

Abstract
Spinal muscular atrophy (SMA) is a neurodegenerative disease caused by the loss of a single gene, Survival Motor Neuron-1 (SMN1). Administration of a self-complementary Adeno-Associated Virus vector expressing full-length SMN cDNA (scAAV-SMN) has proven an effective means to rescue the SMA phenotype in SMA mice, either by intravenous (IV) or intracerebroventricular (ICV) administration at very early time points. We have recently shown that ICV delivery of scAAV9-SMN is more effective than a similar dose of vector administered via an IV injection, thereby providing an important mechanism to examine a timeline for rescuing the disease and determining the therapeutic window in a severe model of SMA. In this report, we utilized a relatively severe mouse model of SMA, SMNΔ7. Animals were injected with scAAV9-SMN vector via ICV injection on a single day, from P2 through P8. At each delivery point from P2 through P8, scAAV9-SMN decreased disease severity. A near complete rescue was obtained following P2 injection while a P8 injection produced a ∼40% extension in survival. Analysis of the underlying neuromuscular junction (NMJ) pathology revealed that late-stage delivery of the vector failed to provide protection from NMJ defects despite robust SMN expression in the central nervous system. While our study demonstrates that a maximal benefit is obtained when treatment is delivered during pre-symptomatic stages, significant therapeutic benefit can still be achieved after the onset of disease symptoms.
INTRODUCTION
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder caused by the mutation of a single gene, Survival Motor Neuron-1 (SMN1) (1). Humans possess a nearly identical gene, SMN2, which can produce fully functioning SMN protein, but only at low levels. A non-polymorphic difference between these transcripts exists. A translationally silent C to T transition located 6 bases into exon 7 of SMN2 (2) results in aberrant splicing in ∼85–90% of SMN2-derived transcripts (3). The alternatively spliced SMN2 transcripts lack exon 7 (SMNΔ7). The SMNΔ7 protein cannot self-associate (4) or form protein/RNA complexes efficiently (5), rendering it less stable and therefore quickly degraded (6). Importantly, however, even with low SMN production, SMN2 can partially compensate for the loss of SMN1; further highlighting SMN2 as an important disease modifier because more copies of SMN2 translate into decreased disease severity (7). SMN is ubiquitously expressed and is intimately involved in the biogenesis of nascent snRNPs, including the major and minor pathways. Downstream targets of SMN depletion have also been identified, such as Stasimon (8), raising the possibility that a discrete subset of cellular genes contribute to the complex SMA pathology. Alternatively, SMN also interacts with a host of non-snRNP proteins within axonal processes, suggesting that a non-snRNP-related function gives rise to disease (9).
Currently, no effective treatment exists for SMA; however, a number of therapeutic strategies have arisen based upon several important criteria: (i) SMA is a monogenic disease; (ii) the disease-causing gene is known; and (iii) SMN2 encodes a wild-type SMN protein and is present in all SMA patients. Recent advances on several fronts have provided encouraging pre-clinical results, including small molecules, antisense oligonucleotides and vector-based gene replacement (10).
As new therapeutics advance, understanding the temporal requirement for SMN will be important, particularly as neonatal screening for SMA is not routinely performed. To examine temporal SMN requirements in SMA mouse models, tet- or Cre-inducible lines have been developed (11,12). In the tet-inducible lines, a P0/P1 induction resulted in SMN increases by P3 and conferred significant protection from disease development, resulting in an average life span of ∼86 days compared with uninduced animals that lived 13 days. P2 induction resulted in P5 SMN expression and an average survival of 25 days. These results suggest that there is a very narrow window to deliver SMN and significantly impact disease severity. In a small cohort in which tetracycline was delivered for the first 28 days and was subsequently withdrawn, five animals died within a few weeks, while two animals lived for longer than 9 months. While the sample size was small, these results suggested that high levels of SMN could be required early during neonatal development, but that a lower maintenance threshold exists that is sufficient for survival. Tamoxifen induction of SMN in a similarly severe model of SMA demonstrated that SMN induction at early time points was more beneficial than later, as tamoxifen delivery at P4, P6, P8 and P10 resulted in life spans averaging ∼139, 50, 20 and 11 days, respectively (12). The genetic approach has clear biological implications, but it is more difficult to extrapolate these results to a therapeutic window. To address this, we have initiated time-point studies with a gene therapy vector shown to provide a dramatic rescue of the SMA phenotype.
SMN gene therapy with a self-complementary adeno-associated virus serotype 9 (scAAV9) has resulted in the greatest extension in survival whether the vector was delivered systemically or directly into the CNS (13–19). AAV9 has broad tissue tropism with a high transduction rate into motor neurons. As SMA is a motor neuron disease, it is appropriate that this serotype be utilized to specifically target the CNS (20–22). We have performed a systematic analysis, utilizing the well-characterized SMNΔ7 model, to determine the effects of delaying scAAV9-SMN administration. We treated each mouse with a single injection of scAAV9-SMN [1 × 1011 viral genomes (v.g.)] at a single time point, P2 through P8. Early SMN induction provided the most benefit, whereas injection at later time points decreased the efficacy of the vector. All treated mice lived significantly longer, but SMN induction at the earliest time point (P2) provided the most robust rescue with the fewest early deaths. While SMN induction in the CNS and periphery was observed, NMJ pathology was largely uncorrected at later time points, indicating that even high levels of SMN protein could not correct the phenotype once the disease has progressed to this advanced stage. Collectively, the earliest administrations provided the greatest degree of phenotypic rescue. Importantly, however, even at late time points, a significant extension in survival was observed, suggesting that even symptomatic delivery of an effective SMN-inducing therapeutic could impact the course of disease.
RESULTS
Intracerebroventricular injection of scAAV9-SMN increases survival of SMNΔ7 mice
A single intracerebroventricular (ICV) injection of 1 × 1011 v.g. was able to significantly extend the life span of all treatment groups (Fig. 1A). As expected, earlier treatment resulted in a more robust life span rescue. Pups injected on P2 experienced no early deaths and all mice lived past 130 days. As treatment was delayed, the average life span steadily declined, with the one exception that P4-injected animals lived longer than the P3 group (Fig. 1A). Overall, mice injected with scAAV9-SMN at earlier time points had a greater extension in survival compared with mice injected at later time points. Importantly, even the late-stage injected mice received therapeutic benefit as mice from all groups lived significantly longer than non-injected SMA controls (Fig. 1B) which lived on average, 13 days with a median survival of 14 days.
Figure 1.

ICV injection of scAAV9-SMN significantly increases the survival of SMNΔ7 mice when administered at early time points. (A) Kaplan–Meier survival curve of untreated SMNΔ7 mice and those injected with 1 × 1011 v.g. of scAAV9-SMN on a single day P2–P8. Mice injected at the earliest time point P2 did not exhibit early deaths and they lived the longest with an average life span of 187 days and a median survival of 204 days. P3-injected mice lived up to 182 days (avg 102, median 75), the P4 group lived up to 211 days (avg 141, median 167), the P5 group lived up to 211 days (avg 76, median 37), the P6 group lived up to 211 days (avg 73, median 34), the P7 group lived up to 70 days (avg 30, median 28), the P8 group lived up to 22 days (avg 18, median 18) and the non-injected group lived up to 15 days (avg 13, median 14). (B) P-table demonstrating statistically significant differences in survival of mice in the treatment and non-treatment groups. All treated mice lived significantly longer than non-injected controls. P-values were calculated using the log-rank Mantel–Cox test and the table illustrates the significance in survival between groups. NI, non-injected. (n.s. no significance, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001).
Intracerebroventricular injection of scAAV9-SMN increases weight of SMNΔ7 mice
Weight gain was analyzed as the average weight for a given group across days (Fig. 2A) and also as the average percent weight gained from birth to peak (Fig. 2B). A statistical comparison of weight gain between groups is described in the P-table (Fig. 2C). At birth, all SMA pups and unaffected pups were similar in weight and there were no obvious phenotypic differences until around Day 7 (Fig. 2D). However, by P11, all treated mice weighed significantly more than non-injected SMA animals. Even the late injected mice (P7 and P8) weighed significantly more, although this difference was not as great as the difference observed between early-injected mice and the non-injected SMA controls. Around P10, the weight of P8 and non-injected SMA mice peaked and subsequently declined. By P14, P8-injected animals weighed significantly less than mice from all other treatment groups (Fig. 2A).
Figure 2.

SMNΔ7 mice treated with scAAV9-SMN gain weight throughout their life span. (A) The average weight per group is plotted across days for the surviving animals in each cohort. ni = initial number of animals in each group. (B) Percent weight gained from birth to peak. SMNΔ7 mice treated with scAAV9-SMN gain more weight than non-injected controls. Percent weight gained from birth to peak for all treated animals is significantly higher compared with non-injected mice. Weight gain for unaffected heterozygous littermates is shown here to provide a visual comparison of treated mice to healthy unaffected individuals. (C) P-table demonstrating statistically significant differences in the average weight gained from birth to peak for all mice in the treatment and non-treated groups. SMNΔ7 mice treated with scAAV9-SMN gain significantly more weight than untreated littermates. Student's t-tests were performed to compare the average percent weight gained from birth to peak between groups. (n.s. no significance, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). (D) Representative images of SMNΔ7 mice. (Left) Phenotypic differences are not apparent at birth and the pups of the various genotypes cannot be distinguished by observation. (Right) At 9 days old, the pup injected at the earliest time point (P2) has gained weight comparable to the unaffected heterozygous littermate; and the pups injected at the latest time points (P7 and P8) are slower to gain weight.
All treated animals exhibited a significantly higher percent weight gain from birth to their peak weight compared with non-injected SMA animals. Treated animals tended to fall into two groups. Those injected on P2, P3 or P4 displayed similar weight gain from birth to peak, and there was no significance when compared with each other. Similarly, animals injected on P5, P6 or P7 had comparable percent weight gain and also did not show significance when compared with one another.
Early SMN replacement improves motor function in mice injected with scAAV9-SMN
Mice have a natural instinct to right themselves onto all four paws immediately after being placed on their back. Young, healthy neonates achieve this ability within the first week of life (23). Thus, observing this ability in mice serves as an early test of motor function, known as the time-to-right test. Mice were given 30 s to perform this test. Over time, a greater percentage of animals from groups P2 through P7 gained the ability to right themselves and to do so at a faster rate (Fig. 3A and B). However, this was not the case for the untreated animals and those injected at the latest time point, P8 (Fig. 3A and B). An analysis of specific trial times at a time at which untreated animals were fully symptomatic, P14, demonstrates that there is still variability in the response to the scAAV9-SMN vector, however, it is clear that earlier treatment results in a greater degree of recovery (Fig. 3C).
Figure 3.

Time-to-right test results shows that SMNΔ7 mice treated with scAAV9-SMN at earlier time points perform substantially better. (A) Percentage of animals able to right themselves. Assessment of motor function shows that SMNΔ7 mice treated with scAAV9-SMN at earlier time points perform substantially better on the time-to-right test. Animals treated at early time points gain the ability to right themselves prior to animals injected at later time points. (B) Average time-to-right for SMNΔ7 mice treated with scAAV9-SMN. Mice injected at later time points exhibit less muscle control and turn slower than mice injected at earlier time points. (C and D) Individual time-to-right on Day 14. At 2 weeks, earlier treated animals perform better on the time-to-right test compared with those injected at later time points. There is a high degree of variability observed within groups on Day 14; where some mice are able to right themselves more quickly than others, and some animals never gain the ability to turn. (D) Mice able to right themselves within 0–5 s was recorded as a success, 6–24 s demonstrated average performance and those from 25 to 30 s were considered failures.
Of the mice that are able to right themselves, the early-injected animals tended to right faster than those injected at later time points and the non-injected SMA controls. On P14, righting time increased with delayed treatment and untreated animals were no longer able to right themselves within the 30 s allotted (Fig. 3D). Overall, earlier treatment produced more robust motor function improvements as measured by time-to-right.
Increased SMN protein was observed in animals treated with scAAV9-SMN
SMNΔ7 mice were injected on P2 or P7 and their tissues harvested on P11. As expected, P2-injected mice had equal or higher levels of SMN protein when compared with unaffected mice due to the large dose of viral particles injected (Fig. 4A). Mice injected on P2 exhibited significantly higher SMN levels in the brain and spinal cord compared with P7-injected mice (Fig. 4A). Both P2- and P7-injected animals had significantly more SMN protein than age-matched, non-injected SMA control mice.
Figure 4.
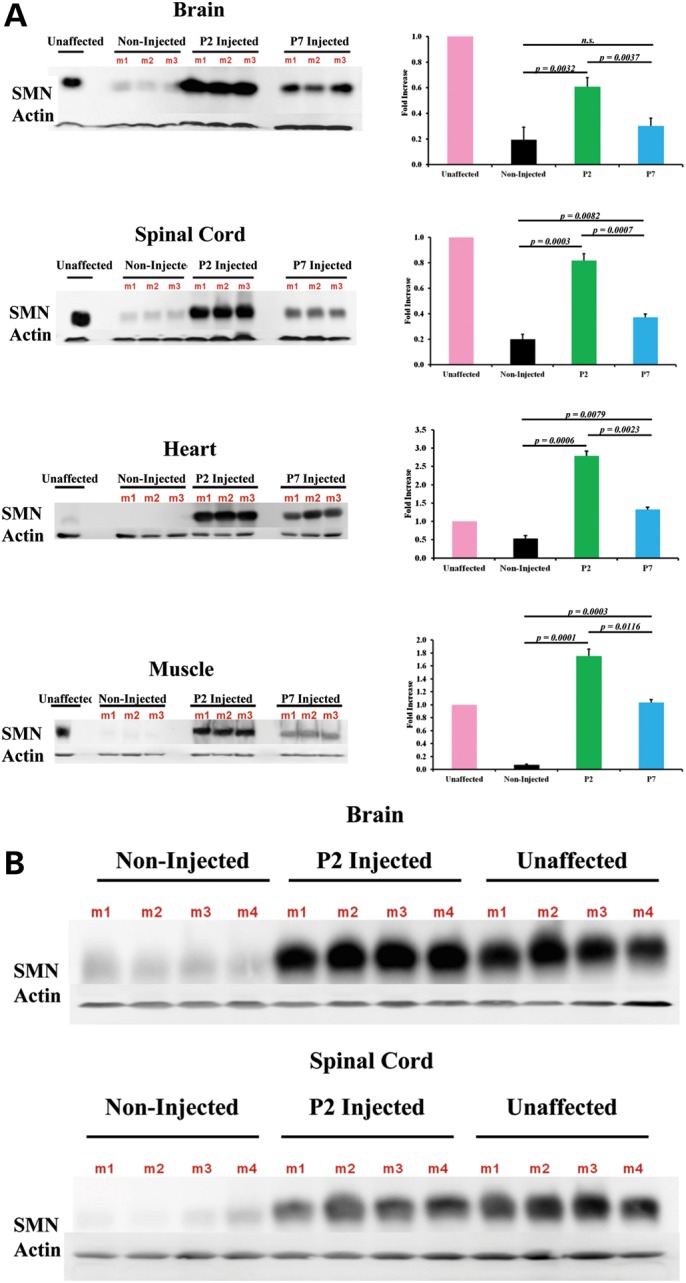
SMN protein induction is evident in SMNΔ7 mice injected at early and late time points. (A) In the brain, SMN was upregulated significantly in the P2 mice compared with both the P7 and non-injected groups, but there was no significance observed between P7 and the non-injected group. There was robust expression observed in the spinal cord where both P2- and P7-injected mice expressed significantly more protein than non-injected controls and the P2 group exhibited a significantly higher level of protein than the P7 group. In the heart, both the P2- and P7-injected mice exhibit a significant induction of SMN. The fold difference of the western blots (n = 3 for each group) is shown as the average SMN/actin ratio after normalization of SMN to β-actin. All tissues were harvested on Day 11. (B) Robust SMN expression is observed 4 days post therapeutic P2 administration, confirming that equal protein induction intensity is consistent with the same time frame as P7 administration. All tissues were harvested on Day 6.
Robust SMN protein induction was also observed in peripheral organs such as the heart and skeletal muscle (Fig. 4A). In the heart tissue, both P2- and P7-injected animals express significantly more SMN than non-injected controls (P = 0.0006 and P = 0.0079, respectively). Furthermore, scAAV9-SMN administration on P2 resulted in significantly higher SMN levels in the heart compared with the P7 group (P = 0.0023). The same pattern of induction was observed in the muscle with P2-injected animals displaying more robust SMN protein levels than P7-treated mice (Fig. 4A).
It was formally possible that the P2-treatment group had more robust protein, when compared with the P7 group, because the virus was allowed to express for 5 days longer. To address this, P2-treated animals were harvested on P6 in order to compare protein levels after equal times of viral expression in both the P2 and P7 groups. The robust protein levels observed in P2-injected animals harvested at P11 were still observed in P2-injected mice harvested at P6 (Fig. 4B).
Timing of protein induction correlates with skeletal muscle pathology
Triceps muscle fiber size from unaffected and non-injected control mice, along with P2- and P7-injected groups were compared. Tissues were harvested on Day 11 and stained with hematoxylin and eosin for visualization. Unaffected mice exhibited healthy muscle tissue with a normal distribution of small and large muscle fibers (Fig. 5A). In contrast, the tissues from non-injected SMA mice displayed smaller fibers. P2- and P7-injected mice exhibit healthy-looking tissue, compared with non-injected animals, although the overall muscle pathology was not completely restored. In the triceps, we observed a significant increase in muscle fiber area of P2 mice compared with P7 (P = 0.0023) and non-injected animals (P = 0.0008); although still smaller than unaffected mice (P = 0.0007). Similarly, P7 muscle fibers were significantly larger than non-injected mice (P = 0.0088), but smaller than unaffected controls (P = 0.0002) (Fig. 5B). Furthermore, restoration of the muscle, as assessed by the distribution of small and large fibers in the P2 and P7 cohorts, correlates with timing of SMN induction (Fig. 5C and Supplementary Material, Fig. S2). These findings suggest that early SMN replacement has the ability to reduce muscle atrophy. To determine the extent to which scAAV9-SMN traverses the blood–brain barrier (BBB) and is distributed throughout the periphery, we utilized the scAAV9-GFP vector to observe virus dispersion. Unaffected mice were injected with scAAV9-GFP on P2 or P7 with the heart, liver, spleen and kidney tissues harvested on P6 or P11 (Supplementary Material, Fig. S1). Based upon extensive GFP expression in all treated animals, the results demonstrate that the virus was well distributed throughout the periphery, especially in the liver and heart tissues.
Figure 5.
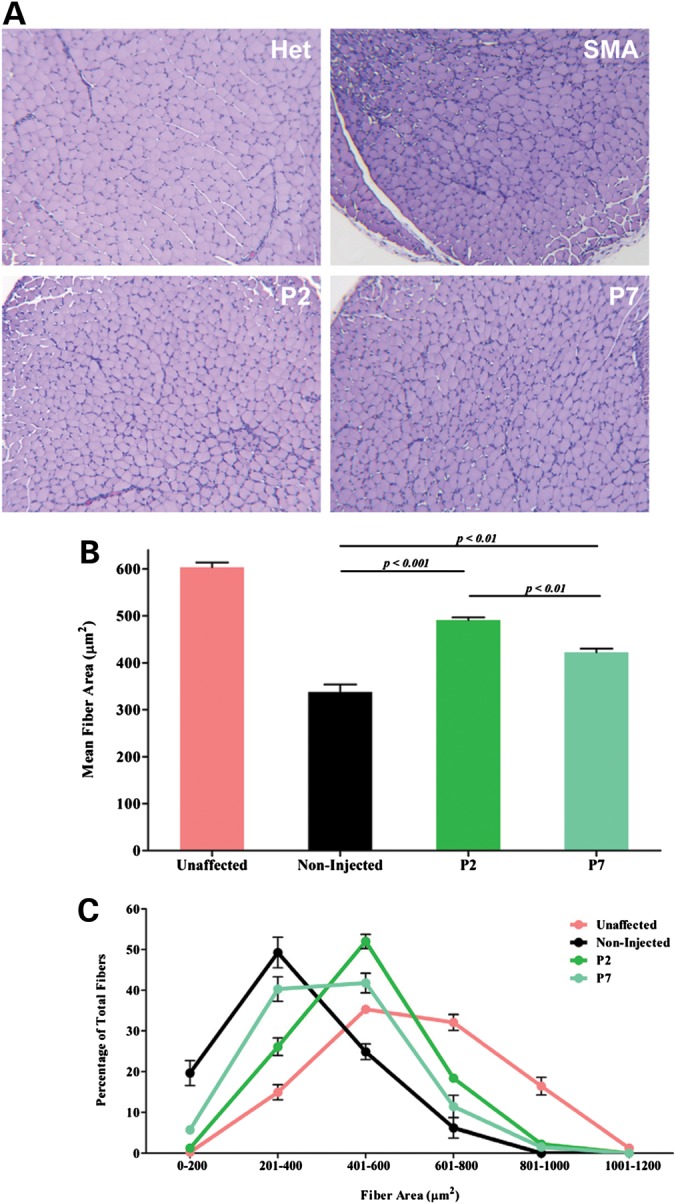
Early SMN restoration correlates with improved muscle pathology. (A) Representative images of triceps muscle cross-sections. Triceps muscles were dissected from P11 mice and stained with hematoxylin and eosin. (B) Mean fiber area was quantified using Metamorph® Software. Data are presented as the mean ± SEM. SMN induction on P2 or P7 leads to a significant increase in muscle fiber area compared with non-injected SMA mice, but both groups were significantly smaller than unaffected controls (Smn+/−;SMN2+/+;SMNΔ7+/+). However, SMN delivery at the earliest time point (P2) resulted in significantly larger muscle fibers compared with mice treated at the later P7 time point. P-values were calculated using Student's t-test. (n = 3 for each group). (C) Muscle fiber size distribution represented in percentage of total fibers.
Early scAAV9-SMN administration results in more profound NMJ phenotype correction
Neuromuscular junction defects have been well documented in SMA models, including the demonstration that a subset of muscles is particularly vulnerable to SMN deficiency, such as the longissimus capitis. Therefore, we investigated the effects of early and late SMN restoration in the longissimus capitis; a proximal muscle required for head stability and movement. SMNΔ7 mice were injected on P2 or P7 and harvested on P12, along with SMA and wild-type mice. Neurofilament accumulation and endplate structure were analyzed by immunofluorescence. While similar numbers of NMJs and axonal projections were observed, the endplate morphology in the P7-treated tissues strongly indicated a developmental delay or inability to appropriately innervate as the majority of endplates lacked the well-defined ‘pretzel-configuration’ consistent with well-developed mature end plates (Fig. 6A). As anticipated, tissue from the untreated SMA cohort was poorly innervated, consistent with previously published observations. The P2-injected NMJs closely resembled the unaffected samples at P14 where endplates were well innervated and largely free of neurofilament accumulations (Fig. 6A). While P7-treated mice exhibited all stages of NMJ development, P7-treated NMJs displayed a greater number of partially innervated endplates and a reduced number of fully innervated endplates compared with P2-treated tissues (Fig. 6B). Our results strongly suggest that there is a time limit in which SMN replacement is most beneficial to NMJs; however, reinstatement after the onset of symptoms (P7) still provides a modest improvement to NMJ pathology.
Figure 6.
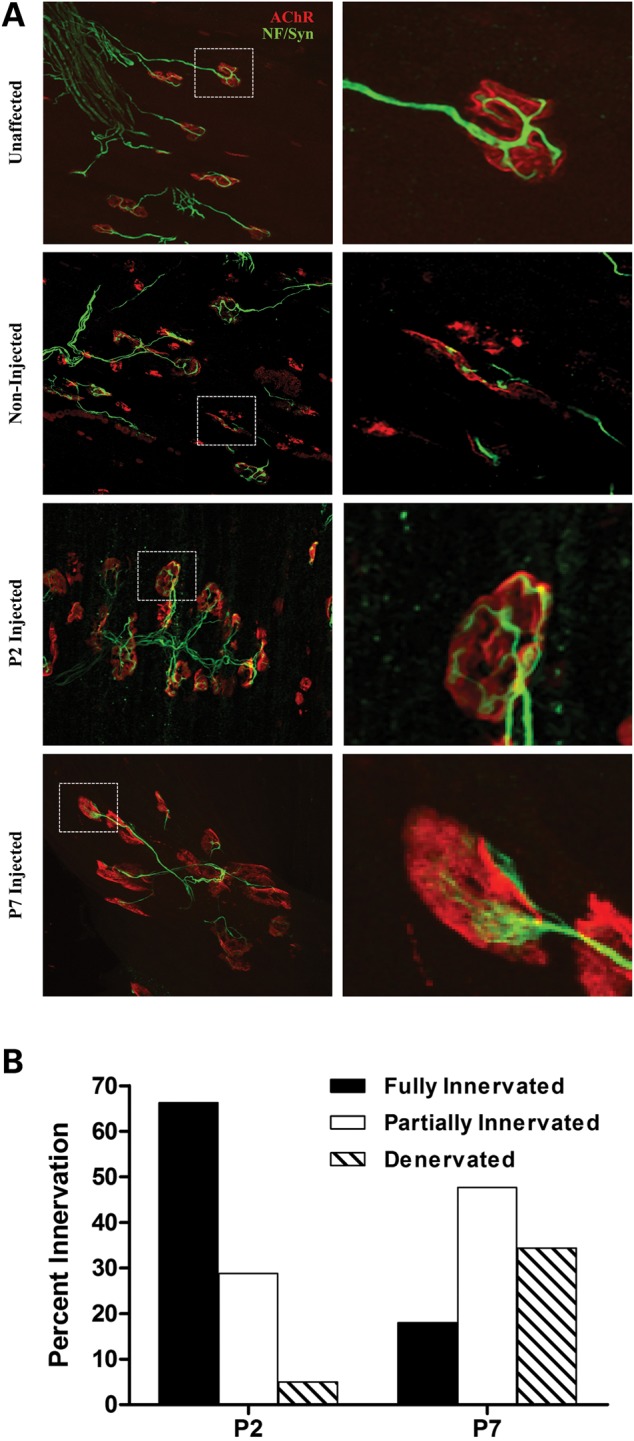
Timing of SMN restoration correlates with improved NMJ pathology. (A) Representative confocal images of NMJ pathology on P12 in the vulnerable muscle, longissimus capitis, of unaffected, non-injected and P2- or P7-treated mice. (Left) ×40 magnification of respective tissues; (Right) Enlargement of inset. Presynaptic components (green) were immunostained with anti-synaptophysin (Syn) and anti-neurofilament (NF). Acetylcholine receptors (AChRs) are labeled with alpha-bungarotoxin (red). Unaffected mice exhibited well-innervated NMJs with mature pretzel-shape endplates. Non-injected SMA mice exhibit a high degree of denervation and an increased number of immature endplates. scAAV9-SMN administration on P2 restores NMJ defects as observed by the fully innervated, mature endplates. NMJ pathology is only partially restored following a delayed delivery (P7). (B) Bar graph representing fully innervated (black), partially innervated (open) and denervated (hatched) for P2- or P7-injected SMA mice harvested on P12. Student's t-test: P2/P7 fully innervated: P < 0.01; partially innervated: P < 0.01; denervated: P < 0.05.
DISCUSSION
SMA is caused by low levels of SMN protein (2). In humans, the SMN2 gene, despite its inability to compensate for the loss of SMN1, is a critical disease modifying gene as it is able to produce low levels of full-length SMN protein (7). Therapeutic approaches for the treatment of SMN can be broadly categorized into two groups: those that modulate SMN2 splicing to generate more full-length transcripts and direct SMN replacement. Designing an effective therapeutic using either strategy requires knowledge of the tissue-specific and temporal requirements for SMN.
In an effort to elucidate the latter, we performed a thorough analysis of SMN replacement via scAAV9 at sequential postnatal time points in the SMNΔ7 mouse model of SMA. First, we confirmed that SMN replacement is able to fully rescue this model when given on P2 as has been previously reported (15). Secondly, we observed that as the treatment is delayed, the resulting phenotypic benefit achieved is diminished. However, even when treatment was administered when overt SMA-like symptoms were present (P8), a significant, albeit lesser, phenotypic improvement was seen. The notable exception was with the P4- and P3-treated animals in which P4-treated provided a greater benefit regarding life span, however, percent weight gain was not statistically significant. The most likely explanation for the difference in life span involves the number of animals in each treatment group (n = 9), although there is a formal possibility that there is a stage in development that responds more favorably to AAV-mediated gene replacement.
It is encouraging to see that our investigation into the therapeutic window produced similar results to transgenic approaches previously described. In an inducible transgenic model, whereby SMN is induced by administration of doxycycline, embryonic or early postnatal induction of SMN resulted in a dramatic rescue of SMA mice. Conversely, later induction (P6) resulted in a significantly reduced rescue suggesting that there is a critical window in which SMN must be expressed in order to effectively rescue (11). Similarly, in a mouse model harboring a Cre-inducible SMN rescue allele, early induction (P4) of SMN resulted in the most profound rescue. Induction at later time points (P6, P8, P10) resulted in a decreasingly substantial phenotypic rescue, with mice receiving SMN induction at P10 not being significantly different than SMA mice in terms of life span (12).
Other therapeutic approaches to investigate temporal requirements of SMN have shown similar results as well. However, those conducted have yielded similar results to our study and the aforementioned transgenic approaches. A study investigating scAAV9-SMN IV delivery at four postnatal time points (P1, P2, P5 and P10) in the SMNΔ7 model showed that injection at P1 and P2 produced a full life span rescue. At P5, the rescue is much less pronounced. By P10, delivery of scAAV9-SMN no longer produces any phenotypic benefit (15). The key difference between this study and ours lies in the injection technique as the same viral dose was used (1 × 1011 v.g.). ICV injection at P5 resulted in ∼30% of mice surviving past 100 days, while IV injection produced a maximum life span of 30 days highlighting the importance of route of delivery (16,17).
In the severe mouse model, in which symptoms are evident at birth, delivery of 2 × 1011 v.g. at P1 produces a significant extension of life span (7.5–17 days) (17). Thus, delivery of scAAV9-SMN after the onset of symptoms in both the SMNΔ7 and severe models is able to modestly improve survival; however, not to the same extent as delivery prior to symptom onset.
Another therapeutic approach, antisense technology, is often utilized in SMA research to increase SMN protein levels by modulating SMN2 splicing. Antisense oligomers (ASOs) to block an intronic splice silencer, ISS-N1, have been demonstrated to significantly increase full-length SMN2 transcript levels and SMN protein in the brain and spinal cord of the SMNΔ7 model (24,25). When the ISS-N1 ASO was administered by ICV injection on P1, survival was extended from 15 to 100 days. However, delaying delivery until P4 decreased the efficacy of the antisense therapy. Furthermore, IV administration on P1 resulted in survival comparable to P1 ICV treatment. However, when delivery was delayed until P4, the IV injected group exhibited decreased survival compared with the ICV-treated group. Taken together, these studies highlight the need for early, pre-symptomatic, therapeutic intervention in order to achieve maximum therapeutic benefit. However, in the extent that these results can be extrapolated to clinic, our study and the others mentioned offer hope for therapeutic benefit even in symptomatic patients.
Understanding why scAAV9-SMN efficiency diminishes when treatment is delayed would shed light on disease pathogenesis and aid in the development of AAV vectors as therapeutics. One possibility is the inability of the virus to escape from the CNS following an ICV injection at later time points. As many peripheral organs and tissues have been reported to be affected in both SMA patients and animal models (reviewed in 26), the ability scAAV9 to cross the BBB is likely necessary to achieve a full life span rescue. It is known that scAAV9 crosses the BBB in P2 neonatal mice (21). However, the ability of scAAV9 to permeate the BBB at later time points is not known. To investigate this, we injected scAAV9-GFP into unaffected animals and observed GFP expression in peripheral organs (Fig. 6). Our findings demonstrate the even when scAAV9-GFP is delivered on P7, robust GFP expression in all peripheral organs examined is observed. Thus, inability of the virus to cross the BBB is not the reason for the lack of rescue observed in mice treated at later time points.
As low levels of SMN result in the specific loss of lower motor neurons, SMN replacement in those cells is critical for AAV-mediated gene therapy. Previous studies have shown that following a P2 IV injection of scAAV9-GFP, ∼42% of lumbar spinal motor neurons were transduced (21). Here we utilized an ICV delivery because our prior work in severe SMA mouse models demonstrated that direct injection into the central nervous system provided an enhanced phenotypic rescue compared with IV delivery (16,17). SMN expression throughout the spinal column in these P2-treated mice was greatly enhanced in the ICV-treated cohort, suggesting elevated transduction compared with IV-treated samples. However, it is expected that transduction levels would be equal to or more than that observed following an IV injection. It is not known if or how transduction levels change as treatment is delayed. Growing evidence suggests that additional cell types in the CNS beyond motor neurons contribute to the overall pathology of SMA, including astrocytes, and Schwann cells (27,28). Therefore, it is possible that the astrogliosis observed in SMNΔ7 mice has progressed to a point that leads to irreparable damage, consistent with the loss of >50% of the total motor neuron population (28). Alternatively, all though not to the exclusion of the astrogliosis, the reduced motor neuron numbers observed by others (28) may simply indicate that sufficient motor neuron targets no longer exist since even a P7 injection would likely not result in robust transgene expression until P9, at which time a percentage of SMNΔ7 mice have already died. Even if motor neurons are present, they may be functionally compromised. Therefore, it remains a possibility that an insufficient population of functional motor neurons is available at later time points, rendering SMN replacement less beneficial.
Another possibility for the lack of rescue may be the accumulation of irreversible neuromuscular junction (NMJ) defects. It has been previously reported that at P2, NMJs in SMA and unaffected littermates are indistinguishable (29,30). However, by P5 ∼25% of all nerve terminals in SMA mice appear thick and swollen with abnormal NF aggregates. As the animals age, there is a progressive increase in the number of such defective terminal axons, reaching ∼90% by P14. Additionally, SMA axons fail to form the fine terminal arbors characteristically found as early as P8 at normal NMJs. There have been varying reports as to the appearance of unoccupied endplates in SMA animals, with some groups reporting significant denervation and other reporting a lack thereof (29,30). However, the absence of anatomically denervated endplates in SMA mice does not suggest that these endplates have maintained their functionality. Further investigation will be needed to determine if these NMJ defects contribute to the inability of sAAV9-SMN to rescue when administration is delayed.
Understanding the underlying reason as to the insufficiency of later therapeutic treatment to fully rescue may aid in the development of new therapeutics and the betterment of existing ones. Nonetheless, early therapeutic intervention may be critical for therapeutic success.
MATERIALS AND METHODS
Animal handling and genotyping
All animals were housed and treated according to the guidelines of the Animal Care and Use Committee at the University of Missouri and the regulations defined in the ‘Guide for the Care and Use of Laboratory Animals,’ (National Research Council, 2011). The SMN delta7 (SMNΔ7) mouse model was utilized for these experiments. SMNΔ7 mice are null for mouse Smn and contain two copies of both transgenes, human SMN2 and the cDNA coding sequence for SMNΔ7. Mice heterozygous for mSmn (Smn+/−; SMN2+/+; SMNΔ7+/+) were bred to generate mSmn knock-out mice (Smn−/−; SMN2+/+; SMNΔ7+/+). Heterozygous (Smn+/−; SMN2+/+; SMNΔ7+/+) mice are used throughout all the experiments as unaffected positive controls and untreated SMA (Smn−/−; SMN2+/+; SMNΔ7+/+) mice were used as negative controls. Pups were genotyped on their day of birth, indicated as postnatal Day 1 (P1), using a small piece of tissue from a tail biopsy; and numbered daily using non-toxic, permanent marker to identify individual pups. Primers were used to amplify the mSmn wild-type allele, forward (5′-TCTGTGTTCGTGCGTGGTGACTTT-3′) and reverse (5′-CCCACCACCTAAGAAAGCCTCAAT-3′) and the lacZ mSmn knockout allele, forward (5′-CCAACTTAATCGCCTTGCAGCACA-3′) and reverse (5′AAGCGAGTGGCAACATGGAAATCG-3′).
Virus production
scAAV9-SMN viral vector was produced as described previously (17). HEK293T cells were triple transfected in the presence of polyethyleneimine (PEI) (1 mg/mL), pH 5.0 (31). The scAAV plasmid was constructed to express the open-reading frame of human SMN1 cDNA (NCBI accession number NM_000344) under the control of the chicken β-actin promoter. Following media change and cell collection at 24 and 48 h post-transfection, respectively, scAAV9-SMN viral vector was purified using three cesium chloride density-gradient centrifugation steps and dialyzed with HEPES buffer.
Real-time PCR (qPCR)
Quantification of viral genomes was performed using SYBR® Green and primers to amplify the chicken β-actin promoter region, forward (5′-CCGGTGGTGGTGCAAATCAAAGAA-3′) and reverse (5′-AGCAGAAGTAACACTTCCGTACAGGC-3′). The absolute quantitation method using a standard curve was utilized on the Applied Biosystems® 7500 Real-Time PCR System using Applied Biosystems 7500 Sequence Detection Software Version 1.3 Viral fractions were diluted 1:1000 and the PCR cycle was as follows: 50°C 2 min,95°C 10 min, 40 cycles (95°C 15 s), 60°C 1 min. A standard curve was obtained using serial dilutions of the transgene-containing plasmid (1010–105) to calculate melting curves of each sample. The viral fractions containing the highest titer were dialyzed with HEPES buffer (100 mm NaCl, 20 mm HEPES). Following dialysis, qPCR was performed to obtain the final titer of the virus-containing solution to be used for injections into the mice.
ICV injection of scAAV9-SMN or scAAV9-GFP
ICV injection was utilized to deliver scAAV9-SMN directly into the CNS. Mice were administered a single injection of 1 × 1011 v.g., on one of the following days, P2 through P8. Briefly, a glass-pulled needle was inserted through the frontal plate (lateral to the metopic suture and rostral to the coronal suture) into the left or right ventricle of the neonatal mouse to deliver a 5 µL bolus of viral vector. Unaffected animals were injected with scAAV9-GFP on P2 or P7 and non-injected mice were used as a negative control.
Phenotypic assessment
All treated and untreated mice were monitored daily to evaluate survival and to record weight gain. Motor function was assessed by the time-to-right test. The time-to-right test consists of placing animals on their back and determining the time it takes for them to right themselves onto all four paws. A mouse unable to right within 30 s is considered to have failed the test for that day.
Tissue collection
Tissues were collected on P11 for all mice treated with scAAV9-SMN and on P6 or P11 for those injected with scAAV9-GFP. Brain, spinal cord, skeletal muscle, heart, kidney, liver and spleen were collected and immediately flash-frozen in liquid nitrogen for protein analysis, submerged in 4% paraformaldehyde (PFA) for histological examination of GFP and muscle fiber examination or perfused in 4% PFA for NMJ analysis.
Western blot analysis
For western blots, tissues were collected at selected time points and immediately frozen in liquid nitrogen. Tissue samples were placed at −80°C until ready for analysis. Roughly 100 mg of tissue was homogenized in JLB buffer (50 mm Tris–HCl pH 7.5, 150 mm NaCl, 20 mm NaH2(PO4), 25 mm NaF, 2 mm EDTA, 10% glycerol, 1% Triton X-100 and protease inhibitors (Roche, Indianapolis, IN, USA). Equal amounts of protein were separated on 12% SDS–PAGE gels. SMN immunoblots were performed using a mouse SMN specific monoclonal antibody (BD Biosciences #610647, San Jose, CA, USA) diluted 1:2000 in 1% dry milk in TBST (Tris-buffered Saline Tween20 (10 mm Tris–HCl, pH 7.5, 150 mm NaCl, 0.2% Tween20) and a secondary anti-mouse HRP-conjugated secondary antibody (1:10 000). Blots were visualized by chemiluminescence on a Fujifilm imager LAS-3000 and the corresponding software. To verify equal loading, the membranes were then stripped using β-mercaptoethanol for 30 min at 50°C and re-probed with anti-β-actin rabbit antibody (Sigma #A5060, St Louis, MO, USA) diluted 1:2000 and anti-rabbit HRP secondary antibody (1:10 000). Western blot analysis was performed in triplicate and representative blots are shown. Probes were visualized by chemiluminescence using the Pierce® SuperSignal Pico reagents.
Muscle fibers
Mouse triceps brachii muscles were harvested on Day 11 and fixed in their natural position with the entire arm or te leg was removed and submerged in 4% PFA and stored at 4° overnight. The muscles were carefully removed from the limb without stretching the muscle and the samples embedded in paraffin and sliced into 4 µm sections. The tissue was stained with hematoxylin and eosin and analyzed using Metamorph Software.
Peripheral transduction analysis
Unaffected animals were injected with scAAV9-GFP on P2 or P7. Unaffected animals not injected with the GFP virus were used as a negative control. All tissues harvested for peripheral transduction analysis were harvested on P11.
Immunohistochemistry of neuromuscular junctions
Mice were transcardially perfused with PBS followed by 4% PFA. Following perfusion, samples were incubated in PFA for an additional 2 h followed by three washes and storage in PBS until relevant muscles were dissected and stained. Whole-mount preparations were used and immunostained with anti-neurofilament (1:2000, Chemicon), anti-synaptophysin (1:200, Invitrogen) and α-bungarotoxin (Invitrogen). Fluorescently labeled NMJs were visualized by confocal microscopy and Z-stack images were obtained in 1 µm sequential focal planes. Representative images are flattened projections of Z-stack images. These methods have been previously described (32).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by an NIH Training Grant (to J.J.G.; M.R.M.) T32 GM008396; and a Research Grant from the Muscular Dystrophy Association (C.L.L.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank John Marston for his assistance with animal husbandry and all the lab members for their critical review of the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Lorson C.L., Hahnen E., Androphy E.J., Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lorson C.L., Androphy E.J. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum. Mol. Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 4.Lorson C.L., Strasswimmer J., Yao J.M., Baleja J.D., Hahnen E., Wirth B., Le T., Burghes A.H., Androphy E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 5.Pellizzoni L., Charroux B., Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc. Natl Acad. Sci. USA. 1999;96:11167–11172. doi: 10.1073/pnas.96.20.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnett B.G., Munoz E., Tandon A., Kwon D.Y., Sumner C.J., Fischbeck K.H. Regulation of SMN protein stability. Mol. Cell. Biol. 2009;29:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldkotter M., Schwarzer V., Wirth R., Wienker T.F., Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lotti F., Imlach W.L., Saieva L., Beck E.S., Hao le T., Li D.K., Jiao W., Mentis G.Z., Beattie C.E., McCabe B.D., et al. An SMN-Dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–454. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fallini C., Bassell G.J., Rossoll W. Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res. 2012;1462:81–92. doi: 10.1016/j.brainres.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorson M.A., Lorson C.L. SMN-inducing compounds for the treatment of spinal muscular atrophy. Future Med. Chem. 2012;4:2067–2084. doi: 10.4155/fmc.12.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le T.T., McGovern V.L., Alwine I.E., Wang X., Massoni-Laporte A., Rich M.M., Burghes A.H. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 2011;20:3578–3591. doi: 10.1093/hmg/ddr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lutz C.M., Kariya S., Patruni S., Osborne M.A., Liu D., Henderson C.E., Li D.K., Pellizzoni L., Rojas J., Valenzuela D.M., et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Invest. 2011;121:3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benkhelifa-Ziyyat S., Besse A., Roda M., Duque S., Astord S., Carcenac R., Marais T., Barkats M. Intramuscular scAAV9-SMN injection mediates widespread gene delivery to the spinal cord and decreases disease severity in SMA mice. Mol. Ther. 2013;21:282–290. doi: 10.1038/mt.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dominguez E., Marais T., Chatauret N., Benkhelifa-Ziyyat S., Duque S., Ravassard P., Carcenac R., Astord S., Pereira de Moura A., Voit T., et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011;20:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 15.Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Glascock J.J., Shababi M., Wetz M.J., Krogman M.M., Lorson C.L. Direct central nervous system delivery provides enhanced protection following vector mediated gene replacement in a severe model of spinal muscular atrophy. Biochem. Biophys. Res. Commun. 2012;417:376–381. doi: 10.1016/j.bbrc.2011.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glascock J.J., Osman E.Y., Wetz M.J., Krogman M.M., Shababi M., Lorson C.L. Decreasing disease severity in symptomatic, Smn(−/−);SMN2(+/+), spinal muscular atrophy mice following scAAV9-SMN delivery. Hum. Gene. Ther. 2012;23:330–335. doi: 10.1089/hum.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Passini M.A., Bu J., Richards A.M., Kinnecom C., Sardi S.P., Stanek L.M., Hua Y., Rigo F., Matson J., Hung G., et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valori C.F., Ning K., Wyles M., Mead R.J., Grierson A.J., Shaw P.J., Azzouz M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2010;2:35–42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- 20.Duque S., Joussemet B., Riviere C., Marais T., Dubreil L., Douar A.M., Fyfe J., Moullier P., Colle M.A., Barkats M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCarty D.M., Fu H., Monahan P.E., Toulson C.E., Naik P., Samulski R.J. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- 23.Butchbach M.E., Edwards J.D., Burghes A.H. Abnormal motor phenotype in the SMNDelta7 mouse model of spinal muscular atrophy. Neurobiol. Dis. 2007;27:207–219. doi: 10.1016/j.nbd.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porensky P.N., Mitrpant C., McGovern V.L., Bevan A.K., Foust K.D., Kaspar B.K., Wilton S.D., Burghes A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osman E.Y., Yen P.F., Lorson C.L. Bifunctional RNAs targeting the intronic splicing silencer N1 increase SMN levels and reduce disease severity in an animal model of spinal muscular atrophy. Mol. Ther. 2012;20:119–126. doi: 10.1038/mt.2011.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamilton G., Gillingwater T.H. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med. 2013;19:40–50. doi: 10.1016/j.molmed.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Hunter G., Aghamaleky Sarvestany A., Roche S.L., Symes R.C., Gillingwater T.H. SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2014;23:2235–2250. doi: 10.1093/hmg/ddt612. [DOI] [PubMed] [Google Scholar]
- 28.McGivern J.V., Patitucci T.N., Nord J.A., Barabas M.E., Stucky C.L., Ebert A.D. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia. 2013;61:1418–1428. doi: 10.1002/glia.22522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kariya S., Park G.H., Maeno-Hikichi Y., Leykekhman O., Lutz C., Arkovitz M.S., Landmesser L.T., Monani U.R. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K., Gillingwater T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 31.Grieger J.C., Choi V.W., Samulski R.J. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 32.Ling K.K., Gibbs R.M., Feng Z., Ko C.P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012;21:185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.