

Abstract
Duchenne muscular dystrophy (DMD) is a common and relentlessly progressive muscle disease. Some interventions have been identified that modestly slow progression and prolong survival, but more meaningful therapies are lacking. The goal of this study is to identify new therapeutic pathways for DMD using a zebrafish model of the disease. To accomplish this, we performed a non-biased drug screen in sapje, a zebrafish line with a recessive nonsense mutation in dystrophin. We identified 6 positive hits (out of 640 total drugs tested) by their ability to prevent abnormal birefringence in sapje. Follow-up analyses demonstrated that fluoxetine, a selective serotonin reuptake inhibitor (SSRI), provided the most substantial benefit. Morpholino-based experimentation confirmed that modulation of the serotonin pathway alone can prevent the dystrophic phenotype, and transcriptomic analysis revealed changes in calcium homeostasis as a potential mechanism. In all, we demonstrate that monoamine agonists can prevent disease in a vertebrate model of DMD. Given the safe and widespread use of SSRIs in clinical practice, our study identifies an attractive target pathway for therapy development.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most common neuromuscular disorder of childhood, affecting ∼1.3–1.8 per 10 000 boys in the USA (1). It is an X-linked condition caused by loss of function mutations in the large scaffolding protein dystrophin (2). Boys with DMD experience progressive weakness and loss of motor function. Ambulation is typically lost between ages 10 and 12, and death, most often from cardiac or respiratory failure, usually occurs between ages 20 and 30 (3, 4). Disease-modifying interventions exist that have modestly impacted outcomes (5). The best studied is glucocorticoid therapy, which has been shown to prolong ambulation by ∼2 years and to improve survival (5). When used in combination with improved respiratory and orthopedic management and aggressive cardiac care, glucocorticoid therapy has shifted life expectancy from the late teens to the mid-to-late 20s (6). However, boys with DMD continue to experience significant and progressive disability along with early mortality, and there remains no cure for this devastating disorder.
At present, there are many efforts underway aimed at developing new therapies for DMD (7). Two prominent strategies are dystrophin gene-manipulation and pathway specific-centered approaches. An example of a gene-based therapy is antisense oligonucleotide-mediated exon skipping, a treatment designed to altered gene splicing and transform out-of-frame DMD gene deletions into in-frame deletions (8). An example of a pathway-based approach is modification of aberrant nitric oxide signaling (NOS) in the disease (9) with phosphodiesterase inhibitors (PDEs, such as sildenafil). PDEs can increase NOS production and improve vascular tone and have been shown to ameliorate aspects of the disease phenotype in pre-clinical models (10, 11). Both exon skipping molecules and PDE inhibitors are currently in clinical trial for DMD, and early trial results are encouraging (12, 13).
Despite these promising advances, there remains a great need for the identification of new therapeutic strategies for DMD. One current barrier in the field relates to the mouse model of the disease. This model, called the mdx mouse, recapitulates the genetics of the disease as well as aspects of its histopathology (14) but does not model the clinical severity. Many potential therapies have initially been identified through studies in the mdx mouse (15). However, to date, none have been successfully translated into therapy. While the reason(s) for this are not certain, it suggests that development of treatment strategies using alternative approaches is important.
The zebrafish is an emerging model system for the study of human disease and for the identification of novel therapies (16, 17). It offers the unique advantage of being a vertebrate model system amenable to large scale, in vivo drug screens (18). Two zebrafish models of DMD, called sapje and sapje-like, have been previously characterized (19, 20). Sapje zebrafish have a recessive nonsense mutation in zebrafish dystrophin. They exhibit severe muscle disorganization, progressive motor dysfunction and early death. The phenotype is first apparent at 3–4 days post fertilization (dpf), and affected zebrafish die between the ages of 10 and 12 days, likely from a failure to feed (normal life span of the zebrafish is 2–4 years). Sapje zebrafish thus not only model the genetic abnormality of DMD but also have a severe phenotype that approximates the disease severity observed in patients. Importantly, Kunkel and colleagues (20) have previously reported a successful drug screen using a zebrafish DMD model. Their study, which tested >1000 compounds, demonstrated the suitability and validity of the model for non-biased therapy identification. The most prominent ‘hits’ provided by the screen were PDE inhibitors, a finding that corroborates the studies referenced above and that supports the utility of zebrafish as a platform for drug discovery in DMD.
In an effort to identify new therapeutic targets in DMD, we performed a large-scale drug screen in sapje zebrafish. We uncovered 6 positive hits out of 640 compounds screened, and identified fluoxetine, a selective serotonin reuptake inhibitor (SSRI), as a promising compound that prevented membrane fragility and promoted survival. We validated the specificity and efficacy of the drug by using a complementary genetic approach, and investigated potential mechanism(s) of action using transcriptomics. In total, our study provides in vivo evidence for a novel and promising pathway for future therapy development.
RESULTS
Birefringence and the drug screening strategy in the sapje zebrafish
The basic strategy for the drug screen is described below and illustrated in Figure 1. Heterozygous (carrier) zebrafish were mated and embryos were pooled, collected and dechorionated at 1 dpf. Sapje zebrafish are not phenotypic at this stage. Embryo pools (n = 20 per well) were placed in individual wells of a 24-well dish. Each well contained either 0.1% dimethyl sulfoxide (DMSO) or one drug from the ENZO drug library diluted to 33 uM in 0.1% DMSO. Drug was changed daily until 4 dpf, at which point fish were screened for abnormal birefringence. Birefringence is the light pattern produced by skeletal muscle when plane-polarized light is applied to it (21). Wild-type embryos have a uniform pattern of birefringence, while sapje zebrafish have an irregular and reduced pattern.
Figure 1.

Schematic depicting the procedural flow for the drug screen. Carrier sapje zebrafish are bred, embryos are collected and dechorionated at 1 dpf, and are then placed into wells containing drugs from the ENZO compound library (diluted at 1/100 in E2 for a final concentration of 33 uM and 0.1% DMSO). Each pool is screened at 4 dpf by birefringence. A positive hit is considered a well with ≤2 fish (out of 20) with abnormal birefringence. Positive hits are secondarily validated by direct DNA sequencing and by testing larger numbers of embryos.
Given that the sapje dystrophin mutation is recessive, an untreated well of 20 embryos should, on average, have 5 embryos (or 25%) with abnormal birefringence. Based on this, we thus considered a positive hit any well where 10% or fewer of the embryos (i.e. 2/20 total) displayed abnormal birefringence. To demonstrate our ability to successfully detect a positive hit, we first treated embryos with MG132, a proteasome inhibitor previously shown to prevent abnormal birefringence in a fraction of sapje zebrafish (22). We tested 3 independent wells with MG132, and detected 5/56 embryos (or 8.9%) with abnormal birefringence, indicating a positive response with this drug, and confirming our ability to successfully identify a positive hit. We then moved forward with a large-scale drug screen, testing 640 total compounds from an Food and Drug Association (FDA) repurposing library. We identified six positive hits (see below). A representative example of both a positive and a negative hit are depicted in Figure 2. Of note, we observed significant non-specific toxicity (affecting all embryos irrespective of genotype) in 18.9% (121/640) of compounds tested.
Figure 2.

Representative images of positive and negative ‘hits’. (Left panel) Example of a negative hit. Depicted is the birefringence pattern from a pool of 20 zebrafish treated with drug. Embryos with abnormal birefringence are circled in red. (Right panel) Example of a positive hit. Depicted is the birefringence pattern of a pool of zebrafish treated with a drug that prevented the development of abnormal birefringence. Note that all of the zebrafish look identical, despite the fact that ∼25% (i.e. 5/20) should be sapje and thus have abnormal birefringence. (Bottom panels) Sequencing chromatograms from DNA extracted from the two embryos from a positive hit pool (treated with fluoxetine). DNA was isolated from each embryo from B and subjected to PCR based Sanger sequencing. The chromatogram on the left depicts wild-type sequence, and the chromatogram on the right depicts sequence from an embryo with normal birefringence but the sapje genotype (** marks the homozygous nonsense mutation A>T).
Drug screen identifies monoamine agonists among group of six positive hits
In our initial screen (using pools of 20 embryos), we identified aminophylline, ergotamine, pergolide, flunarizine, ropinirole and fluoxetine as drugs where 10% or less (i.e. ≤2/20) of embryos developed the sapje phenotype (Table 1 and Fig. 3). Aminophylline was previously identified by Kunkel and colleagues (20) in a drug screen of sapje zebrafish using a different chemical library, and we thus did not pursue it further. Flunarizine is a calcium channel antagonist, and calcium channel antagonists have previously been examined in depth as potential modifiers of DMD disease course (23). We thus focused our attention on pergolide, ergotamine, ropinirole and fluoxetine.
Table 1.
Positive hits from a large-scale drug screen in the sapje zebrafish
| No. | Chemical name | Total number treated | Number of phenotypic Sapje (%) | CAS s# | MW (g/mol) | Drug action |
|---|---|---|---|---|---|---|
| 1 | Aminophylline | 20 | 1 (5%) | 317-34-0 | 180.1 | Non-selective PDE inhibitor |
| 2 | Ergotamine | 20 | 0 (0%) | 379-79-3 | 581.3 | Monoamine agonist |
| 3 | Pergolide | 20 | 1 (5%) | 66104-23-2 | 410.2 | Monoamine agonist |
| 4 | Fluoxetine | 20 | 0 (0%) | 56296-78-7 | 309.1 | Selective serotonin reuptake inhibitor |
| 5 | Flunarizine | 20 | 2 (10%) | 30484-77-6 | 477.4 | Selective calcium entry blocker |
| 6 | Ropinirole | 20 | 0 (0%) | 91374-20-8 | 296.84 | Monoamine agonist |
Figure 3.

Positive hits from a non-biased drug screen in the sapje zebrafish. Positive hits from the drug screen are represented as the percentage of fish with abnormal birefringence (red) compared with the percentage of fish examined with normal birefringence (blue). 0.1% DMSO was used as a negative control (25 embryos with abnormal birefringence out of 100 total screened). MG-132 was our positive control (5 out of 56 with abnormal birefringence). Six drugs were found to give ≤2/20 fish with abnormal birefringence: pergolide (1/20), aminophylline (1/20), ergotamine (0/20), fluoxetine (0/20), flunarizine (2/20) and ropinirole (0/20).
We attempted to validate these hits using two subsequent lines of experimentation. The first was to test a greater number of embryos to exclude non-random segregation of wild-types and sapjes (i.e. wells where there was not ∼25% sapje embryos). We screened 334 embryos with pergolide (finding 35/334 with abnormal birefringence, or 10%), 181 embryos with ergotamine (16/181 or 8.8%), 242 embryos with ropinirole (42/242 or 16%), and 1091 embryos with fluoxetine (3/1091 or 0.3%) (Table 2). Thus, all but ropinirole continued to show a positive effect when larger numbers of embryos were treated.
Table 2.
Results of a secondary screen of monoamine agonists in the sapje zebrafish
| No. | Chemical name | Total number treated | Number of phenotypic sapje (%) | CAS # | MW (g/mol) | Drug action |
|---|---|---|---|---|---|---|
| 1 | Serotonin | 146 | 0 (0%) | 153-98-0 | 212.68 | Monoamine neurotransmitter |
| 2 | Ropinirole | 242 | 42 (16%) | 91374-20-8 | 296.84 | Monoamine agonist |
| 3 | Domperidone | 40 | 9 (22.5%) | 57808-66-9 | 425.91 | Monoamine antagonist |
| 4 | Bromocriptine | 40 | 9 (22.5%) | 22260-51-1 | 750.70 | Monoamine agonist |
| 5 | Pramipexole | 40 | 8 (20%) | 104632-26-0 | 211.33 | Monoamine agonist |
| 6 | Paroxetine | 59 | 10 (17%) | 110429-49-8 | 329.37 | Selective serotonin reuptake inhibitor |
| 7 | Venlafaxine | 78 | 19 (24%) | 99300-78-4 | 277.41 | Serotonin–norepinephrine reuptake inhibitor |
The second method of validation was to take all individual embryos from a treated pool of 20 and perform gene sequencing on each after measuring birefringence. For pergolide-, ergotamine- and fluoxetine-treated pools, we have found 3–5/20 embryos with normal birefringence but with a homozygous mutation consistent with the sapje genotype (Fig. 2). In other words, we successfully documented phenotypically normal embryos that are genetically sapje. These embryos appeared normal in all respects (morphology, motor behavior, etc.) and were indistinguishable from wild-type littermates (see also Fig. 7).
Figure 7.
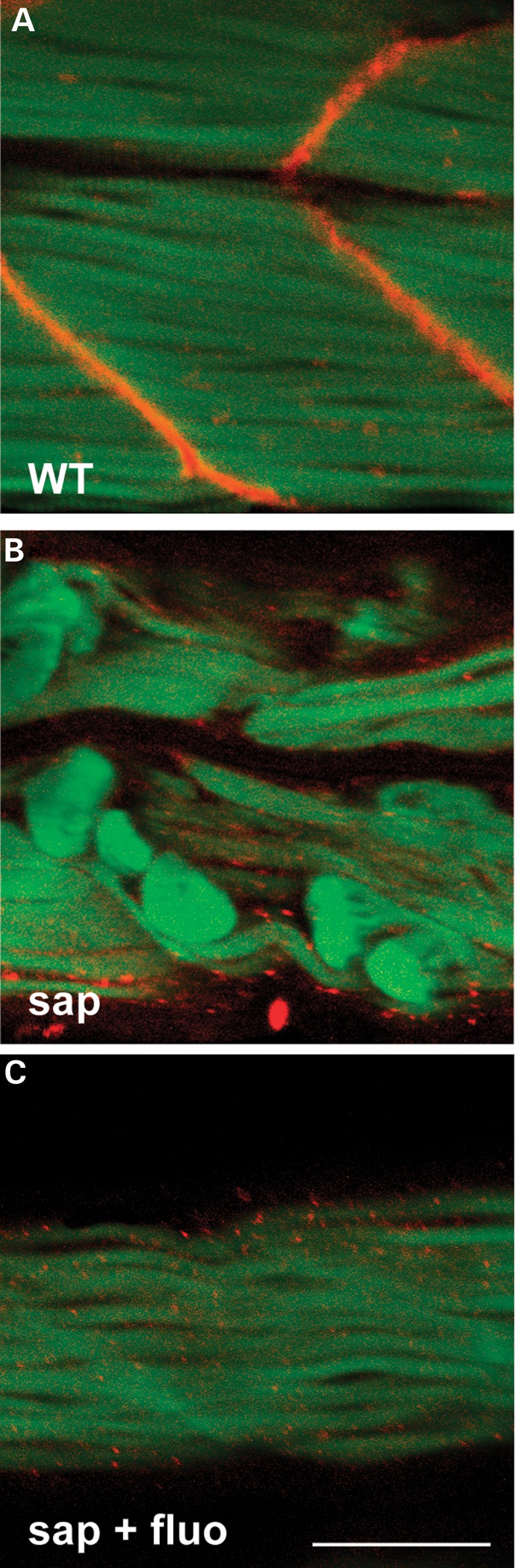
Fluoxetine does not restore dystrophin expression in sapje zebrafish. Embryos were treated with either 0.1% DMSO (i.e. untreated) or 1× fluoxetine from 1 dpf until 4 dpf. They were then genotyped, fixed in 4% paraformaldehyde, and analyzed by whole mount immunofluorescence using anti-dystrophin (red) and phalloidin-FITC (green). (A) Wild-type sibling (WT) showing normal dystrophin expression at the myotendon and a normal pattern of phalloidin staining at the sarcomere. (B) Untreated sapje (sap) with absent dystrophin expression and abnormal phalloidin expression in many fibers. (C) Fluoxetine-treated sapje (sap + fluo) also have absent dystrophin expression, but exhibit normal phalloidin staining. Scale bar = 100 um.
Pergolide, ergotamine and fluoxetine are drugs in the broad class of monoamine agonists. We thus tested the following additional monoamine agonists (Table 2): domperidone (9/40 or 22.5%), bromocriptine (9/40 or 22.5%), venlafaxine (serotonin–norepinephrine reuptake inhibitor; 19/78 or 24%), pramipexole (8/40 or 20%), paroxetine (selective serotonin reuptake inhibitor, 10/59 or 17%) and serotonin (0/146). Of this group, only serotonin prevented the development of abnormal birefringence in ≤ 10% of embryos. In fact, out of 146 embryos treated presymptomatically with serotonin, we did not find a phenotypically abnormal embryo. We thus concluded that the serotonin pathway (either directly through serotonin or else through reuptake inhibition) was the common element among the monoamine agonist positive hits.
Genetic manipulation of the serotonin transporter prevents the development of abnormal birefringence in sapje zebrafish
Fluoxetine's main mechanism of action is the prevention of serotonin reuptake by inhibiting the serotonin transporter. It additionally can act on other monoamine transporters more weakly. To confirm that serotonin reuptake inhibition is the mechanism via which fluoxetine prevents the development of the sapje phenotype, we used morpholinos to target the normal expression of slc6a4 (the serotonin transporter) during embryonic development. There are two orthologs of slc6a4 in the zebrafish, and we thus designed morpholinos to each. Morpholinos were injected at the one cell stage into embryos from sapje carrier crosses (n = 3 trials, minimum 20 embryos per group per trial). Injected embryos were allowed to develop, and birefringence was examined at 4 dpf. As expected, >10% of embryos injected with a control morpholino developed abnormal birefringence (18.3% ± 1.3, P = 0.18 when compared with uninjected embryos). Embryos injected with morpholino to slc6a4a also had >10% of embryos appear phenotypically sapje (15.0% ± 7.6, P = 0.27 when compared with uninjected). On the other hand, only 2.3% (±2.3, P = 0.003 when compared with uninjected and P = 0.004 when compared with control morpholino) of embryos injected with morpholino to slc6a4b developed abnormal birefringence (Fig. 4). This indicates that gene knockdown of an ortholog of the serotonin transporter can prevent phenotype development in a subset of sapje, and supports a specific effect on the serotonin pathway of fluoxetine as a modifier of phenotype development.
Figure 4.
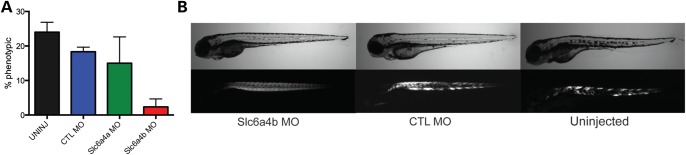
Morpholino knockdown of slc6a4b prevents the sapje birefringence phenotype. (A) Graph depicting the results from three independent injections of morpholinos into 1-cell stage embryos. Embryos are the result of sapje carrier intercrosses, which should result in ∼25% sapje embryos (with abnormal birefringence). Embryos were screened at 4 dpf for abnormal birefringence. Results were: uninjected = 24.0 ± 2.9% with abnormal birefringence, control morpholino (CTL MO) = 18.3 ± 1.3% (P = 0.18), slc6a4a MO = 15.0 ± 7.6 (P = 0.20), and slc6a4b MO = 2.3 ± 22.3 (P = 0.004 compared with CTL MO). (B) Brightfield and birefringence images from the embryos reported in the above graph. Representative examples are given of an slc6a4b morpholino injected embryo, a control morpholino (CTL MO) injected embryo and an uninjected embryo.
Presymptomatic treatment with fluoxetine improves membrane integrity
Based on the nearly complete prevention of phenotype development provided by pre-symptomatic treatment with fluoxetine, we focused the remainder of our analysis on this compound. We first established that fluoxetine did not exert its effect by limiting movement (and thus injury stimulus) of zebrafish embryos. We treated wild-type embryos starting at 1 dpf until 4 dpf, or starting at 3 dpf and treated until 7 dpf, and measured swim velocity and distance traveled. We saw no subjective difference in embryo movement, and observed no objective difference in swim parameters between treated and untreated wild-type zebrafish (Supplementary Material, Fig. S1). This indicated to us that fluoxetine does not prevent dystrophic changes by decreasing spontaneous movement.
The fact that fluoxetine prevents abnormal birefringence suggests that it may improve membrane integrity in sapje mutant zebrafish. To study this more specifically, we examined Evans Blue dye (EBD) uptake in untreated and treated sapje embryos. EBD, when injected into the systemic circulation, is normally excluded from healthy, intact skeletal muscle, but is taken up into myofibers where membrane integrity and/or stability are compromised. As has been previously shown, wild-type embryos do not permit EBD uptake in skeletal muscle, while untreated sapje abundantly do (Fig. 5) (19). When we injected EBD into a mixed pool (i.e. phenotypically normal but dystrophin mutation status unknown) of fluoxetine-treated 4 dpf embryos, however, we did not detect dye uptake in any fish examined. Post-injection genotyping of the injected embryos revealed that three of the embryos were homozygous for the dystrophin mutation, indicating that fluoxetine prevented an interruption in membrane integrity (represented by dye uptake) in these sapje embryos (representative images shown in Fig. 5).
Figure 5.

Prevention of EBD uptake in sapje embryos by fluoxetine treatment. Four dpf embryos were co-injected in the peri-cardial space with EBD (to detect impaired membrane continuity) and dextran-FITC (as a marker for successful systemic injection) and then examined 2 h later. Wild-type littermate control embryos displayed no EBD uptake (n = 25, left panel). Untreated sapje embryos contained abundant areas of EBD positive myofibers (arrow) (n = 6, middle panel). Treated sapje embryos (confirmed by genotyping after treatment) were comparable to wild-type, with no EBD uptake in skeletal muscle (n = 3 sapje confirmed by genotyping, right panel). Scale bar = 180 um.
We were interested to understand to what extent membrane integrity in sapje was restored by pre-symptomatic fluoxetine treatment. We thus subjected untreated and treated sapje to a significant mechanical stress in the form of body axis pinning. Untreated sapje showed abnormal birefringence before pinning that persisted with the pin placement procedure (Fig. 6). Sapje treated for 3 days with fluoxetine had normal birefringence prior to pinning, but then developed patches of abnormal birefringence upon pin placement (Fig. 6). This indicated to us that fluoxetine improves but does not completely restore membrane stability in embryos lacking dystrophin.
Figure 6.

Fluoxetine does not protect against mechanical stress induced injury in sapje. Embryos were treated with 0.1% DMSO or fluoxetine for 3 days. Birefringence (as a marker of membrane stability) was examined before after a significant mechanical stress (pinning of the anterior and posterior body axis). (Left column) wild-type siblings (sibs) demonstrated normal birefringence both before and after pinning. This was regardless of treatment. (Right column) untreated sapje embryos had abnormal birefringence both before and after pinning, while fluoxetine-treated sapje embryos had normal birefringence before pinning that became abnormal within 1 min after pinning.
Presymptomatic exposure to fluoxetine improves survival in sapje zebrafish
We next investigated whether fluoxetine could improve survival in sapje zebrafish. Sapje zebrafish typically die between ages 10 and 12 dpf. We treated a mixed pool of 80 embryos with daily fluoxetine starting at 1 dpf. We examined birefringence starting at day 4 and monitored survival. The expected number (n = 20) of sapje embryos was detected at Day 4 in the untreated (0.1% DMSO) pool, and all untreated sapje were dead by age 12. In contrast, no embryos with abnormal birefringence were detected in the fluoxetine-treated group, and all embryos survived to Day 17, at which point the experiment was terminated (n = 10 trials and >400 total embryos treated). Of note, fluoxetine-treated sapje zebrafish were indistinguishable from wild-types, and exhibited no differences in motor function, with swim velocities and swim distances at wild-type levels (n = 10 independent trials). Thus, while we stopped the survival analysis at 17 days (a time when animal size dictates much larger drug volumes), and determination of true overall survival benefit requires additional study, we demonstrate that fluoxetine not only prevents the development of abnormal birefringence but also significantly prolongs survival.
We next tested the effect of different treatment strategies using fluoxetine. We first performed a dose–response trial using treatment starting at 1 dpf and ending at 4 dpf. We tested the following doses: 0.25×, 0.5×, 1× (30 μM), 2×, 5× and 10×. The sapje phenotype was best suppressed at 1× dosing, though was also suppressed at 0.5× and 2× (Supplementary Material, Fig. S2). All embryos (regardless of genotype) treated at 5× and 10× died with 24 h of exposure to the drug (n = 100 for each).
We then examined temporally limited treatments with fluoxetine. Specifically, we treated embryos from Days 1–4 (n = 57) and Days 4–7 (n = 60). In the Days 1–4 group, no embryos with abnormal birefringence were noted during the treatment period. None were detected in the day following treatment as well. However, the expected frequency (25%) of embryos with abnormal birefringence, i.e. phenotypic sapjes, was found 2 days after treatment ceased. Once phenotypic, the sapje embryos had a disease phenotype that was indistinguishable from that of similarly aged untreated sapje embryos. For the 4–7 day treatment (n = 5 trials), we tested the effect of fluoxetine only on embryos that had already developed the sapje phenotype. We treated the embryos either continuously or twice daily for 3 days total (dpf 4–7), and then quantitatively measured birefringence and swim velocity at dpf 7 (22). We observed no significant difference in abnormal birefringence (Supplementary Material, Fig. S2) or swim velocity between untreated and fluoxetine-treated sapjes, indicating no positive effect of post-symptomatic treatment with fluoxetine.
Fluoxetine does not qualitatively alter dystrophin expression
One potential explanation for the ability of fluoxetine to improve the sapje phenotype is that it may somehow restore dystrophin expression. To test this, we performed whole mount immunofluorescence on pools of embryos either not treated or treated for 3 days with fluoxetine. As expected, embryos in the untreated group with abnormal birefringence (i.e. phenotypic sapje) had undetectable expression of dystrophin. In the treated group, there was also no restoration of dystrophin expression, though in this case the observation of absent expression occurred in embryos with normal birefringence (Fig. 7). The effect of fluoxetine on the development of the sapje phenotype is, therefore, not due to re-expression of dystrophin.
We also stained embryos with phalloidin, a dye, which bind filamentous actin and that, provides a marker of skeletal muscle organization. Phalloidin staining was altered in untreated sapje, often appearing in clumps within detached fibers (Fig. 7B). In fluoxetine exposed sapje, on the other hand, the pattern of phalloidin staining was similar to that observed in wild-type embryos (Fig. 7C). This indicates that internal muscle architecture, at least at the level of light microscopic examination, is essentially normal in treated sapje embryos.
Transcriptome analysis reveals changes in calcium homeostasis with fluoxetine treatment
Lastly, to begin to define the potential mechanism(s) via which fluoxetine prevents the development of the dystrophic phenotype, we performed a gene expression microarray analysis on treated versus untreated sapje. To accomplish this, we incubated 3 pools of 20 embryos (derived from a sapje +/− × +/− crosses) from days 1–4 in either 0.1% DMSO or fluoxetine. At day 4, embryos were screened for birefringence and then processed by taking the anterior portion of the embryos for genotyping and the posterior portion (which is primarily muscle) for RNA extraction. Microarray analysis was subsequently performed independently on RNA from three embryos per condition, with the conditions being: untreated littermate, treated littermate, untreated sapje (genetically confirmed) and treated sapje (genetically confirmed).
We discovered 1019 differentially expressed genes (DEGs) between untreated littermates and untreated sapje, of which 226 (∼22%) were also differentially expressed between untreated sapje and treated sapje (Fig. 8A). The expression of all but one common DEG (225 out of 226) was reversed by the treatment (Supplementary Material, Table S1). Figure 8B is a heat-map of the five most significantly over-represented biological functions among the DEGs in Figure 8A, demonstrating that the common DEGs were highly associated with receptor complex and calcium ion binding functions. Figure 8B also suggests that genes related to functions such as oxidative phosphorylation, structural and metabolic processes were not reversed by fluoxetine treatment.
Figure 8.

Microarray analysis of fluoxetine-treated sapje zebrafish. Comparative microarray analysis was performed on untreated and fluoxetine-treated embryos using the Affymetrix Zebrafish Gene 1.1 array platform. (A) Venn diagram of DEGs. There were 1019 DEGs between untreated wild-type and sapje embryos. Two hundred and twenty-six of these were reversed with fluoxetine treatment. An additional 704 DEGs were uncovered between untreated and treated sapje. (B) Heat map showing the most significantly over-represented biological functions in the differentially expressed transcripts. (C) Examination of the most significantly changed transcripts revealed enrichment for gene products associated with calcium homeostasis. (D) Quantitative RT-PCR was performed to validate several of the transcriptional changes observed by microarray analysis. Depicted graphically are the results for calsequestrin 1b and calsequestrin 2. Specific fold changes were as follows (with untreated wild-type set nominally as 1.0): calsequestrin 1b = 0.8-fold (wild-type treated with fluoxetine), 0.4-fold (untreated sapje), and 0.7-fold (treated sapje); calsequestrin-2 = 1.4-fold (WT plus fluoxetine), 0.4-fold (untreated sapje) and 1.1-fold (sapje plus fluoxetine).
Among the gene products most significantly altered (Fig. 8C), we were drawn to the fact that this group included several genes products involved in calcium homeostasis. This was noteworthy to us because (1) there is previous data to support an effect of SSRIs on calcium dynamics and (2) altered intracellular calcium homeostasis has been implicated as an aspect of disease pathogenesis in DMD. We sought to validate these transcriptional changes using real time PCR. Quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) confirmed several of the changes seen by microarray, including especially the alterations in calsequestrin 1b and calsequestrin 2 levels (Fig. 8D). These data thus support an overall impression that one potential mechanism of action for fluoxetine is improved calcium homeostasis.
DISCUSSION
In this study, we have performed a large-scale (n = 640) chemical screen to identify drugs that prevent the development of the dystrophic phenotype in the sapje zebrafish model. We found 6 positive hits using our screening methodology. One hit (aminophylline) has been found previously in a drug screen of a zebrafish model of DMD (20), and has further been shown in mammalian models to be a positive modifier of disease (10). The identification of aminophylline in our screen thus supports the validity and utility of our methodology. The major novel finding from our study is the identification of fluoxetine and serotonin as drugs that can prevent the development of disease in the sapje zebrafish.
The identification of serotonin pathway modulators as positive hits in the sapje zebrafish is somewhat surprising. Serotonin and its regulators are most typically associated with the central nervous system, where they factor prominently in the regulation of homeostatic processes such as appetite and mood (24). Serotonin is also highly expressed in the gut, where it participates in intestinal peristalsis (25). It additionally functions as a regulator of vascular tone (26). It is therefore possible that the positive impact of serotonin and fluoxetine on the sapje zebrafish is due to modulation of non-muscle pathways. Regulation of blood flow to the muscle, an area clearly shown to be important for DMD pathogenesis, is one potential possibility. In fact, fluoxetine has previously been shown to dilate skeletal muscle arterioles in rat skeletal muscle (27).
Conversely, there is data instead to support a primary role for serotonin in skeletal muscle. Serotonin receptors are expressed in skeletal muscle (28), and serotonin has been shown to directly increase glucose transport through these receptors (28). It has also been shown to have additional direct metabolic effects on muscle, including dose-dependent activation of the glycolytic pathway (29). At present, as suggested by our data, we favor a model whereby fluoxetine (and serotonin) act directly on skeletal muscle to promote/stabilize membrane integrity. Based on our transcriptome analysis, we postulate that this affect is mediated by improved/restored intracellular calcium homeostasis, which in turn may improve calcium-dependent membrane repair processes necessary for membrane stabilization in the setting of absent dystrophin (30). Further experimentation will be necessary to test these concepts.
Importantly, there are previous associations between serotonin and its modifiers and the pathogenesis of muscular dystrophy. Based on data from mouse studies (31, 32) prior to the identification of dystrophin mutations as the genetic cause of DMD, there was an investigation of serotonin levels in the serum of patients with DMD. In a small cohort of patients, serotonin levels were found to be comparable to controls in the plasma but lower than controls in platelets (33). This finding was corroborated by a subsequent study that demonstrated decreased serotonin uptake in platelets from DMD patients (34). The significance of these data remains unclear. Later studies using the dystrophic chicken model system [genetic cause unknown (35)] showed that several drugs that block serotonin reuptake (including fluoxetine) improved aspects of the disease phenotype, including improvement of motor function (36). More recently, several serotonergic drugs were identified as suppressors of skeletal muscle degeneration in a drug screen using a Caenorhabditis elegans model of DMD (37).
Using their positive hits in C. elegans as a springboard, Ségalat and colleagues (38) studied 21 modulators of monoamines in the mdx mouse model of DMD. They did not uncover meaningful overall improvement with any of the 21 drugs tested, though they did find that fluoxetine reduced CPK levels and that imipramine improved some aspects of motor function and force generation. They found only marginal improvement with prednisone as well, a finding in keeping with other recent data that have failed to show significant benefit with glucocorticoids in the mdx mouse model (39). In addition, the authors did not measure blood levels or bioactivity of the compounds, and thus it is unclear whether meaningful levels of the drugs in question were obtained.
Based on our results, as well as these previous associations between serotonin and muscular dystrophy, it is tempting to predict that SSRIs may be a viable therapeutic strategy for DMD. SSRIs are currently used in many DMD and BMD patients to treat neurocognitive aspects of the disease, especially depression. However, there has not been a systematic examination of motor function in patients treated with these compounds. The question of whether it makes sense to first more rigorously test SSRIs in the mdx mouse model is an open one. On one hand, the mdx model largely remains the gold standard for pre-clinical drug testing in DMD. On the other hand, the true relationship between a drug's ability to modulate disease in mdx and its ability to ameliorate disease in patients is uncertain. The fact that prednisone, the one proven therapy for DMD, does not provide substantial benefit in the mdx mouse should raise concerns about results based on drug testing in the mdx mouse.
CONCLUSION
In summary, we have used a non-biased drug screen of FDA-approved compounds in the sapje zebrafish model to demonstrate (1) the validity of the zebrafish as a powerful model for drug discovery and (2) the potential therapeutic benefit of serotonin pathway modulation for treating DMD. Future experimentation is obviously required to test whether serotonin or SSRIs are viable treatments for this disease, and the question of how best to do this testing (further examination in a mammalian model versus human myotube model versus direct testing via clinical trial in patients) remains to be resolved.
MATERIALS AND METHODS
Zebrafish husbandry
Heterozygous sapje zebrafish were obtained from University of Tubingen (kind gift of C. Nusslein Volhard) and then subsequently housed and bred under UCUCA approved conditions and specifications (University of Michigan and The Hospital for Sick Children).
Zebrafish genotyping
Zebrafish were genotyped using both Sanger sequencing methodology as well as by Taqman assay. Primers for direct DNA sequencing have been previously published (19). The Taqman assay utilized the following oligonucleotides:
Reporter#1F: 5-′TTGCAATGGATGCTCAAAGTTCATTT-3′FAM
Reporter#1R: 5-′GGGAGTGCACTCGAGTGAAG-3′
Reporter#2F: 5-′CACGTTCTTTAACCTGC-3′
Reporter#2R: 5-′ACGTTCTTAAACCTGC-3′
Drug screen
Embryos from heterozygous matings were pooled and dechorionated at 1 dpf. Embryos were placed 20 per well in 24-well dishes. Each well contained either an experimental compound or else 0.1% DMSO. Drug was changed daily until the termination of the experiment. Embryos were screened at 4 dpf for abnormal birefringence (see Results section).
Birefringence
Birefringence was measured by light microscopy using a plane-polarizing filter attached to either an Olympus dissecting microscope (equipped with a Firefly camera) or at Nikon AZ-100 Macroscope (equipped with a Nikon EZ-snap camera).
Drug library
For the screen, we utilized a library of 640 FDA-approved drugs (ENZO Biomol). Each compound was diluted 1/100 into E2 media (final concentration of 33 uM) and then added to a single well of a 24-well dish. The dose used was based on a previously published drug screen that employed this library in the zebrafish (40).
Morpholinos
Splice site morpholinos were designed to the two zebrafish orthologs of the serotonin transporter (slc6a4A and slc6a4B). Optimal MO concentration was determined as the dose at which splicing was effectively inhibited but minimal toxicity was observed. For slc6a4A MO, the concentration was 0.5 mM, and for slc6a4B, the concentration was 0.3 mM. Standard control morpholino (41) (GeneTools) was used at 0.5 mM. Morpholino sequences were as follows:
Slc6a4A: 5′-ACGCACTTACATGCACTTACACATA-3′
Slc6a4B: 5′-CAGCCACTTACATGCACTTACGTGT-3′
EBD injections
EBD was injected into the peri-cardial space of 4 dpf zebrafish embryos using a procedure adapted from Currie and colleagues (42). Dye uptake into skeletal muscle was determined 2 h after injection using fluorescence microscopy (Nikon AZ-100 macroscope).
Pin analysis
Embryos at 4 dpf were anesthetized in tricaine and examined for their pattern of birefringence on a Sylgard dish background. Selected embryos were then pinned to a same Sylgard coated petri dish with tungsten wire pins (Scientific Instrument Services W406 diameter) placed approximately around the 5th and 18th somites. Pinned embryos were re-analyzed for birefringence.
Whole mount immunofluorescence
Immunolabeling was carried out as previously described (41). In brief, embryos were fixed in 4% paraformaldehyde, incubated with blocking solution, and then stained with anti-dystrophin (Sigma 1:500) followed by alexa594 anti-mouse secondary (1:1000). Embryos were additionally labeled with fluorescein isothiocyanate (FITC)-conjugated phalloidin (Invitrogen), mounted on glass slides, and then visualized using a Nikon AZ-100 macroscope.
Activity monitoring
Zebrafish motor activity (distance traveled and swim velocity) was determined using the Noldus activity monitoring system using our previously established methodology (43).
Microarray analysis
Microarray analysis was performed as previously described (43). Briefly, RNA was extracted from three zebrafish embryos per condition using RNeasy kit (Qiagen), with the conditions being: untreated littermate, treated littermate, untreated sapje (genetically confirmed), and treated sapje (genetically confirmed). Extracted RNA from each condition was amplified, biotin labeled and hybridized to Affymetrix Zebrafish Gene 1.1 ST Arrays containing 59 302 transcripts (Affymetrix). The raw image files were analyzed using a local version of the GenePattern genomic analysis platform from the Broad Institute (http://www.broadinstitute.org/cancer/software/genepattern) (44). The samples were Robust Multi-array Average normalized using the BrainArray Custom CDF version 17 optimized for NCBI Entrez Gene dataset (http://brainarray.mhri.med.umich.edu) (45). Transcripts with a minimum fold expression change of 1.5-fold and intensity-based moderated T-test (IBMT) P-value <0.05 were selected as DEGs (46). A local implementation of the Database for Annotation, Visualization and Integrated Discovery (DAVID) (47, 48) was used to identify enriched biological functions in terms of Gene Ontology terms (http://www.geneontology.org/) and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways (http://www.genome.jp/kegg/).
Quantitative RT-PCR
qPCR was performed as previously described. Primer sequences are listed in the Supplementary Material.
SUPPLEMENTARY MATERIAL
FUNDING
This study was funded in part by the Team Joseph Foundation. It was additionally supported by the Department of Pediatrics and the Taubman Medical Institute at the University of Michigan. J.J.D. is supported in part by National Institutes of Health K08AR054835.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Angela Busta for assistance with zebrafish husbandry, and Eva Feldman, MD, PhD for critical reading of the manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Centers for Disease Control and Prevention. Prevalence of Duchenne/Becker muscular dystrophy among males aged 5–24 years—four states, 2007. MMWR Morb. Mortal Wkly Rep. 2009;58:1119–1122. [PubMed] [Google Scholar]
- 2.Engel A., Franzini-Armstrong C. Myology: Basic and Clinical. New York: McGraw-Hill, Medical Pub. Division; 2004. [Google Scholar]
- 3.Bushby K., Finkel R., Birnkrant D.J., Case L.E., Clemens P.R., Cripe L., Kaul A., Kinnett K., McDonald C., Pandya S., et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9:177–189. doi: 10.1016/S1474-4422(09)70272-8. [DOI] [PubMed] [Google Scholar]
- 4.Verma S., Anziska Y., Cracco J. Review of Duchenne muscular dystrophy (DMD) for the pediatricians in the community. Clin. Pediatr. (Phila) 2010;49:1011–1017. doi: 10.1177/0009922810378738. [DOI] [PubMed] [Google Scholar]
- 5.Moxley R.T., 3rd, Pandya S., Ciafaloni E., Fox D.J., Campbell K. Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J. Child Neurol. 2010;25:1116–1129. doi: 10.1177/0883073810371004. [DOI] [PubMed] [Google Scholar]
- 6.Manzur A.Y., Kinali M., Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis. Child. 2008;93:986–990. doi: 10.1136/adc.2007.118141. [DOI] [PubMed] [Google Scholar]
- 7.Malik V., Rodino-Klapac L.R., Mendell J.R. Emerging drugs for Duchenne muscular dystrophy. Expert Opin. Emerg. Drugs. 2012;17:261–277. doi: 10.1517/14728214.2012.691965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A. Overview on DMD exon skipping. Methods Mol. Biol. 2012;867:97–116. doi: 10.1007/978-1-61779-767-5_7. [DOI] [PubMed] [Google Scholar]
- 9.Heydemann A., McNally E. NO more muscle fatigue. J. Clin. Invest. 2009;119:448–450. doi: 10.1172/JCI38618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Percival J.M., Adamo C.M., Beavo J.A., Froehner S.C. Evaluation of the therapeutic utility of phosphodiesterase 5A inhibition in the mdx mouse model of Duchenne muscular dystrophy. Handb. Exp. Pharmacol., 2011:323–344. doi: 10.1007/978-3-642-17969-3_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Percival J.M., Whitehead N.P., Adams M.E., Adamo C.M., Beavo J.A., Froehner S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012;228:77–87. doi: 10.1002/path.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cirak S., Arechavala-Gomeza V., Guglieri M., Feng L., Torelli S., Anthony K., Abbs S., Garralda M.E., Bourke J., Wells D.J., et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goemans N.M., Tulinius M., van den Akker J.T., Burm B.E., Ekhart P.F., Heuvelmans N., Holling T., Janson A.A., Platenburg G.J., Sipkens J.A., et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 14.Banks G.B., Chamberlain J.S. The value of mammalian models for Duchenne muscular dystrophy in developing therapeutic strategies. Curr. Top. Dev. Biol. 2008;84:431–453. doi: 10.1016/S0070-2153(08)00609-1. [DOI] [PubMed] [Google Scholar]
- 15.De Luca A. Pre-clinical drug tests in the mdx mouse as a model of dystrophinopathies: an overview. Acta Myol. 2012;31:40–47. [PMC free article] [PubMed] [Google Scholar]
- 16.Zon L.I., Peterson R.T. In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov. 2005;4:35–44. doi: 10.1038/nrd1606. [DOI] [PubMed] [Google Scholar]
- 17.Ingham P.W. The power of the zebrafish for disease analysis. Hum. Mol. Genet. 2009;18:R107–R112. doi: 10.1093/hmg/ddp091. [DOI] [PubMed] [Google Scholar]
- 18.Bowman T.V., Zon L.I. Swimming into the future of drug discovery: in vivo chemical screens in zebrafish. ACS Chem. Biol. 2010;5:159–161. doi: 10.1021/cb100029t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bassett D.I., Bryson-Richardson R.J., Daggett D.F., Gautier P., Keenan D.G., Currie P.D. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development. 2003;130:5851–5860. doi: 10.1242/dev.00799. [DOI] [PubMed] [Google Scholar]
- 20.Kawahara G., Karpf J.A., Myers J.A., Alexander M.S., Guyon J.R., Kunkel L.M. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U S A. 2011;108:5331–5336. doi: 10.1073/pnas.1102116108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger J., Sztal T., Currie P.D. Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem. Biophys. Res. Commun. 2012;423:785–788. doi: 10.1016/j.bbrc.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 22.Winder S.J., Lipscomb L., Angela Parkin C., Juusola M. The proteasomal inhibitor MG132 prevents muscular dystrophy in zebrafish. PLoS Curr. 2011 doi: 10.1371/currents.RRN1286. November 17; 3, RRN1286. PMID 22130468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips M.F., Quinlivan R. Calcium antagonists for Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2008 doi: 10.1002/14651858.CD004571.pub2. October 8; 4: CD004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kroeze W.K., Kristiansen K., Roth B.L. Molecular biology of serotonin receptors structure and function at the molecular level. Curr. Top. Med. Chem. 2002;2:507–528. doi: 10.2174/1568026023393796. [DOI] [PubMed] [Google Scholar]
- 25.Gershon M.D. Review article: serotonin receptors and transporters—roles in normal and abnormal gastrointestinal motility. Aliment Pharmacol. Ther. 2004;20(Suppl 7):3–14. doi: 10.1111/j.1365-2036.2004.02180.x. [DOI] [PubMed] [Google Scholar]
- 26.Ramage A.G., Villalon C.M. 5-Hydroxytryptamine and cardiovascular regulation. Trends Pharmacol. Sci. 2008;29:472–481. doi: 10.1016/j.tips.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Pacher P., Ungvari Z., Kecskemeti V., Koller A. Serotonin reuptake inhibitor, fluoxetine, dilates isolated skeletal muscle arterioles. Possible role of altered Ca2+ sensitivity. Br. J. Pharmacol. 1999;127:740–746. doi: 10.1038/sj.bjp.0702571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hajduch E., Dombrowski L., Darakhshan F., Rencurel F., Marette A., Hundal H.S. Biochemical localisation of the 5-HT2A (serotonin) receptor in rat skeletal muscle. Biochem. Biophys. Res. Commun. 1999;257:369–372. doi: 10.1006/bbrc.1999.0471. [DOI] [PubMed] [Google Scholar]
- 29.Coelho W.S., Costa K.C., Sola-Penna M. Serotonin stimulates mouse skeletal muscle 6-phosphofructo-1-kinase through tyrosine-phosphorylation of the enzyme altering its intracellular localization. Mol. Genet. Metab. 2007;92:364–370. doi: 10.1016/j.ymgme.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 30.Lek A., Evesson F.J., Lemckert F.A., Redpath G.M., Lueders A.K., Turnbull L., Whitchurch C.B., North K.N., Cooper S.T. Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair. J. Neurosci. 2013;33:5085–5094. doi: 10.1523/JNEUROSCI.3560-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendell J.R., Engel W.K., Derrer E.C. Duchenne muscular dystrophy: functional ischemia reproduces its characteristic lesions. Science. 1971;172:1143–1145. doi: 10.1126/science.172.3988.1143. [DOI] [PubMed] [Google Scholar]
- 32.Parker J.M., Mendell J.R. Proximal myopathy induced by 5-HT-imipramine simulates Duchenne dystrophy. Nature. 1974;247:103–104. doi: 10.1038/247103b0. [DOI] [PubMed] [Google Scholar]
- 33.Murphy D.L., Mendell J.R., Engel W.K. Serotonin and platelet function in Duchenne muscular dystrophy. Arch. Neurol. 1973;28:239–242. doi: 10.1001/archneur.1973.00490220047006. [DOI] [PubMed] [Google Scholar]
- 34.Arora R.C., Kuncl R.W., Morgan J., Cohen L., Meltzer H.Y. Serotonin uptake in blood platelets of Duchenne muscular dystrophy patients. Muscle Nerve. 1987;10:359–362. doi: 10.1002/mus.880100413. [DOI] [PubMed] [Google Scholar]
- 35.Saito F., Blank M., Schroder J., Manya H., Shimizu T., Campbell K.P., Endo T., Mizutani M., Kroger S., Matsumura K. Aberrant glycosylation of alpha-dystroglycan causes defective binding of laminin in the muscle of chicken muscular dystrophy. FEBS Lett. 2005;579:2359–2363. doi: 10.1016/j.febslet.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 36.Hudecki M.S., Pollina C.M., Bhargava A.K., Hudecki R.S. Screening of antiserotoninergic drugs with the genetically dystrophic chicken. Arch. Neurol. 1980;37:545–550. doi: 10.1001/archneur.1980.00500580041005. [DOI] [PubMed] [Google Scholar]
- 37.Carre-Pierrat M., Mariol M.C., Chambonnier L., Laugraud A., Heskia F., Giacomotto J., Segalat L. Blocking of striated muscle degeneration by serotonin in C. elegans. J. Muscle Res. Cell Motil. 2006;27:253–258. doi: 10.1007/s10974-006-9070-9. [DOI] [PubMed] [Google Scholar]
- 38.Carre-Pierrat M., Lafoux A., Tanniou G., Chambonnier L., Divet A., Fougerousse F., Huchet-Cadiou C., Segalat L. Pre-clinical study of 21 approved drugs in the mdx mouse. Neuromuscul. Disord. 2011;21:313–327. doi: 10.1016/j.nmd.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 39.Sali A., Guerron A.D., Gordish-Dressman H., Spurney C.F., Iantorno M., Hoffman E.P., Nagaraju K. Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse. PLoS One. 2012;7:e34204. doi: 10.1371/journal.pone.0034204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaufman C.K., White R.M., Zon L. Chemical genetic screening in the zebrafish embryo. Nat. Protoc. 2009;4:1422–1432. doi: 10.1038/nprot.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dowling J.J., Vreede A.P., Low S.E., Gibbs E.M., Kuwada J.Y., Bonnemann C.G., Feldman E.L. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5:e1000372. doi: 10.1371/journal.pgen.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall T.E., Bryson-Richardson R.J., Berger S., Jacoby A.S., Cole N.J., Hollway G.E., Berger J., Currie P.D. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc. Natl. Acad. Sci. U S A. 2007;104:7092–7097. doi: 10.1073/pnas.0700942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dowling J.J., Arbogast S., Hur J., Nelson D.D., McEvoy A., Waugh T., Marty I., Lunardi J., Brooks S.V., Kuwada J.Y., et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain. 2012;135:1115–1127. doi: 10.1093/brain/aws036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J.P. GenePattern 2.0. Nat. Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 45.Dai M., Wang P., Boyd A.D., Kostov G., Athey B., Jones E.G., Bunney W.E., Myers R.M., Speed T.P., Akil H., et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sartor M.A., Tomlinson C.R., Wesselkamper S.C., Sivaganesan S., Leikauf G.D., Medvedovic M. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinformatics. 2006;7:538. doi: 10.1186/1471-2105-7-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.