mouse mammary tumor model, treatment with the PPARδ agonist GW501516 accelerated tumor formation, while a PPARγ agonist GW7845 delayed tumor growth (Yin et al., 2005). This observation suggests that there are distinct mechanistic differences between PPARγ and PPARδ in regulating tumor progression. Since the beneficial effect of PPARγ agonists in clinical trials for patients with CRC and in ApcMin/+ mice has not been observed, a potential explanation for these observations could be related to PPARδ. Here we present evidence demonstrating that overexpression of PPARδ or activation of endogenous PPARδ counteracts the pro-apoptotic effect of PPARγ ligands, while disruption of PPARδ restores the ability of PPARγ to induce apoptosis in colorectal carcinoma cells (Figure 2-3). These results may explain the discrepancies of PPARγ anti-tumor effects in different intestinal tumor animal models and confirm that PPARγ may actually serve as a tumor suppressor. Furthermore, our data also provides important evidence that could enable the design of novel strategies for CRC prevention and/or treatment. For example, combinational treatment of PPARγ agonists with PPARδ antagonists will overcome resistance to cell death and restore the sensitivity to PPARγ agonists. Understanding the mechanism(s) by which colorectal carcinoma cells are resistant to PPARγ agonist is of great interest and potential clinical value.
Although it has been established that activation of PPARγ induces tumor cell apoptosis, the downstream mediators of these effects are not well defined. Recent emerging data indicates that PPARγ agonists can modulate the expression of several apoptotic activators and suppressors, such as PTEN, Bcl-2, and NF-κB. For example, PPARγ agonists induce PTEN expression in pancreatic cancer, non-small cell lung carcinoma, and breast cancer cells (Farrow and Evers, 2003; Han and Roman, 2006; Teresi et al., 2006), while these agonists inhibit Bcl-2 expression in neuroblastoma and lung cancer cells (Kim et al., 2003; Li et al., 2005) and NF-κB expression in thyroid cancer (Kato et al., 2006). Microarray studies with colorectal carcinoma cells have led to the identification of a number of PPARγ target genes that could serve in the regulation of cell growth, differentiation, and adhesion (Gupta et al., 2001). However, the function of these genes in regulating colorectal tumor cell apoptosis has not been fully investigated. Here, we present data showing that PPARγ agonists at a relative low dose do not significantly affect PTEN and Bcl-2 expression in CRC cells (Figure 4a), although a high dose of PPARγ agonists do inhibit Bcl-2 expression (data not shown).
Survivin is one of eight members of the IAP family, including the most studied XIAP, c-IAP1, c-IAP2, and survivin. The IAP gene family encodes proteins which inhibit cellular apoptosis by binding to caspases and inhibiting their activity (Salvesen and Duckett, 2002). Survivin expression is elevated in almost every human tumor that has been studied and is associated with poor clinical outcome with reduced tumor cell apoptosis in patients with CRC, but is barely detectable in most normal adult tissues (Altieri, 2003; Kawasaki et al., 1998; Sarela et al., 2001). Although increased survivin expression is an important event in tumorigenesis, the mechanisms responsible for survivin regulation are not fully understood. Specifically, it is not clear which chemotherapeutic agents might target survivin. We show here for the first time that survivin is a downstream target of PPARγ agonists (Figure 4) and it mediates PPARγ-induced apoptosis (Figure 5A).
The dysregulation of survivin in cancer cells depends on its gene transcription and post-translational modification that affect its stability, such as phosphorylation. For example, survivin transcription is repressed by wild-type p53 (Hoffman et al., 2002) and likely upregulated by Wnt-β-catenin signaling (Zhang et al., 2001). However, activation of PPARγ did not affect survivin mRNA levels (Figure 4c), suggesting that these effects occur post-translationally. During cell cycle progression, rapid degradation of survivin protein is controlled by ubiquitylation and proteasome-dependent destruction in interphase cells (Zhao et al., 2000), while phosphorylation of survivin on Thr34 by CDC2–cyclin-B1 has been associated with increased protein stability at metaphase (O'Connor et al., 2002). Our data reveals that inhibition of protease activity PPARγ reduces survivin phosphorylation at Thr34 and its expression in CRC cells (Figure 4d), indicating that activation of PPARγ may affect survivin degradation. Understanding the precise mechanism by which PPARγ affects the phosphorylation state of survivin will be addressed in future studies.
A more complete understanding of the underlying molecular mechanism(s) by which activation of PPARδ protects against apoptosis induced by PPARγ is of critical importance. PPARδ has been shown to promote cell survival in the kidney following hypertonic stress (Hao et al., 2002) and the skin following wound injury (Di-Poi et al., 2003; Di-Poi et al., 2002). Our previous studies also showed that activation of PPARδ protects against colorectal carcinoma cell apoptosis induced by serum deprivation (Gupta et al., 2004). In this study, our results present the first evidence showing that ligand-activated PPARδ attenuates the pro-apoptotic effect of PPARγ (Figure 3). More importantly, we reveal that activation of PPARδ partially reverses the effect of PPARγ on survivin phosphorylation and expression (Figure 4f). Furthermore, PPARδ agonists inhibit PPARγ-enhanced caspase-3 activity (Figure 5c-d), which is dependent on the presence of the PPARδ receptor (Figure 5e). In addition, there is a positive correlation between PPARδ and survivin expression in human colorectal tumor tissues and cell lines (Figure 6). Thus, the precise mechanism(s) by which PPARδ inhibits the pro-tumor effect of PPARγ requires further investigation.
In summary, data obtained from this study supports a concept that differential activation of PPARγ or PPARδ tilts the balance between cell death and cell survival. PPARδ confers resistance to PPARγ-induced apoptosis. Thus, activation of PPARδ promotes survival advantages and supports tumor progression, while inhibition of PPARδ would overcome resistance and restore sensitivity to agents that stimulate cell death.
Materials and Methods
Cell culture and Reagents
LS-174T, HCT-116, HCA-7, HCT-15, HT-29, LoVo, SW480 and SW620 cells were maintained in McCoy's 5A medium with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT). Charcoal/Dextran treated FBS (fat-free) was obtained from Hyclone. PPARδ null HCT-116 cells were a gift from Dr. Kinzler (Johns Hopkins School of Medicine) and generated by targeted homologous recombination. The detailed information for generating PPARδ null cell line has been previously described by Dr. Kinzler's group (Park et al., 2001). GW7845 and GW501516 were obtained from Ramidus AB (Sweden). Carbaprostacyclin (cPGI2) and 15-deoxy-Δ12,Δ14PGJ2 (15-PGJ2) were purchased from Cayman Chemical (Ann Arbor, MI). MG-132 is obtained from Enzo Life Sciences International (Plymouth Meeting, PA).
Real-time quantitative PCR
The mRNA levels of PPARδ, PPARγ, and survivin in CRC cells were quantified by real-time quantitative PCR using iCycler (BIO-RAD, Hercules, CA) and iQ™ SYBR Green Supermix (BIO-RAD, Hercules, CA). The real-time PCR assay was conducted previously described (Wang et al., 2006a).
Retroviral virus infection
Human PPARδ cDNA was cloned into retroviral expression vector pBMN-IRES-EGFP at Xho I and Not I sites. The pBMN-IRES-EGFP was obtained from Dr. Arteaga (Vanderbilt University School of Medicine) (Ueda et al., 2004). The sequence of PPARδ insert was confirmed before transfection. Retroviral expression vector MIEG3 and human survivin-MIEG3 plasmid were obtained from Dr. Pelus (Indiana University School of Medicine). Phoenix eco cells (ATCC, Manassas, VA) were transfected with 20 μg of MIEG3 vector, survivin, pBMN-IRES-EGFP vector, or PPARδ plasmid by FuGENE 6 according to the manufacturer's protocol (Roche Diagnostics Corp., Indianapolis, IN). Virus-containing medium was collected 48-72 h later and passed through a 45-μm filter. LS-174T cells were infected with the retrovirus supernatant containing MIEG3 vector, human survivin (MIEG3), pBMN-IRES-EGFP vector, or human PPARδ (pBMN-IRES-EGFP) as described (Fukuda et al., 2002). After 5 passages, cells stably expressing GFP were sorted and collected by flow cytometry. GFP expression was monitored with an inverted fluorescent microscope and maintained at 100% throughout all experiments.
Whole cell extracts and western blot analysis
Whole cell extracts were prepared from cells treated with either vehicle, GW7845 or/and GW501516 at the indicated times and dose in medium with 0.5% fat-free FBS for 24 h. Western blots were performed as described previously (Wang et al., 2005). The membrane was blocked with 5% dry milk in TBS-T buffer for 1 h and then incubated for 12-16 h at 4 °C in a 1:200 dilution of a PPARδ (Rockland, Gilbertsville, PA), PPARγ (Santa Cruz), survivin (Santa Cruz), Bcl-2 (Santa Cruz), PTEN (Santa Cruz), phospho-survivin (Thr 34) antibody (Santa Cruz), or cleaved PARP antibody (Cell Signaling, Beverly, MA) in TBS-T buffer containing 5% BSA. The blots were stripped and then reprobed with β-actin antibody.
Apoptosis assays
The cells (2.5 × 105/each well) were plated in 6-well plates. After culture overnight the cells were washed twice with PBS and then incubated in 0.5% fat-free FBS medium containing either 10% FBS, vehicle, GW7845, 15-PGJ2, or/and GW501516 or cPGI2 for indicated days. The percent of apoptotic cells was determined by flow cytometry using TACS™ Annexin V-FITC Apoptosis Detection Kit according to the manufacturer's instructions (R&D System, Inc., Minneapolis, MN). The combination of Annexin V-FITC and propidium iodide allowed for the differentiation between early apoptotic cells (Annexin V-FITC positive), late apoptotic cells (Annexin V-FITC and propidium iodide positive), necrotic cells (propidium iodide positive), and viable cells (both negative).
DNA fragment assays
The cells (1 × 105) were plated in 6-well plates and treated with vehicle or GW7845 as described earlier. For caspase inhibitor experiments, the cells were pretreated with indicated dose of caspase inhibitor I (R&D, Minneapolis, MN) and then treated with 5 μM of GW7845 for 2 days. The mono- and oligonucleosomes in apoptotic cells were determined by Cell Death Detection ELISA kit according to the manufacturer's instructions (Roche Diagnostics Corp., Indianapolis, IN).
Cell viability and clonogenic cell survival assays
The LS-174T cells (4 × 105/each well), HCT-116 (1.3 × 105/each well) were plated in 6-well plates. After culture 24 h the cells were washed twice with PBS and then incubated in 0.5% fat-free FBS medium containing either vehicle or GW7845 for indicated days. The viable cells were determined by trypan blue exclusion assays using Vi-Cell™XR (Beckman Coulter, Brea, CA). The colonies were formed in 9 days and stained by crystal violet.
Caspase-3 activity
The caspase-3 activity was measured by using a Caspase-3 Colorimetric Assay kit (R&D, Minneapolis, MN) according to the manufacturer's instructions. Briefly, the cells (2 × 106) were treated indicated concentration of GW7845, 15-PGJ2, or/and GW501516 or cPGI2 in 0.5% fat-free medium for 1 day and then harvested. Cell lysates were subjected to Caspase-3 colorimetric assay.
Supplementary Material
Supplemental Fig. 1. HCT-116 cells are more resistant to PPARγ agonist than LS-174T cells
(a-b) PPARγ agonist reduces viable cells of LS-174T and HCT-116 cells. The cells were cultured in media with 0.5% fat-free FBS and treated with GW7845 at 10 μM for indicated times. The viable cells were determined by trypan blue exclusion assays (a) and the colonies were stained by crystal violet (b).
Acknowledgments
We also thank the National Colorectal Cancer Research Alliance (NCCRA) for its generous support (RND).
This work is supported, in part, by the NIH MERIT award R37 DK47297, RO1 DK 62112, NCI P01 CA77839, and CPRIT RP100960.
Footnotes
Conflict of Interest: All authors declare no conflict of interest.
References
- Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- Asanuma K, Moriai R, Yajima T, Yagihashi A, Yamada M, Kobayashi D, et al. Survivin as a radioresistance factor in pancreatic cancer. Jpn J Cancer Res. 2000;91:1204–9. doi: 10.1111/j.1349-7006.2000.tb00906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat. 2003;79:391–7. doi: 10.1023/a:1024038127156. [DOI] [PubMed] [Google Scholar]
- Cellai I, Benvenuti S, Luciani P, Galli A, Ceni E, Simi L, et al. Antineoplastic effects of rosiglitazone and PPARgamma transactivation in neuroblastoma cells. Br J Cancer. 2006;95:879–88. doi: 10.1038/sj.bjc.6603344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di-Poi N, Michalik L, Tan NS, Desvergne B, Wahli W. The anti-apoptotic role of PPARbeta contributes to efficient skin wound healing. J Steroid Biochem Mol Biol. 2003;85:257–65. doi: 10.1016/s0960-0760(03)00215-2. [DOI] [PubMed] [Google Scholar]
- Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell. 2002;10:721–33. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- Farrow B, Evers BM. Activation of PPARgamma increases PTEN expression in pancreatic cancer cells. Biochem Biophys Res Commun. 2003;301:50–3. doi: 10.1016/s0006-291x(02)02983-2. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Foster RG, Porter SB, Pelus LM. The antiapoptosis protein survivin is associated with cell cycle entry of normal cord blood CD34(+) cells and modulates cell cycle and proliferation of mouse hematopoietic progenitor cells. Blood. 2002;100:2463–71. doi: 10.1182/blood.V100.7.2463. [DOI] [PubMed] [Google Scholar]
- Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, et al. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc Natl Acad Sci U S A. 2002;99:13771–6. doi: 10.1073/pnas.162480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Brockman JA, Sarraf P, Willson TM, DuBois RN. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J Biol Chem. 2001;276:29681–7. doi: 10.1074/jbc.M103779200. [DOI] [PubMed] [Google Scholar]
- Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat Med. 2004;10:245–7. doi: 10.1038/nm993. [DOI] [PubMed] [Google Scholar]
- Han S, Roman J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol Cancer Ther. 2006;5:430–7. doi: 10.1158/1535-7163.MCT-05-0347. [DOI] [PubMed] [Google Scholar]
- Hao CM, Redha R, Morrow J, Breyer MD. Peroxisome proliferator-activated receptor delta activation promotes cell survival following hypertonic stress. J Biol Chem. 2002;277:21341–5. doi: 10.1074/jbc.M200695200. [DOI] [PubMed] [Google Scholar]
- Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-delta attenuates colon carcinogenesis. Nat Med. 2004;10:481–3. doi: 10.1038/nm1026. [DOI] [PubMed] [Google Scholar]
- He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–45. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–57. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, et al. PPARgamma insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-kappaB signaling pathway. Oncogene. 2006;25:2736–47. doi: 10.1038/sj.onc.1209299. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998;58:5071–4. [PubMed] [Google Scholar]
- Kim EJ, Park KS, Chung SY, Sheen YY, Moon DC, Song YS, et al. Peroxisome proliferator-activated receptor-gamma activator 15-deoxy-Delta12,14-prostaglandin J2 inhibits neuroblastoma cell growth through induction of apoptosis: association with extracellular signal-regulated kinase signal pathway. J Pharmacol Exp Ther. 2003;307:505–17. doi: 10.1124/jpet.103.053876. [DOI] [PubMed] [Google Scholar]
- Kulke MH, Demetri GD, Sharpless NE, Ryan DP, Shivdasani R, Clark JS, et al. A phase II study of troglitazone, an activator of the PPARgamma receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J. 2002;8:395–9. doi: 10.1097/00130404-200209000-00010. [DOI] [PubMed] [Google Scholar]
- Kuo PC, Liu HF, Chao JI. Survivin and p53 modulate quercetin-induced cell growth inhibition and apoptosis in human lung carcinoma cells. J Biol Chem. 2004;279:55875–85. doi: 10.1074/jbc.M407985200. [DOI] [PubMed] [Google Scholar]
- Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, et al. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–7. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- Li M, Lee TW, Mok TS, Warner TD, Yim AP, Chen GG. Activation of peroxisome proliferator-activated receptor-gamma by troglitazone (TGZ) inhibits human lung cell growth. J Cell Biochem. 2005;96:760–74. doi: 10.1002/jcb.20474. [DOI] [PubMed] [Google Scholar]
- Nadra K, Anghel SI, Joye E, Tan NS, Basu-Modak S, Trono D, et al. Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor beta/delta. Mol Cell Biol. 2006;26:3266–81. doi: 10.1128/MCB.26.8.3266-3281.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor DS, Wall NR, Porter AC, Altieri DC. A p34(cdc2) survival checkpoint in cancer. Cancer Cell. 2002;2:43–54. doi: 10.1016/s1535-6108(02)00084-3. [DOI] [PubMed] [Google Scholar]
- Olie RA, Simoes-Wust AP, Baumann B, Leech SH, Fabbro D, Stahel RA, et al. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 2000;60:2805–9. [PubMed] [Google Scholar]
- Osawa E, Nakajima A, Wada K, Ishimine S, Fujisawa N, Kawamori T, et al. Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology. 2003;124:361–7. doi: 10.1053/gast.2003.50067. [DOI] [PubMed] [Google Scholar]
- Panigrahy D, Huang S, Kieran MW, Kaipainen A. PPARgamma as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biol Ther. 2005;4:687–93. doi: 10.4161/cbt.4.7.2014. [DOI] [PubMed] [Google Scholar]
- Park BH, Vogelstein B, Kinzler KW. Genetic disruption of PPARdelta decreases the tumorigenicity of human colon cancer cells. Proc Natl Acad Sci U S A. 2001;98:2598–603. doi: 10.1073/pnas.051630998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, et al. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta) Mol Cell Biol. 2000;20:5119–28. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino MV, Kelley MF, Jayyosi Z. Promotion of colon tumors in C57BL/6J-APC(min)/+ mice by thiazolidinedione PPARgamma agonists and a structurally unrelated PPARgamma agonist. Toxicol Pathol. 2004;32:58–63. doi: 10.1080/01926230490261320. [DOI] [PubMed] [Google Scholar]
- Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, et al. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat Med. 1998;4:1058–61. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–10. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- Sarela AI, Scott N, Ramsdale J, Markham AF, Guillou PJ. Immunohistochemical detection of the anti-apoptosis protein, survivin, predicts survival after curative resection of stage II colorectal carcinomas. Ann Surg Oncol. 2001;8:305–10. doi: 10.1007/s10434-001-0305-0. [DOI] [PubMed] [Google Scholar]
- Sarraf P, Mueller E, Jones D, King FJ, DeAngelo DJ, Partridge JB, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med. 1998;4:1046–52. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- Takayama O, Yamamoto H, Damdinsuren B, Sugita Y, Ngan CY, Xu X, et al. Expression of PPARdelta in multistage carcinogenesis of the colorectum: implications of malignant cancer morphology. Br J Cancer. 2006;95:889–95. doi: 10.1038/sj.bjc.6603343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teresi RE, Shaiu CW, Chen CS, Chatterjee VK, Waite KA, Eng C. Increased PTEN expression due to transcriptional activation of PPARgamma by Lovastatin and Rosiglitazone. Int J Cancer. 2006;118:2390–8. doi: 10.1002/ijc.21799. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Wang S, Dumont N, Yi JY, Koh Y, Arteaga CL. Overexpression of HER2 (erbB2) in human breast epithelial cells unmasks transforming growth factor beta-induced cell motility. J Biol Chem. 2004;279:24505–13. doi: 10.1074/jbc.M400081200. [DOI] [PubMed] [Google Scholar]
- Wang D, Buchanan FG, Wang H, Dey SK, DuBois RN. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Ras-mitogen-activated protein kinase cascade. Cancer Res. 2005;65:1822–9. doi: 10.1158/0008-5472.CAN-04-3671. [DOI] [PubMed] [Google Scholar]
- Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, et al. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006a;203:941–951. doi: 10.1084/jem.20052124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Wang H, Guo Y, Ning W, Katkuri S, Wahli W, et al. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc Natl Acad Sci U S A. 2006b;103:19069–74. doi: 10.1073/pnas.0607948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Wang H, Shi Q, Katkuri S, Walhi W, Desvergne B, et al. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004;6:285–95. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Williams CW, Luongo C, Radhika A, Zhang T, Lamps LW, Nanney LB, et al. Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology. 1996;111:1134–1140. doi: 10.1016/s0016-5085(96)70083-5. [DOI] [PubMed] [Google Scholar]
- Yin Y, Russell RG, Dettin LE, Bai R, Wei ZL, Kozikowski AP, et al. Peroxisome proliferator-activated receptor delta and gamma agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Res. 2005;65:3950–7. doi: 10.1158/0008-5472.CAN-04-3990. [DOI] [PubMed] [Google Scholar]
- Zaffaroni N, Daidone MG. Survivin expression and resistance to anticancer treatments: perspectives for new therapeutic interventions. Drug Resist Updat. 2002;5:65–72. doi: 10.1016/s1368-7646(02)00049-3. [DOI] [PubMed] [Google Scholar]
- Zhang T, Otevrel T, Gao Z, Gao Z, Ehrlich SM, Fields JZ, et al. Evidence that APC regulates survivin expression: a possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res. 2001;61:8664–7. [PubMed] [Google Scholar]
- Zhao J, Tenev T, Martins LM, Downward J, Lemoine NR. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J Cell Sci. 2000;113 Pt 23:4363–71. doi: 10.1242/jcs.113.23.4363. [DOI] [PubMed] [Google Scholar]
- Zuo X, Peng Z, Moussalli MJ, Morris JS, Broaddus RR, Fischer SM, et al. Targeted genetic disruption of peroxisome proliferator-activated receptor-delta and colonic tumorigenesis. J Natl Cancer Inst. 2009;101:762–7. doi: 10.1093/jnci/djp078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. HCT-116 cells are more resistant to PPARγ agonist than LS-174T cells
(a-b) PPARγ agonist reduces viable cells of LS-174T and HCT-116 cells. The cells were cultured in media with 0.5% fat-free FBS and treated with GW7845 at 10 μM for indicated times. The viable cells were determined by trypan blue exclusion assays (a) and the colonies were stained by crystal violet (b).