

Abstract
Mitochondrial electron transport chain (ETC) disorders cause severe neurological disease, typically in the context of fatal encephalomyelopathies. Neuronal cell autonomous energy deficiency due to reduced mitochondrial adenosine triphosphate production is currently the leading hypothesis to explain the neurotoxicity in ETC disorders. To define the mechanisms underlying neuropathology in ETC disorders, we have modeled the most common type of ETC disorder, complex I deficiency, in Drosophila. Our model recapitulates important clinical features of the disease including neuronal loss, mitochondrial enlargement, motor dysfunction and early death. Using cell-type specific gene knockdown, we find that both neurons and glia contribute to the disease phenotype and that glia play a critical non-cell autonomous role in the development of neuronal toxicity. Our results open up an unexpected avenue of research, and could lead to the development of new treatment strategies.
INTRODUCTION
With an estimated prevalence of at least 1 in 5000 (1), mitochondrial electron transport chain (ETC) disorders are one of the most common human genetic diseases. The spectrum of ETC disorders includes both dysfunction limited to individual complexes and, through a variety of molecular mechanisms, global ETC dysfunction involving multiple complexes (2). Isolated complex I deficiency is the most common type of mitochondrial ETC disorder (3). Neurological dysfunction and neuropathologies, including strokes and spongiform degeneration, are predominant features of this group of diseases. Most cases of complex I deficiency present in early childhood and result in progressive disability and early death. While a number of hypotheses have been proposed to explain cellular dysfunction and death in mitochondrial ETC disorders, the mechanistic basis of neuronal dysfunction and loss remains poorly understood. The paucity of tractable animal models has been a fundamental barrier to progress. Recently Drosophila has been proven to be a useful model system to understand human neurological diseases (4,5).
Here we describe a model of complex I deficiency using transgenic RNAi expression and the Drosophila bipartite UAS/GAL4 expression system (6). While the effect of reducing complex I function on longevity (7) and in a Parkinson's disease model (8) has been examined in Drosophila, the neuropathology of complex I deficiency has not previously been studied in flies. Here we recapitulate key features of human ETC disorders in Drosophila, including shortened lifespan, impaired locomotion, progressive neuronal loss and abnormal mitochondrial morphology. By taking advantage of cell-type specific complex I knockdown we demonstrate that glia make an important contribution to neuronal toxicity in complex I deficiency.
RESULTS
Knockdown of the core complex I subunit ND75, results in complex I deficiency
Of the various subunits of mitochondrial complex I, 14 are highly conserved and considered to be the core subunits (9). Seven of these 14 core subunits are encoded by the mitochondrial genome, and 7 by the nuclear genome. To select the subunit of complex I to manipulate we used the following criteria: it should be one of the seven core subunits encoded by the nuclear genome, it should not have paralogs in Drosophila, and mutations of the subunit should be known to cause humans disease. We targeted ND75 because it fulfilled all of these criteria. Mutations in NDUFS1, the human homolog of ND75, cause a severe form of complex I deficiency in children characterized by psychomotor retardation, leukodystrophy, Leigh syndrome and death in infancy (10,11).
We reduced ND75 levels globally by using the ubiquitous driver da-GAL4 to express transgenic RNAi (VDRC 52047) directed against ND75. We verified reduction of ND75 protein expression by western blot, using an antibody to NDUFS1, the human homolog of ND75. The protein is highly conserved with 62% amino acid identity between Drosophila and humans, and the antibody exhibited the expected cross-species reactivity. We saw a significant reduction of ND75 expression in knockdown animals (Fig. 1A).
Figure 1.
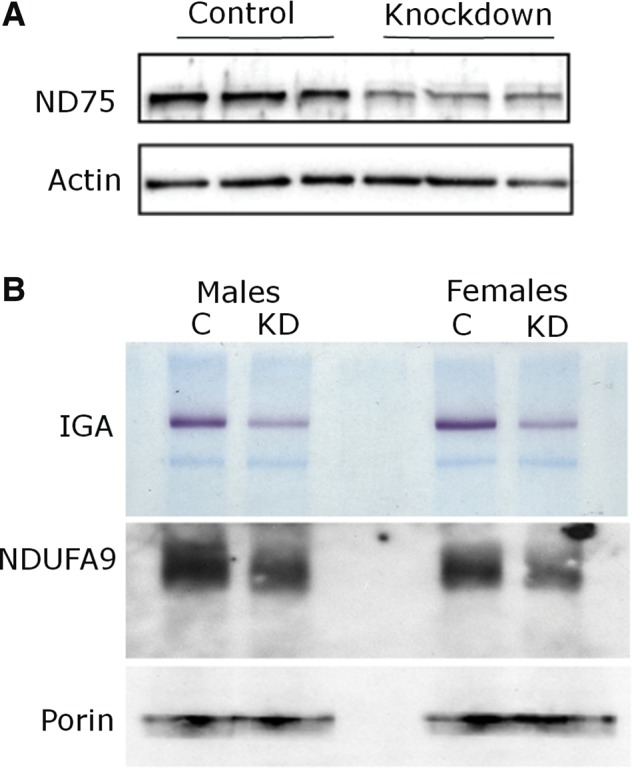
Reduction of ND75 and complex I in knockdown animals. (A) Western blot shows reduced ND75 levels (upper panel). The blot is reprobed for actin to illustrate equivalent protein loading (lower panel). (B) Native gel electrophoresis with complex I in-gel activity assay (IGA) shows reduction of complex I activity (upper panel). Immunoblotting of the same shows reduction in a distinct component of complex I (NDUFA9/CG6020 middle panel), and equivalent total amount of mitochondrial loading as illustrated by porin (lower panel). Flies are 10 days old. Driver is da-GAL4; control is da-GAL4/+.
To further explore complex I levels and function, native gel electrophoresis and in-gel NADH oxidase-ferrireductase activity of complex I was assayed on purified mitochondria (12). The native gel was subsequently blotted, and the blot probed with antibodies to NDUFA9, another subunit of complex I (Fig. 1B, middle panel), and to porin, a mitochondrial membrane protein (Fig. 1B, lower panel). As seen in Figure 1B, both NADH oxidase–ferrireductase activity and levels of complex I were reduced in flies expressing transgenic RNAi targeting ND75.
Complex I deficiency in Drosophila
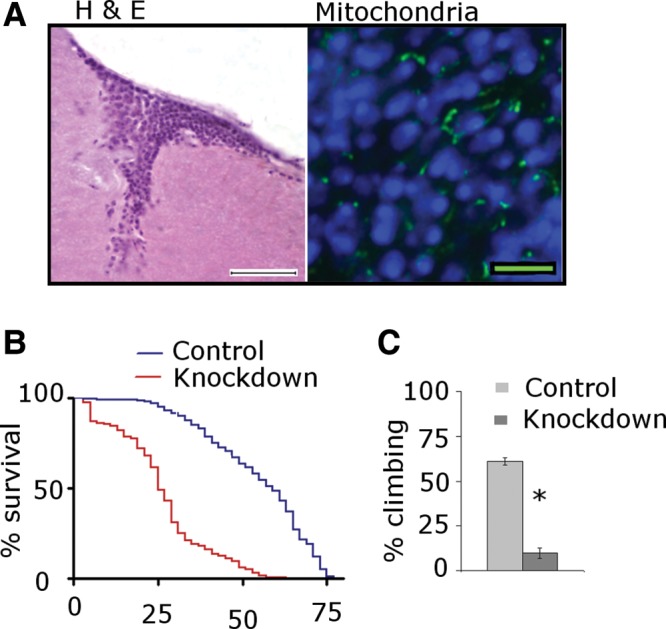
Complex I deficiency and other mitochondrial ETC disorders typically cause progressive disease and early death. We thus assessed the effect of complex I knockdown on lifespan of our animals. Kaplan–Meyer analysis showed that lifespan of knockdown animals was significantly reduced in mutant flies (Fig. 2A) with median lifespans of 27 and 49 days in knockdown and control flies, respectively. To determine if knockdown flies display progressive functional disability, we tested their climbing ability, a commonly used behavioral task in Drosophila (13). Significant and progressive motor disability was present in the complex I deficient flies (Fig. 2B). By 5 days of age, climbing in the knockdown group was about half that of the control group.
Figure 2.

Complex I knockdown phenotypes. (A) Kaplan–Meyer survival curves show truncated lifespan in knockdown animals. N = 300 per genotype, P < 0.001, log rank test. (B) Climbing assays show progressive motor dysfunction in knockdown animals. N = 60 per genotype. *P < 0.05, Student's t-test. (C) Brain histology is normal in control animals at 1 and 10 days, while marked neuronal loss is seen in mutants at 10 days. Scale bars are 20 µm. (D) Caspase activation is detected by cleaved PARP in neurons (upper panel, arrows) but not in glia (lower panel, arrows). Scale bar is 5 µm. (E) Significantly higher number of cleaved PARP positive cells in brains of knockdown flies when compared with control flies. Flies are 10 days old. N = 5 per genotype. *P < 0.001, Student's t-test. Driver is da-GAL4; control is da-GAL4/+.
Patients with complex I deficiency develop a variety of neuropathological abnormalities including neuronal loss and spongiform degeneration of the gray and white matter (14). To determine if complex I knockdown flies developed neuropathology we examined hematoxylin and eosin (H&E) stained sections of the adult brain. Brains from 1-day-old control and knockdown animals showed no histological differences on H&E analysis (Fig. 2C, arrows). In contrast, by Day 10 experimental flies demonstrated significant cortical neuronal loss (Fig. 2C, arrowheads).
To further evaluate neuropathology in our model, we used a transgenic fusion protein containing the cleavage site of a caspase substrate, human poly ADP ribose polymerase-1 (PARP) (15). During cell death the transgenic reporter is cleaved by activated caspase. Proteolysis of the reporter construct is detected by using an antibody specific to cleaved human PARP. To quantify caspase activation we counted the total number of cleaved PARP positive cells per fly brain (Fig. 2E). At 10 days experimental flies showed an average of 111 cells while control flies showed an average of 15.6 cells with caspase activation. To determine if the dying cells were neurons or glia, we double stained for cleaved PARP (Fig. 2D, arrows) and either the neuronal marker elav or the glial marker repo (n = 4 brains per marker). We found that the majority of cells (99%) with caspase activation were neurons. Thus we find that there is significant difference in caspase activation between control and knockdown flies, and that caspase activation occurs predominantly in neurons.
Mitochondrial enlargement in complex I deficiency
A variety of mitochondrial morphological abnormalities have been described in patients with respiratory chain diseases (16). Using an antibody to the alpha subunit of complex V [adenosine triphosphate (ATP) synthase], we observed striking mitochondrial enlargement (Fig. 3A), which was widespread in cortex of the brains of knockdown flies. Similar mitochondrial enlargement was also seen using transgenic flies expressing mitochondrially targeted green fluorescent protein (GFP), UAS-mito-GFP, confirming the observed mitochondrial enlargement phenotype (Supplementary Material, Fig. S1). This widespread mitochondrial enlargement was confined to, and completely penetrant in, knockdown flies.
Figure 3.

Mitochondrial enlargement in complex I deficiency. (A) Staining with an antibody to a subunit of complex V shows abnormal mitochondrial enlargement in the cortex of knockdown flies 10 days. Low power scale bar is 20 µm and high power scale bar is 5 µm. (B) Costaining with antibodies to complex V and either the neuronal marker elav or the glial marker repo, demonstrates colocalization of mitochondrial enlargement with both cell types. Scale bar is 3 µm. (C) Electron microscopy shows normal mitochondrial size with normal structure of the cristae (arrows) in control neurons (1,2). In contrast, knockdown flies (3,4,5,6) display mitochondrial enlargement and morphological abnormalities such as honeycombing, effacement of cristae and concentric lammelations. Flies are 30 days old in EM experiments. Scale bars are 500 nm in the EM images. Driver is da-GAL4; control is da-GAL4/+.
To determine the cell type showing mitochondrial enlargement we double stained for ATP synthase and either the neuronal marker elav or the glial marker repo. We observed that enlarged mitochondria were present in both neurons and in glia (Fig. 3B). To characterize mitochondrial morphology at the ultrastructural level, electron microscopy was performed on the brains of 30-day-old control and knockdown flies. Control animals showed morphologically normal mitochondria with appropriate internal cristae structure (Fig. 3C, arrows). In contrast, complex I mutants had increased number and size of mitochondria, sometimes to the extent of almost filling the cytoplasm. These mitochondria displayed a variety of morphological abnormalities including effacement of cristae and concentric lamellations of internal mitochondrial membrane (Fig. 3C, arrows).
Neuronal complex I knockdown and organismal toxicity
The predilection of mitochondrial disorders for the nervous system is often attributed to neuronal dependence on oxidative phosphorylation for energy production (17). While the mechanisms of cell death in ETC disorders are not known, cell-autonomous processes such as energy deficiency and reactive oxygen species (ROS) production are thought to be most important. To determine if cell-autonomous mechanisms alone are sufficient to cause the neurotoxicity seen in our model, we used the pan-neuronal driver elav-GAL4 to study the effects of neuronal complex I knockdown specifically in neurons. Histological and immunofluorescent studies of the brain (Fig. 4A) did not demonstrate the neuronal loss, vacuolation, or mitochondrial enlargement we had previously seen with ubiquitous complex I knockdown. By comparing levels of ND75 knockdown we determined that elav-GAL4 was at least as strong a driver as da-GAL4 (Supplementary Material, Fig. S2). While neuronal-specific complex I knockdown did not lead to observable neuropathological changes, neuronal knockdown did have substantial effects on behavioral phenotypes. Lifespan of knockdown flies was significantly shortened (Fig. 4B) with median survival of 25 days in knockdown and 59 days in control. The climbing assay also demonstrated significant dysfunction in the knockdown flies at 5 days, with 61% of control and 10% of knockdown flies demonstrating the ability to climb (Fig. 4C).
Figure 4.
Neuronal complex I knockdown. (A) Selective knockdown of ND75 in neurons produces normal brain histology (left) and morphologically normal mitochondria (right). Scale bars are 20 µm in the H&E section and 5 μm in the immunostained section. Flies are 10 days old. (B) Kaplan–Meyer curves show significantly truncated lifespan with neuronal-selective ND75 knockdown animals. N = 300 per genotype; P < 0.001, log rank test. (C) Climbing assays show significant motor dysfunction in knockdown animals at 5 days. N = 60 per genotype. *P < 0.001, Student's t-test. Driver is elav-GAL4; control is elav-GAL4/+.
Glial complex I knockdown and neurotoxicity
Because neuronal-specific knockdown did not demonstrate the neuropathologic changes of ubiquitous complex I knockdown, and because glia are known to promote non-cell autonomous neurodegeneration in other disorders (18,19), we determined if glial complex I deficiency played a role in our model. Using the glial-specific driver repo-GAL4 we were able to recapitulate the neuronal loss, cortical vacuolation and diffuse mitochondrial enlargement (Fig. 5) we had previously seen with ubiquitous knockdown (Fig. 2C). To determine if the mitochondrial enlargement we observed with complex I knockdown was specific, we expressed mutant human ataxin-3 (20) in glia, to assess the non-specific effect of glial toxicity (21). Despite producing significant pathological changes in the brain including large vacuoles (Fig. 5A5 arrows), ataxin-3 expression did not produce the mitochondrial enlargement seen with ND75 knockdown (Fig. 5A6). To ensure that the neuropathological effects of glial ND75 knockdown were not due to off-target or driver-specific effects, we tested a second, non-overlapping RNAi line (VDRC 100733) and an additional glial driver, EAAT1-GAL4. We observed vacuolation, neuronal loss and mitochondrial enlargement with both the second RNAi line and the glial driver EAAT1-GAL4 (Supplementary Material, Fig. S3).
Figure 5.

Glial contributions to neurotoxicity. (A) Selective knockdown of ND75 in glia produces cortical cell loss and vacuolation in the brain (A3). Mitochondrial staining of the glial ND75 knockdown line (A4) demonstrates mitochondrial enlargement similar to that seen in ubiquitous complex I knockdown. H&E sections of the brains from flies expressing mutant human ataxin-3 in glia shows marked cortical and neuropil tissue disruption (A5), but normal mitochondrial size (A6). Scale bar is 20 µm in the H&E sections and 5 µm in the immunostained sections. (B) Co-staining with antibodies to complex V and either the neuronal marker elav or the glial marker repo, demonstrates mitochondrial enlargement in both cell types. Scale bar is 3 µm. (C) Kaplan–Meyer curves show truncation of lifespan in glial ND75 knockdown flies N = 300 per genotype; P < 0.001, log rank test. (D) Climbing assays show no motor dysfunction in knockdown animals. N = 60 per genotype. *P < 0.001, Student's t-test. Flies are 10 days old. Driver is repo-GAL4; control is repo-GAL4/+.
To determine the cell type affected by mitochondrial enlargement with glial-specific complex I knockdown, we conducted double-label immunofluorescence studies using antibodies to mitochondrial ATP synthase and either the glial marker repo or the neuronal marker elav. We observed that enlarged mitochondria were present in both neurons and glia. Thus, we demonstrate that glial complex I knockdown is critical for the development of neuropathological changes of complex I deficiency, and that glial complex I knockdown leads to mitochondrial changes both in glia, and through non-cell autonomous mechanisms, in neurons. The observed changes are neither off-target effects, nor are they non-specific results of glial toxicity. While glial-specific knockdown caused neuropathological changes in the fly brain, it did not lead to the significant behavioral dysfunction seen with ubiquitous and neuron specific knockdown. The lifespan of knockdown flies was moderately truncated (Fig. 5C) with median survival of 41 days in knockdown and 47 days in control animals. Climbing assays demonstrated no significant difference at 5 days (Fig. 5D), with 57.3% of control and 58.6% of knockdown flies demonstrated the ability to climb.
DISCUSSION
As whole-genome sequencing becomes routine, the essential challenge of medicine lies in translating the power of genetic diagnosis into devising effective treatments. Understanding the mechanisms of genetic diseases is fundamental to that end. While mitochondrial respiratory chain dysfunction is one of the most common genetic diseases (1), little is known about its mechanisms of cellular toxicity. Isolated complex I deficiency is the most common type of mitochondrial disorder. To facilitate comprehensive genetic analysis of cellular and molecular pathways involved in the pathogenesis of this disease, we have developed a Drosophila of mitochondrial complex I deficiency. We use the UAS/GAL4 system to create dominantly inherited mutants as it simplifies genetic crosses, and thus enables the use of the large collection of genetic tools available in Drosophila. This system also facilitates complex I knockdown in a tissue specific manner, allowing us to investigate the roles of different cell types in disease pathogenesis. In this study we demonstrate that knockdown of complex I in Drosophila recapitulates key features of mitochondrial disease in patients. Our complex I deficiency flies develop progressive functional disability, neuronal loss, mitochondrial morphological changes and early death. Thus, by recapitulating clinically important aspects, we demonstrate that Drosophila can be used to model mitochondrial complex I deficiency.
Neuropathologies such as stoke and progressive neurodegeneration are common in mitochondrial complex I deficiency (22), and cause profound and devastating disability, ultimately leading to early death. Because mitochondrial disease manifests predominantly in neurons and muscles, tissues dependent on oxidative phosphorylation, it is commonly assumed that neurotoxicity of mitochondrial ETC disorders occurs through cell autonomous processes such as ATP deficiency and ROS production (17). By using tissue specific knockdown, we are directly able to dissect the contributions of neurons and glia towards the disease phenotype. We demonstrate that neuronal complex I deficiency causes organismal toxicity resulting in locomotor dysfunction and early death. Importantly, we find that glial complex I deficiency can independently cause neuronal toxicity and pathological mitochondrial changes in a non-cell autonomous manner. These results, we believe, do not represent a dichotomy between neuronal cell death and its functional consequence, but rather represent early phenotypic manifestations of different pathways of neurotoxicity. Indeed, disruption of any fundamental metabolic process will most likely result in at least two effects, an accumulation of proximal substrates and a deficiency of the end product. As neurons and glia have interdependent but separate metabolic functions (23), it is possible that complex I deficiency activates different pathways of toxicity in the two cell types.
Previous studies in Drosophila have shown significant morphological, biochemical, molecular and functional similarities between mammalian and fly glia (24). A well characterized mouse model of complex I deficiency has shown glial activation in neurotoxicity of mitochondrial complex I deficiency (25). Additionally, the areas of the brain with the highest glia/neuron ratios such as the basal ganglia and brainstem (26), are the predominant sites of subacute degeneration in patients with mitochondrial ETC dysfunction. Taken together, these factors lead us to believe that our model and findings are relevant to, and representative of, the disease process in humans. Because glia play many roles in the nervous system including neuronal trophic support, immune surveillance, blood brain barrier and neurotransmitter metabolism, disturbance of any of these vital functions could account for its role in the mechanisms of toxicity in mitochondrial disease. Our model facilitates robust genetic studies to test the significance of these and other pathways in complex I deficiency. Such studies could identify novel mechanisms, thus provide us with insights into neurobiology, and identify new targets for treatment of these diseases.
MATERIALS AND METHODS
Drosophila genetics
Transgenic RNAi lines 52047 and 100733 were obtained from the Vienna Drosophila RNAi Center. The EAAT1-GAL4 driver was obtained from Serge Birman. RNAi was expressed using da-GAL4, elav-GAL4, repo-GAL4 or EAAT1-GAL4 drivers. All crosses and larval development involving da-GAL4 were performed at 23°C to minimize organismal toxicity in developmental stages. All other crosses and development were conducted at 25°C. All flies were aged at 25°C.
Behavioral analysis
For lifespan analyses 300 sex matched flies were assayed per genotype, with 30 flies placed in each vial. Flies were transferred into new vials every second day. Mortality was plotted and Kaplan–Meyer analysis performed using GraphPad Prism 3 software. For climbing assays, 10 flies were placed in each vial, tapped down to the bottom of the vial, and the number of flies climbing above 5 cm or more, within 10 s was recorded. Six vials per genotype for a total of 60 flies of each genotype were used. Each climbing assay was repeated five times and the mean and standard errors calculated. Student's t-test was used for statistical analysis.
Immunohistochemistry and immunofluorescence
For paraffin histology and immunostaining, adult flies were fixed in formalin and embedded in paraffin. Four micrometers thick serial frontal sections were prepared through the entire fly brain. Antigen retrieval by boiling in sodium citrate was performed before immunostaining. Slides were blocked in PBS containing 0.3% Triton X-100 and 2% milk for 1 h. The following primary antibodies were used at the specified dilutions: anti-ATP synthase subunit alpha (1:500, Abcam ab14748), anti-repo (1:25, Developmental Studies Hybridoma Bank 8D12), anti-elav (1:10, Developmental Studies Hybridoma Bank 9F89A), PARP (1:1500, Abcam ab32064). For immunohistochemistry, biotin-conjugated secondary antibodies (1:200; Southern Biotechnology) and avidin–biotin–peroxidase complex (Vectastain Elite; Vector Laboratories) staining was performed using 3,3′-diaminobenzidine (DAB) (Sigma-Aldrich) as a chromogen. For double-labeling studies, secondary antibodies coupled to Alexa Fluor 488 or Alexa Fluor 555 were used.
Western blots
For standard western blot analysis, adult Drosophila heads were homogenized in 10 μl of Laemmli's buffer (Sigma-Aldrich). Samples were boiled for 5 min, briefly centrifuged, and subjected to SDS-PAGE (PAGEr Gold pre-cast gels). Proteins were transferred to nitrocellulose membranes (Bio-Rad), blocked in 2% milk in PBS with 0.05% Tween 20, and immunoblotted using one of the following antibodies used at the specified dilutions: anti-NDUFS1 (1:2000 Sigma N6039), anti-actin (1:5000, Developmental Studies Hybridoma Bank JLA20), anti-porin (1:2000, Abcam ab14734) and anti-NDUFA9 (1:1000, Abcam ab14713). The appropriate anti-mouse or anti-rabbit horseradish peroxidase-conjugated secondary antibody (Southern Biotechnology) was applied, and signal was detected by chemiluminescence (Alpha Innotech). Band density was quantified using ImageJ. Ponceau S staining was used to monitor protein transfer.
In-gel Assay
Native gel electrophoresis and in-gel complex I activity assays were performed using standard protocols (26). Thirty age and sex matched fly heads were used to obtain the mitochondrial pellet. Mitochondrial pellets were then solubilized in ABCT buffer with 2% dodecyl beta-d-maltoside and equal amount of protein was loaded and run on a non-denaturing gel. In-gel activity was assayed by using nitro tetrazolium blue reduction. The native gel was subsequently blotted and probed with antibodies to NDUAF9, and porin.
Electron microscopy
Brains from 30-day-old ND75 knockdown and control flies were dissected out of the cuticle and fixed in 2.5% glutaraldehyde (Polysciences). Brains were then incubated in 1% osmium tetroxide (Electron Microscopy Sciences)/1.5% potassium ferrocyanide (MP Biomedicals) for 1 h, 1% uranyl acetate for 30 min, and then processed through 70, 90 and 100% ethanol solutions. Brains were then incubated in propyleneoxide for 1 h, embedded in Epon, and allowed to polymerize for 2 days at 60°C. Thin sections were cut and examined with a Tecnai G2 Spirit BioTWIN transmission electron microscope at an accelerating voltage of 80 kV.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health [HL00727-25 to V.R.H., NS082818 to M.B.F.] and the Energy4All foundation (to R.V.). The Vienna Drosophila RNAi Center provided transgenic Drosophila stocks. Antibodies obtained from the Developmental Studies Hybridoma Bank (DSHB) were developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA 52242. Confocal imaging was performed at the Harvard NeuroDiscovery Center Enhanced Neuroimaging Core Facility.
Supplementary Material
Acknowledgments
Conflicts of Interest statement. The authors declare no competing financial interests.
REFERENCES
- 1.Schaefer A.M., Taylor R.W., Turnbull D.M., Chinnery P.F. The epidemiology of mitochondrial disorders—past, present and future. Biochim. Biophys. Acta. 2004;1659:115–120. doi: 10.1016/j.bbabio.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Chinnery P.F. Mitochondrial disorders overview. In: Pagon R.A., Bird T.D., Dolan C.R., Stephens K., Adam M.P., editors. GeneReviewsTM. Seattle, Seattle (WA): University of Washington; 1993. [PubMed] [Google Scholar]
- 3.Scaglia F., Towbin J.A., Craigen W.J., Belmont J.W., Smith E.O., Neish S.R., Ware S.M., Hunter J.V., Fernbach S.D., Vladutiu G.D., et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925–931. doi: 10.1542/peds.2004-0718. [DOI] [PubMed] [Google Scholar]
- 4.Bilen J., Bonini N.M. Drosophila as a model for human neurodegenerative disease. Annu. Rev. Genet. 2005;39:153–171. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- 5.Ambegaokar S.S., Roy B., Jackson G.R. Neurodegenerative models in Drosophila: polyglutamine disorders, Parkinson disease, and amyotrophic lateral sclerosis. Neurobiol. Dis. 2010;40:29–39. doi: 10.1016/j.nbd.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 7.Copeland J.M., Cho J., Lo T., Jr, Hur J.H., Bahadorani S., Arabyan T., Rabie J., Soh J., Walker D.W. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Wu Z., Sawada T., Shiba K., Liu S., Kanao T., Takahashi R., Hattori N., Imai Y., Lu B. Tricornered/NDR kinase signaling mediates PINK1-directed mitochondrial quality control and tissue maintenance. Genes Dev. 2013;27:157–162. doi: 10.1101/gad.203406.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandt U. Energy converting NADH:quinone oxidoreductase (complex I) Annu. Rev. Biochem. 2006;75:69–92. doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- 10.Bénit P., Chretien D., Kadhom N., de Lonlay-Debeney P., Cormier-Daire V., Cabral A., Peudenier S., Rustin P., Munnich A., Rötig A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am. J. Hum. Genet. 2001;68:1344–1352. doi: 10.1086/320603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martín M.A., Blázquez A., Gutierrez-Solana L.G., Fernández-Moreira D., Briones P., Andreu A.L., Garesse R., Campos Y., Arenas J. Leigh syndrome associated with mitochondrial complex I deficiency due to a novel mutation in the NDUFS1 gene. Arch. Neurol. 2005;62:659–661. doi: 10.1001/archneur.62.4.659. [DOI] [PubMed] [Google Scholar]
- 12.Calvaruso M.A., Smeitink J., Nijtmans L. Electrophoresis techniques to investigate defects in oxidative phosphorylation. Methods. 2008;46:281–287. doi: 10.1016/j.ymeth.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 13.Jones M.A., Grotewiel M. Drosophila as a model for age-related impairment in locomotor and other behaviors. Exp. Gerontol. 2011;46:320–325. doi: 10.1016/j.exger.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanji K., Kunimatsu T., Vu T.H., Bonilla E. Neuropathological features of mitochondrial disorders. Semin. Cell Dev. Biol. 2001;12:429–439. doi: 10.1006/scdb.2001.0280. [DOI] [PubMed] [Google Scholar]
- 15.Williams D.W., Kondo S., Krzyzanowska A., Hiromi Y., Truman J.W. Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- 16.Hirano M. Mitochondrial neurology I: encephalopathies. In: DiMauro S., Schon E.A., editors. Mitochondrial Medicine. Informa Healthcare; 2006. [Google Scholar]
- 17.Wallace D.C. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 18.Lobsiger C.S., Cleveland D.W. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 2007;10:1355–1360. doi: 10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilieva H., Polymenidou M., Cleveland D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warrick J.M., Paulson H.L., Gray-Board G.L., Bui Q.T., Fischbeck K.H., Pittman R.N., Bonini N.M. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 21.Colodner K.J., Feany M.B. Glial fibrillary tangles and JAK/STAT-mediated glial and neuronal cell death in a Drosophila model of glial tauopathy. J. Neurosci. 2010;30:16102–16113. doi: 10.1523/JNEUROSCI.2491-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeffen J.L., Smeitink J.A., Trijbels J.M., Janssen A.J., Triepels R.H., Sengers R.C., van den Heuvel L.P. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum. Mutat. 2000;15:123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 23.Magistretti P.J. Neuron-glia metabolic coupling and plasticity. J. Exp. Biol. 2006;209:2304–2311. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- 24.Freeman M.R., Doherty J. Glial cell biology in Drosophila and vertebrates. Trends Neurosci. 2006;29:82–90. doi: 10.1016/j.tins.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Quintana A., Kruse S.E., Kapur R.P., Sanz E., Palmiter R.D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10996–11001. doi: 10.1073/pnas.1006214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azevedo F.A.C., Carvalho L.R.B., Grinberg L.T., Farfel J.M., Ferretti R.E.L., Leite R.E.P., Jacob Filho W., Lent R., Herculano-Houzel S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009;513:532–541. doi: 10.1002/cne.21974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.