

Abstract
Purpose
Although there are effective HER2-targeted agents, novel combination strategies in HER2-overexpressing breast cancers are needed for patients whose tumors develop drug resistance. To develop new therapeutic strategy, we investigated the combinational effect of entinostat, an oral isoform-selective histone deacetylase type I inhibitor, and lapatinib, a HER2/EGFR dual tyrosine kinase inhibitor, in HER2+ breast cancer cells.
Methods
We assessed the combinational synergistic effect and its mechanism by CellTiter Blue assay, flow cytometry, anchorage-independent growth, quantitative real-time PCR, small interfering RNA, Western blotting, and mammary fat pad xenograft mouse models.
Results
We found that compared with entinostat or lapatinib alone, the two drugs in combination synergistically inhibited proliferation (P < 0.001) and reduced in vitro colony formation (P < 0.05) and resulted in significant in vivo tumor shrinkage or growth inhibition in two xenograft mouse models (BT474 and SUM190, P < 0.001). The synergistic anti-tumor activity of the entinostat/lapatinib combination was due to downregulation of phosphorylated Akt, which activated transcriptional activity of FOXO3, resulting in induction of Bim1 (a BH3 domain-containing pro-apoptotic protein). Furthermore, entinostat sensitized trastuzumab/lapatinib-resistance-HER2-overexpressing cells to the trastuzumab/lapatinib combination and enhanced the anti-proliferation effect compare with single or double combination treatment.
Conclusions
This study provides evidence that entinostat has enhanced anti-tumor effect in combination with HER2-targeted reagent, lapatinib, and resulting induction of apoptosis by FOXO3-mediated Bim1 expression. Our finding justifies for conducting a clinical trial of combinational treatment with entinostat, lapatinib and trastuzumab in patients with HER2-overexpressing breast cancer resistant to trastuzumab-based treatment.
Keywords: Entinostat, Lapatinib, HER2-overexpressing breast cancer, FOXO3, Bim1
Introduction
Gene amplification or overexpression of HER2 has been reported in 15-20% of invasive breast carcinomas, and this abnormal expression is associated with an aggressive phenotype and poor prognosis [1]. Currently, four therapeutic options have been approved by the U.S. Food and Drug Administration (FDA) for patients with HER2-overexpressing (HER2+) breast cancer: trastuzumab, pertuzumab, lapatinib, and TDM-1 [2]. Trastuzumab (Roche) is a humanized monoclonal antibody that binds to the extracellular segment of the HER2 receptor [2,3]. Trastuzumab inhibits HER2 dimerization and PI3K signaling pathway, resulting in reduced cell proliferation by G1-phase arrest of the cell cycle [4], and induces cell death by antibody-dependent cell-mediated cytotoxicity [5]. Lapatinib (GlaxoSmithKline, NC) is a dual epidermal growth factor receptor (EGFR) and HER2 tyrosine kinase inhibitor that was approved specifically for treatment of patients with HER2+ advanced-stage breast cancer [6]. Lapatinib reversibly inhibits auto-phosphorylation of the C-terminus intracellular kinase domain of both EGFR and HER2 and thereby suppresses its downstream targets, by inhibiting the PI3K-AKT and MAPK-ERK1/2 pathways, resulting in induction of G1 phase arrest of the cell cycle and apoptosis [7-9]. Although it has been successful in prolong survival, both trastuzumab or lapatinib generally develop resistance 1 year after initiating treatment, with rapid progression of disease [6,10]. Such resistance may be overcome by combining anti-cancer drugs that work by different mechanisms.
To overcome drug resistance and thereby increase therapeutic potential, histone deacetylase (HDAC) inhibitors are being studied as potential combinatory agents [11]. Recent studies have shown that HDAC inhibitors are effective as epigenetic targeted anti-cancer drugs [12,13]. Entinostat (formerly MS-275, Syndax Pharmaceuticals Inc., MA), a selective class I HDAC inhibitor with low toxicity to normal cells, is a synthetic benzamide derivative that has shown both in vitro and in vivo anti-cancer effects against various human cancers [14]. In breast cancer, entinostat induces TRAIL-mediated apoptosis and mediates chemosensitization [15]. In a randomized phase II study, entinostat with an aromatase inhibitor significantly prolonged the median progression-free survival and reduced the risk of disease progression compared with the aromatase inhibitor alone in patients with metastatic estrogen receptor-positive (ER+) breast cancer [16]. Entinostat was shown to sensitize ER-negative tumors to aromatase inhibitors by functional activation of ER-α and aromatase [17] and to restore responsiveness of letrozole-resistant cells to aromatase inhibitors in a breast cancer xenograft model [18]. However, it is not known whether entinostat can reverse resistance to anti-HER2 targeting drugs and/or enhance the anti-tumor effect of anti-HER2 drugs in HER2+ breast cancer cells.
The purpose of this study was to investigate the anti-tumor effect of the combination of entinostat and lapatinib in HER2+ breast cancer cell lines and a xenograft mouse model. We also elucidated the mechanism of the toxicity induced by the combination. We found that combined treatment with entinostat and lapatinib had synergistic anti-tumor effects both in vitro and in vivo. We also found that this synergistic mechanism involves AKT, FOXO3a, and Bim1; our data indicate that Bim1 is a major molecule involved in the synergistic anti-tumor effect of entinostat/lapatinib in HER2+ breast cancer cells.
Materials and Methods
Detailed information regarding In vitro cell proliferation assay, Cell-cycle distribution and apoptosis analysis, Soft agar assay, Transfection, Western blot analysis, Immunohistochemistry (IHC), and Nuclear and cytosolic protein fractions are included in Electronic supplementary material.
Cell lines
Human breast cancer cell lines BT20, MDA-MB-231, MDA-MB-468, SKBR3, and BT474 were purchased from American Type Culture Collection (ATCC, Manassas, VA). SUM190 was purchased from Asterand, Inc. We authenticated all tested cell lines by genotyping through MD Anderson Cancer Center's Characterized Cell Line Core Facility.
Reagents and antibodies
Entinostat was provided by Syndax Pharmaceuticals, Inc. Lapatinib was purchased from ChemieTek. Small interfering RNA (siRNA) targeting FOXO3 and Bim1 were purchased from Sigma-Aldrich. The following antibodies were purchased from Cell Signaling Technology (Beverly, MA): pEGFR-Tyr1173, EGFR, pHER2-Tyr1248, HER2, pHER3-Tyr1289, HER3, pERK-Thr202/Tyr204, ERK, pAKT-Ser473, AKT, Bim1. We obtained β-actin (clone AC-15; Sigma-Aldrich, St Louis, MO), U1 snRNP70 (Santa Cruz Biotechnology, Santa Cruz, CA), Alexa Fluor 680 and 800 (Invitrogen, Carlsbad, CA), and horseradish peroxidase (HRP)-conjugated antibodies (Thermo Scientific, Rockford, IL). The following small interfering RNA oligos (Sigma-Aldrich) were used for depletion of FOXO3a or Bim1: FOXO3a #1, 5′CGAAUCAGCUGACGACAGU[dT][dT]3′; FOXO3a #2, 5′CGAUUCAUGCGGGUCCAGA[dT][dT]3′; FOXO3a #3, 5′GAAUGAGGGCUGACUGAA[dT][dT]3′; Bim1 #1, 5′GAAUGGUUAUCUUACGACU[dT][dT]3′; Bim1 #2, 5′CAGAUAUGCGCCCAGAGAU[dT][dT]3′; Bim1 #3, 5′CAUGAGUUGUGACAAAUCA[dT][dT]3′. Knockdown efficiency of single siRNAs was tested by Western blotting (Supplementary Fig. S1), and we used pooled siRNA of three target siRNAs for knockdown experiments. The scrambled siRNA was purchased from Thermo Scientific (ON-TARGETplus Non-targeting Control Pool, part number D-001810-10).
In vivo xenograft animal model
Four- to eight-week-old female athymic BALB/c nu/nu mice were purchased from Harlan Laboratories for the BT474 experiment and MD Anderson's Department of Veterinary Medicine & Surgery for the SUM190 experiment. Mice were housed under specific pathogen-free conditions and treated in accordance with National Institutes of Health guidelines. To establish breast cancer xenografts in nude mice, BT474 (1×107 cells/100 μl) or SUM190 (2×106 cells/100 μl) cell suspensions were injected into one site in the abdominal mammary fat pad area of each mouse. We observed 100% tumor incidence for both the BT474 and SUM190 cell lines. Drug treatment was started when the tumors were approximately 70-150 mm3. Tumor volume (V=0.52×W×L2) and body weight were measured twice weekly. We used the following vehicles for drug preparation: HP-β-CD solution (30% w/v, 51 mM NaCl, pH 5.0) for entinostat, and PEG400 solution (40% v/v, pH 5.0) for lapatinib. Drug treatment continued for 70 days (BT474) or 25 days (SUM190), and then all mice were euthanized, and samples of tumors were collected at biopsy and analyzed for immunohistochemical staining.
Quantitative real-time PCR
Total RNA was purified using the PureLink® RNA Mini Kit (Invitrogen), and real-time qRT-PCR was performed using the iScript™ One-Step RT-PCR Kit with SYBR® Green (Bio-Rad, Hercules, CA) according to the manufacturer's instruction. Equal amounts of total RNA (15 ng for each sample) were mixed, and target genes were amplified with a specific primer set using the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). The following primers (Sigma-Aldrich) were used for detection: Bim1 primers were 5′CAGCACCCATGAGTTGTGAC3′ (forward) and 5′CCTCATGGAAGCCATTGCAC3′ (reverse), and 7SL scRNA primers were 5′ATCGGGTGTCCGCACTAAGTT3′ (forward) and 5′CAGCACGGGAGTTTTGACCT3′ (reverse). 7SL scRNA levels were used as an endogenous control. The real-time PCR data were calculated using the comparative threshold cycle method and iCycler CFX96 analyzer software (Bio-Rad).
Isobologram analysis
To evaluate the effect of the drug combination, we used isobologram analysis of IC50 values [19,20]. Fractional inhibitory concentration (FIC) was calculated for each on the basis of the following equation: FIC Drug , in which IC50A (combination) is the 50% inhibitory concentration of drug A in combination with drug B. Isobologram analysis (FICs index, sum of FIC index for drug A and drug B) indicated a synergistic (< 0.5), additive (0.5 - 2.0), or antagonistic (> 2.0) effect of the two-drug combination.
Statistical analysis
For experimental outcomes, descriptive statistics (mean and standard deviation) were summarized for each group. An analysis of variance (ANOVA) model was used to compare the mean outcome values among the tested groups. Statistical analyses were performed using an unpaired t-test with Prism version 5 (GraphPad Software, La Jolla, CA). P values of <0.05 were considered statistically significant.
Results
Targeted inhibition of HER2 and EGFR reduces the proliferative capacity of HER2+ breast cancer cells in a synergistic manner
We first investigated whether entinostat can enhances lapatinib efficacy in HER2+ breast cancer cells, we screened a panel of low HER2-expressing (HER2-) breast cancer cell lines (BT20, MDA-MB-231, and MDA-MB-468) and HER2+ breast cancer cell lines (SUM190, SKBR3, and BT474). The combination of entinostat and lapatinib synergistically induced an increase in the sub-G1 population, indicating apoptosis, in HER2+ cell lines (SUM190, 40%; SKBR3, 45%; and BT474, 80% compared with untreated cells), but had no effect in HER2- cell lines (Fig. 1a). To further study the combinational effect of entinostat and lapatinib in HER2+ cells, we selected the BT474 and SUM190 cell lines. We then evaluated the synergistic anti-proliferation index of entinostat and lapatinib using an ATP-based cell viability assay, WST-1. When cells were treated with the combination of entinostat and lapatinib for 72 hours, we observed a significant shift in the IC50 value of lapatinib concentration (BT474, from 0.13 μM to 0.026 μM, FIC index = 0.469; SUM190, from 1.01 μM to 0.021 μM, FIC index = 0.319) (Fig. 1b). These data indicated that entinostat acts as a sensitizer for lapatinib in HER2+ breast cancer cells.
Fig. 1.
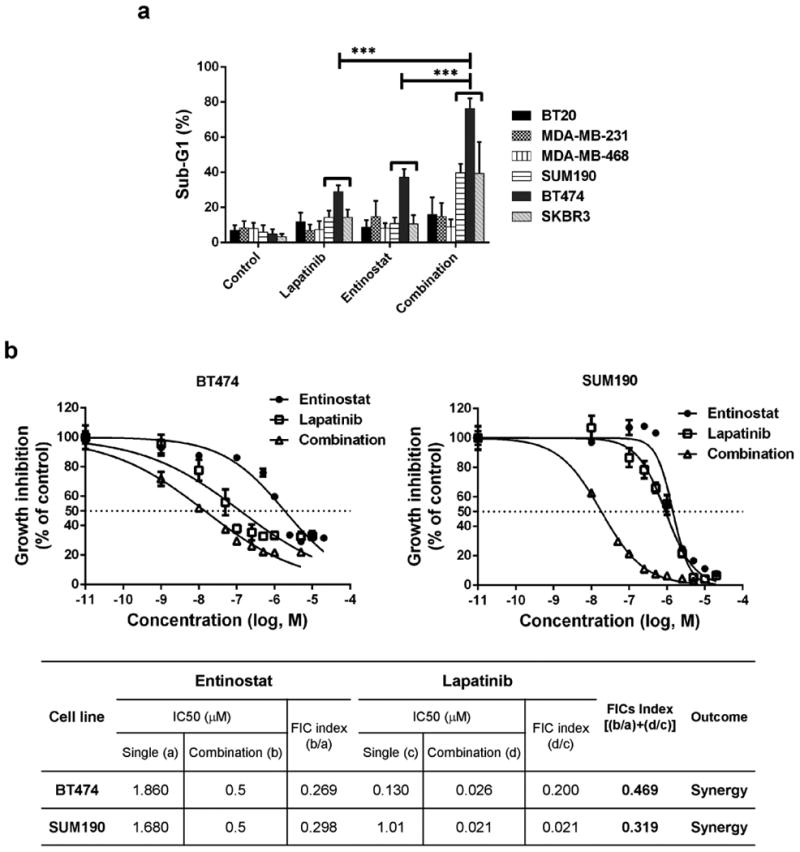
The combination of entinostat and lapatinib inhibited proliferation of HER2+ breast cancer cells. a Cells (2×105) were placed on a 6-well plate overnight, treated with or without the drugs (entinostat, 5.0 μM for all cell lines; lapatinib, 1.0 μM for HER2+ cell lines and 5.0 μM for HER2- cell lines) for 72 hours, and then stained with propidium iodide (PI) for cell cycle analysis using flow cytometry. Each bar represents the mean of 3 independent experiments; error bars, SD. *** p <0.001 combination compared with either entinostat or lapatinib. b Cells were treated with entinostat or lapatinib or both in combination for 72 hours. For combination, entinostat was fixed with 0.5 μM and mixed with various range of lapatinib (0.01 – 20 μM). And then a WST-1 proliferation assay was performed. For analysis, the non-linear fit curve method was used via GraphPad Prism software. The table represents the synergistic inhibitory effect of entinostat and lapatinib. The fractional inhibitory concentration (FIC) for the combination is the sum of the FICs of the 2 drugs and was interpreted as follows: < 0.5, synergy; 0.5 – 2.0, additive; > 2.0, antagonistic.
We next analyzed the effect of entinostat and lapatinib on cell cycle distribution and cell apoptosis with a clinically relevant (≤ 1 μmol/L) dose. After 48 hours of treatment, lapatinib increased the sub-G1 fraction in both BT474 and SUM190 cells; however, entinostat strongly increased G1 arrest in BT474 cells and G2 arrest in SUM190 cells. Both cell lines showed an increased sub-G1 fraction for the combination treatment compared with the single treatments (Fig. 2a). To further confirm apoptosis, we measured the Annexin V-positive cells following treatment with each agent alone and both in combination. As shown in Fig. 2b, lapatinib alone induced apoptotic cells by 12.06 ±2.92% (BT474) and by 17.79 ±3.03% (SUM190) compared with untreated cells, whereas entinostat alone induced apoptosis by 15.49 ±2.13% in SUM190 cells. However, when cells were treated with both entinostat and lapatinib, apoptotic cells were significantly increased by 22.01 ±3.54% (BT474, P<0.01) and by 31.1 ±4.36% (SUM190, P<0.01) compared with untreated cells. These data indicate that the combination of entinostat and lapatinib was more effective in inducing enhanced apoptosis in HER2+ breast cancer cells.
Fig. 2.
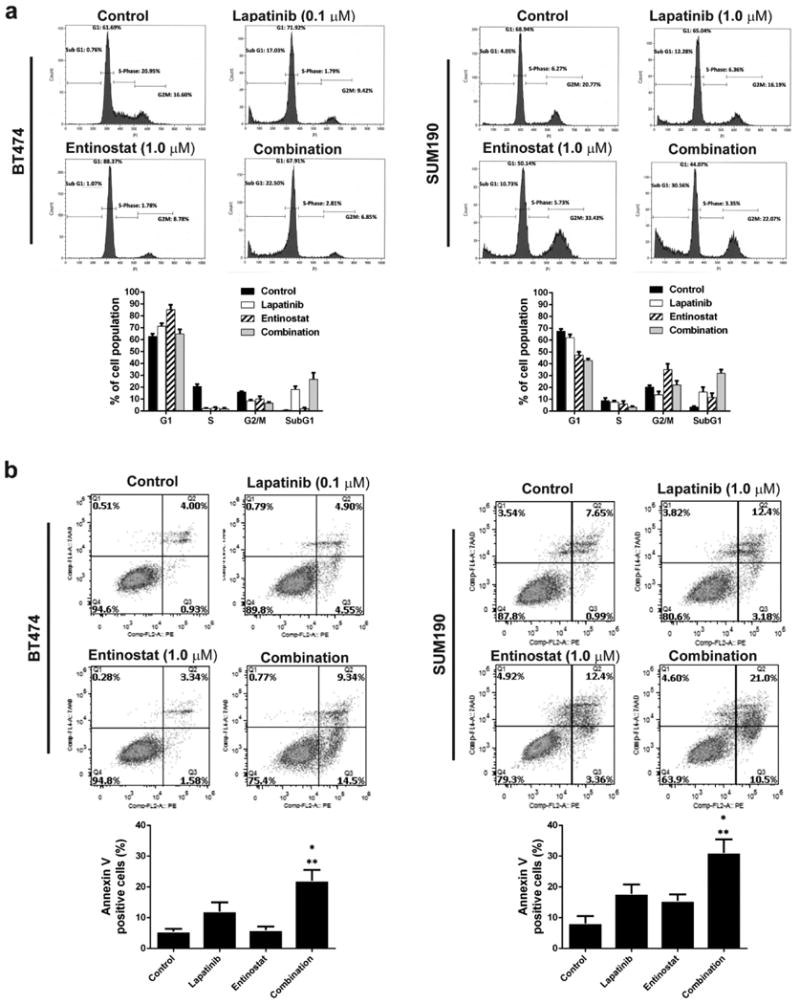
Combined entinostat and lapatinib at clinically relevant doses inhibited the cell cycle and induced apoptosis. a Cells (2×105) were placed on a 6-well plate overnight and then treated with or without the drugs (entinostat, 1.0 μM for all cell lines; lapatinib, 1.0 μM for SUM190 and 0.1 μM for BT474) for 48 hours. After the cells were collected, a PI staining assay was performed for cell-cycle analysis using flow cytometry. b Cells (2×105) were placed on a 6-well plate overnight, and then treated with or without the drugs (entinostat, 1.0 μM for all cell lines; lapatinib, 1.0 μM for SUM190 and 0.1 μM for BT474) for 48 hours. An Annexin V/7AAD staining assay was performed for detection of apoptosis using flow cytometry. * p <0.05 combination compared with lapatinib. ** p <0.01 combination compared with either control or entinostat. Data shown are representative of 3 experiments with similar results.
Combination treatment of entinostat and lapatinib effectively suppressed in vitro colony formation ability and tumor growth in a breast cancer xenograft
To determine whether the combination of entinostat and lapatinib enhances anti-tumorigenic effect in HER2+ breast cancer cells over that of single agents, we performed an in vitro (soft-agar colony formation) tumorigenicity assay. Preliminary studies indicated that the IC50 dose for both drugs could totally ablate colony formation (data not shown). For this reason, we selected lower doses than the IC50s of entinostat and lapatinib (see methods). When BT474 and SUM190 cells were treated with both drugs, the number of colonies was significantly reduced compared with those in cells treated with either drug alone (P<0.05), and a similar reduction was seen for colony size compare with either drug alone (P<0.05) (Fig. 3a, b).
Fig. 3.
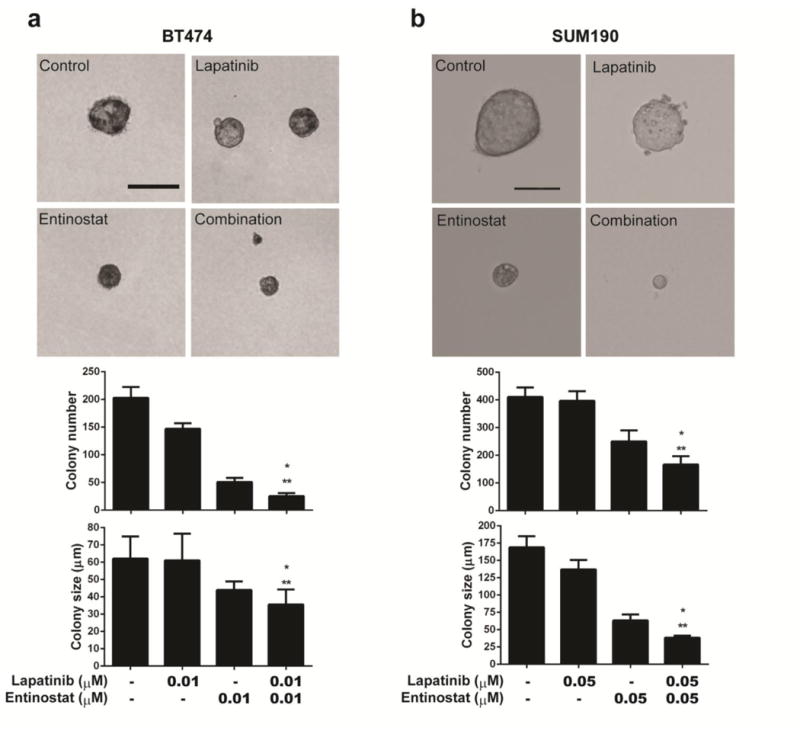
The combination of entinostat and lapatinib enhanced drug efficacy in terms of in vitro colony formation in a soft agar assay. For visualizing or counting, colonies were stained with 200 μL of MTT (1 mg/mL) solution for 2 hours and counted using the GelCount colony-counting system according to the manufacturer's instructions. Statistical significance was evaluated by t test using GraphPad Prism software. a BT474. b SUM190. Cells (2×103) were seeded into soft agar with the indicated drug(s) and incubated for 3 weeks. Scale bars, 100 μm. Bars in the graph represent means; error bars, standard deviation. * p <0.05 combination compared with entinostat. ** p <0.01 combination compared with either control or lapatinib. Data shown are representative of 3 experiments with similar results.
After confirming that the combination of entinostat and lapatinib reduced cell proliferation and anchorage-independent growth in vitro, we examined whether these two drugs singly or together would inhibit tumors in a xenograft animal model of breast cancer. Mice (8 per group) were treated with lapatinib (75 mg/kg/day for BT474, 60 mg/kg/day for SUM190), entinostat (15 mg/kg/day), or a combination of both drugs for a period of 70 days (BT474) and 25 days (SUM190), respectively. Compared with mice treated with vehicle control, the mice that were treated with entinostat/lapatinib showed tumor growth suppression of 90% (P <0.001) and 45% (P <0.001) in BT474 and SUM190, respectively (Fig. 4a, b). In contrast with in vitro proliferation data (Figs 1, 2), growth inhibition efficacy was moderate in the SUM190 xenograft model compared to the BT474 xenograft model. We speculate that this effect is due to the type of cancer. The SUM190 cell line is derived from inflammatory breast cancer, an aggressive and fast-growing breast cancer in which cancer cells infiltrate the skin and lymph vessels of the breast. When increased the amounts of lapatinib (100 mg/kg/day) and entinostat (20 mg/kg/day) in the SUM190 xenograft model, combination treatment enhanced tumor growth suppression (69%, P <0.0001) (Supplementary Fig. 2a).
Fig. 4.
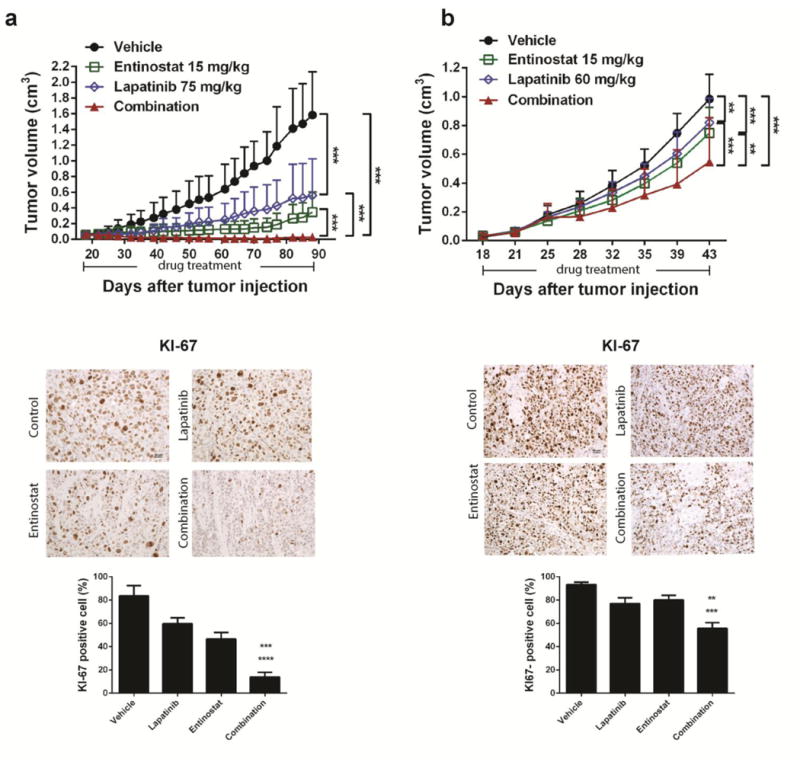
The combination of entinostat and lapatinib effectively suppressed tumor growth in breast cancer xenograft model. Cells (1×107 for BT474 or 2×106 for SUM190) in 50% Matrigel solution were transplanted into the mammary fat pads of 8 female nude mice per group. a BT474. b SUM190. Treatment started on day 18 after tumor cell implantation. The administered volume was 0.2 ml/30 g mouse body weight. Treatment was continued for 70 days for the BT474 group, and 25 days for the SUM190 group. IHC staining represents quantification of KI-67-positive cells in tumor tissue samples from each treatment group. Graphs and statistical significance were evaluated by an ANOVA using GraphPad Prism software. Error bars, standard deviation; magnification, ** p <0.01, *** p <0.001, ×20; scale bar, 50 μm.
After the initial response to lapatinib or entinostat, the mice harboring the BT474 tumors developed signs of resistance after a mean of 40 days of single-agent treatment. In contrast, the mice treated with the combination of entinostat and lapatinib had long-lasting tumor regression, until the endpoint of the experiment (90 days). In this group, 6 of the 8 mice showed significant tumor shrinkage (Supplementary Fig. 2b). These data support our in vitro data showing an enhanced anti-tumor effect of the lapatinib and entinostat combination. The anti-tumor effect of the combination was accompanied by low in vivo toxicity, evaluated based on the weight measurements of the mice during drug treatment (Supplementary Fig. 3).
Efficacy of combination treatment in HER2+ breast cancer cells involved FOXO3 -mediated BIM1 expression
To identify the mechanism of the synergistic anti-tumor effect of entinostat and lapatinib, we first focused on the MEK-ERK and PI3K-AKT pathways, well-established downstream signaling pathways of the HER2/HER1 pathway [21]. As shown in Supplementary Fig. 4a, lapatinib inhibited phosphorylation of EGFR, HER2, HER3, and their downstream effector molecules, ERK and AKT. However, lapatinib increased expression levels of HER2, as expected from previous studies [22]. Compared with either drug alone, combination treatment showed enhanced inhibition effect on both phosphorylation and expression levels of EGFR, HER2, and HER3 in tested cells. Indeed, we observed enhanced inhibition of phospho-AKT after combination treatment. We also observed the same inhibition effect on AKT phosphorylation in low-dose combination conditions (Supplementary Fig. 4b). To further confirm the involvement of AKT in the anti-proliferation effect of both lapatinib and trastuzumab, we transiently overexpressed the constitutively active form of AKT (AKT-CA) in BT474 and SUM190 cells and then treated them with lapatinib and entinostat together. AKT-CA increased the cell viability in the presence of lapatinib and entinostat (Supplementary Fig. 5a, b). These data indicate that AKT is a key molecule involved in the synergistic anti-tumor effect of lapatinib and entinostat. Because AKT inhibition activates transcriptional activity of FOXO3 by relieving the suppression pathway [22-24], we also observed enhanced inhibition of AKT phosphorylation with combination treatment (Supplementary Fig. 4a, b).
We next assessed whether combination treatment induces transcriptional activity of FOXO3a and leads to apoptosis of BT474 and SUM190 cells. We observed reduced phosphorylation of FOXO3a under lapatinib- or combination-treatment conditions (Fig. 5a), and nucleus-cytosol fractionation analysis showed increased translocalization of FOXO3 under combination-treatment conditions (Fig. 5b). Using qRT-PCR analysis, we found that the expression levels of FOXO3-targeted genes related to apoptosis or cell cycle arrest—Bim1 (Fig. 5c, d), GADD45, and p21Waf (Supplementary Fig. 6)—were significantly increased by combination treatment, whereas expression of receptor tyrosine kinase receptor HER3 was increased by lapatinib single treatment (Supplementary Fig. 6). To assess how the entinostat/lapatinib combination induces apoptosis, we focused on Bim1, an apoptosis inducer. Using siRNA targeting FOXO3a, we identified that entinostat/lapatinib-induced Bim1 expression is mediated by FOXO3a (Fig. 5e). To confirm whether entinostat/lapatinib-induced apoptosis depends on Bim1 expression, we first knocked down Bim1 expression using siRNA, and then treated the cells with lapatinib and entinostat together (Fig. 5f, g). We observed significantly increased cell viability after Bim1 siRNA treatment compared with siControl at a high-dose range of lapatinib and entinostat combination in both BT474 and SUM190 cell lines (40% vs. 10% viability, P <0.001). For low-dose combination treatment (both drugs having concentrations ≤ 0.5 μM), cell viability was not significantly increased by Bim1 siRNA treatment (Supplementary Fig. 7). These data indicated that cells undergo cell-cycle inhibition under low-dose treatment conditions; however, to approach an effective apoptosis level, threshold doses are required (> 0.5 μM for BT474 and > 1.25 μM for SUM190). In accordance with the in vitro data, IHC staining of combination-treatment BT474 xenograft tumor samples revealed higher Bim1 levels (Fig. 6a), and we also observed dose-dependent Bim1 expression levels in SUM190 xenograft tumor samples (Fig. 6b, c). These results suggest that the combination of entinostat and lapatinib enhanced apoptosis through FOXO3-mediated Bim1 expression in HER2+ breast cancer cells.
Fig. 5.
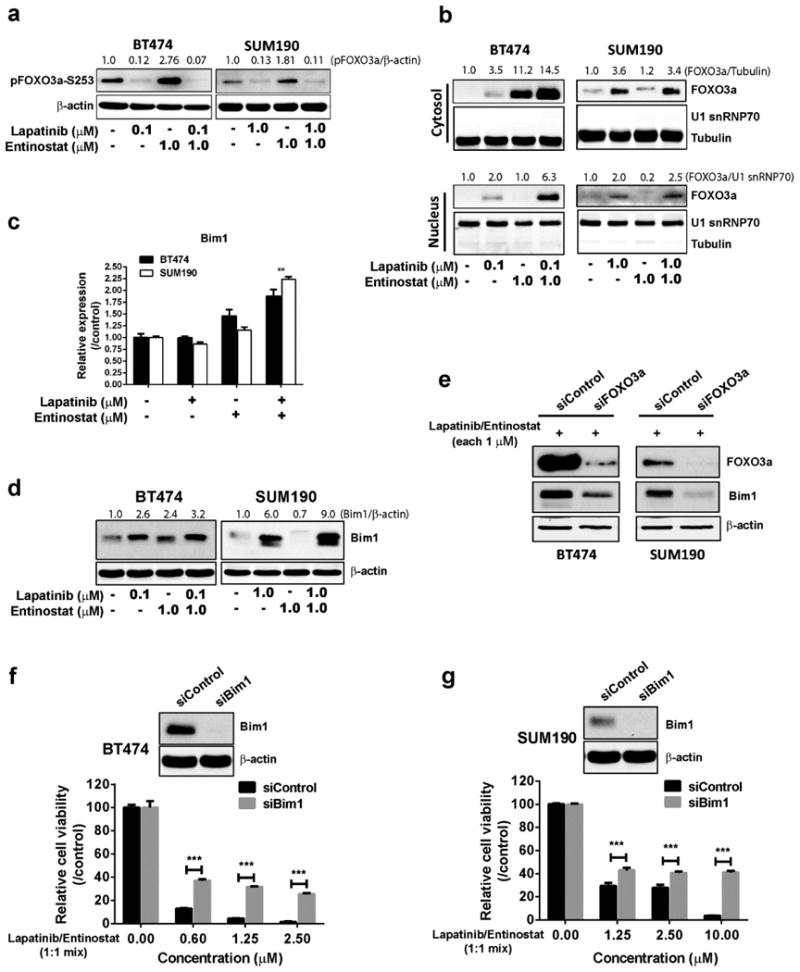
The enhanced anti-tumor effect of the entinostat/lapatinib combination depends on FOXO3a-mediated Bim1 expression. a Combination treatment or single-agent lapatinib treatment inhibited pFOXO3a phosphorylation at the Ser253 position. Cells were treated with drugs for 24 hours, and then total cell lysates were collected for Western blotting. The ratios of pFOXO3a-S253are shown above the blots. b Cytosol-nucleus fractionation. Cells (2×106) were placed on a 6-well plate overnight and then treated with or without the drugs for 24 hours. U1 snRNP 70, a measure of FOXO3a, was assessed by Western blotting. The ratios of FOXO3a are shown above the blots. c Quantitative RT-PCR. Cells (2×105) were placed on the 6-well plate overnight and then treated with or without the drugs (entinostat, 1.0 μM for both cell lines; lapatinib, 0.1 μM for BT474 and 1.0 μM for SUM190) for 24 hours. Equal amounts of total RNA (20 ng for each sample) were mixed, and target genes were amplified with a specific primer set. ** p <0.01 compared combination with either control or single treatment. d Western blot assay. Cells (2×105) were placed on a 6-well plate overnight, and then treated with or without the drugs for 48 hours. e Effect of siFOXO3 on Bim1 expression. Cells were pre-treated with scrambled siRNA or siFOXO3a for 48 hours, followed by entinostat/lapatinib treatment for 48 hours. Cell lysates were harvested and Western blotting performed. The ratios of Bim1are shown above the blots. f, g Depletion of Bim1 by siBim1 protected entinostat/lapatinib-induced cell death in BT474 and SUM190. Cells were pre-treated with scrambled siRNA or siBim1 for 48 hours, followed by entinostat/lapatinib treatment for 72 hours. Cell viability was assessed using a WST-1 assay, *** p <0.001 compared siControl with siBim1.
Fig. 6.
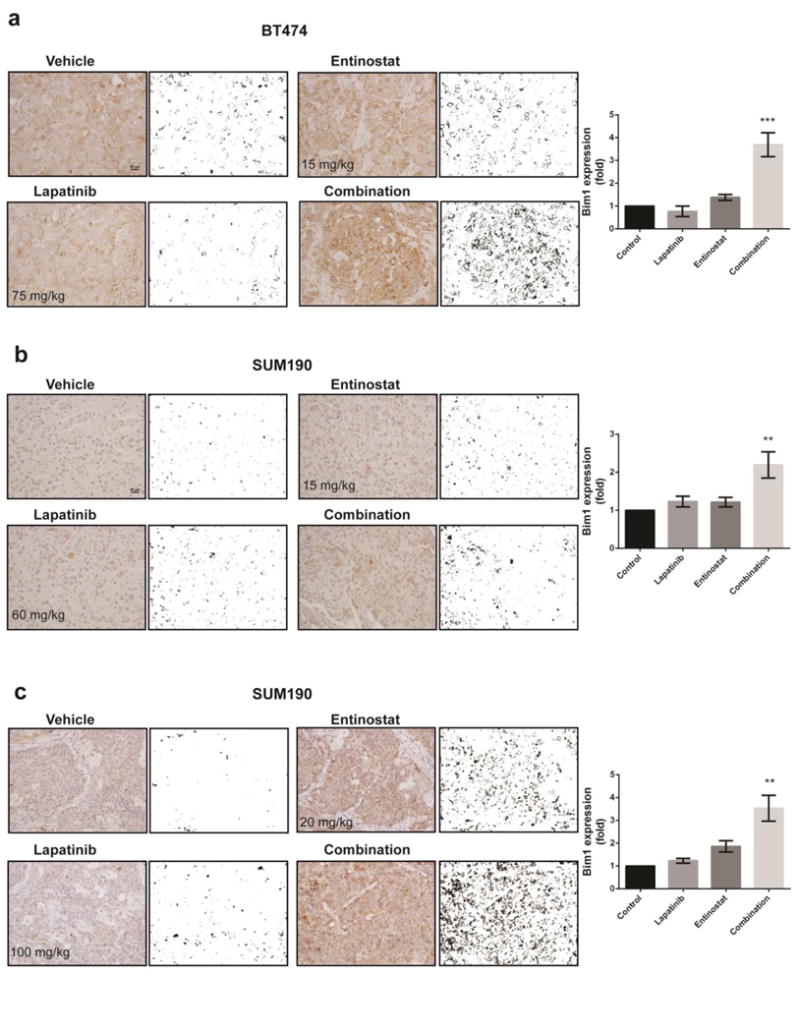
Immunohistochemical staining and quantification of Bim1 expression in representative tumor samples from each treatment group. a BT474. b SUM190 with low-dose treatment. c SUM190 with high-dose treatment. The images were converted by ImageJ software to accomplish quantification of Bim1 expression. Data shown are representative of 3 IHC staining experiments with similar results. Magnification, ×20; scale bar, 50 μm. Bars in graph represent means; error bars, standard deviation. ** p <0.01, *** p <0.001 compared combination with either control or single treatment.
Entinostat restores responsiveness of trastuzumab/lapatinib resistance-HER2+ cells to trastuzumab/lapatinib combination treatment
Lapatinib is commonly used in combination with trastuzumab for HER2+ breast cancer treatment; however, acquired resistance to both lapatinib and trastuzumab remains a substantial clinical problem. Thus, we next aimed to investigate whether entinostat can overcome trastuzumab/lapatinib resistance. To do this, we used a preclinical model, a SUM190 cell line with acquired resistance to both lapatinib and trastuzumab (SUM190-TLR). As shown in Fig. 7, there was no significant anti-proliferation effect with either single treatment or a trastuzumab/lapatinib combination, whereas entinostat sensitized SUM190-TLR cells to the trastuzumab/lapatinib combination and enhanced the anti-proliferation effect. Further, our data suggests that an effective dose of the entinostat/trastuzumab/lapatinib combination in trastuzumab-resistant cancer cells can be achieved at concentrations lower than 1 μM.
Fig. 7.
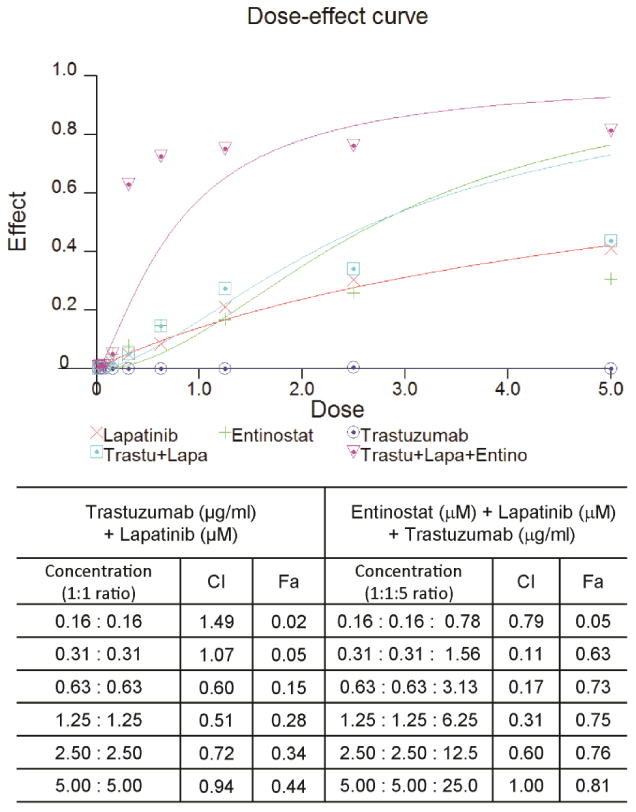
Entinostat enhances trastuzumab/lapatinib efficacy in trastuzumab/lapatinib-resistant HER2+ breast cancer cells. Trastuzumab/lapatinib-resistant SUM190 cells (SUM190-TLR) were obtained by continuously exposing SUM190 cells (which are known to be trastuzumab resistant) to lapatinib (5 μg/ml) for 6 months. Dose-effect curve was analyzed using CalcuSyn software (Biosoft, Cambridge, UK). Combination index (CI) of entinostat, lapatinib and trastuzumab in SUM190-TLR cells: A quantitative measure of the degree of drug interaction. CI value: <0.1 indicates very strong synergism; 0.10-0.30, strong synergism; 0.30-0.70, synergism; 0.70-0.85, moderate synergism; 0.85-0.90, slight synergism; 0.90-1.10, nearly additive; 1.10-1.20, slight antagonism; 1.20-1.45, moderate antagonism; 1.45-3.3, antagonism; 3.3-10, strong antagonism; >10, very strong antagonism. Fractional index (Fa), the fraction of cells affected by the dose.
Discussion
Our findings revealed that the combination of entinostat/lapatinib promotes FOXO3 translocalization into the nucleus and increases its target genes' expression. We showed that the synergistic effect of lapatinib/entinostat-induced growth inhibition and death of cancer cells are mediated through FOXO3-induced Bim1 expression. Further, we observed for the first time that entinostat re-sensitized lapatinib/trastuzumab-resistant cells to lapatinib/trastuzumab treatment at low-dose concentration. These data indicate that entinostat could be a potential addition to the HER2-targeted therapies employed to treat trastuzumab-resistance breast cancer.
Several studies demonstrated that activation of the PI3K-AKT-mTOR pathway results in low efficacy of both trastuzumab and lapatinib [25,26]. In addition, the activated AKT pathway also plays a major role in HER2-mediated resistance to tamoxifen and paclitaxel [27,28]. Here we observed that the drug-induced inactivation of AKT phosphorylation was a critical mediator of HER2+ breast cancer cell death. Thus, our current data imply that entinostat could be effective for trastuzumab- or tyrosine kinase inhibitor-resistant HER2+ breast cancer cells that show activated AKT status. Indeed, a similar synergy was observed by combining entinostat and trastuzumab, which exhibited potential for overcoming trastuzumab resistance via disrupting HER2/HER3 interactions and downstream kinases [29].
It has been demonstrated that activated AKT induces phosphorylation of FOXOs and induces proteasomal degradation, thereby inhibiting the apoptosis pathway [30,31]. Previous studies demonstrated that FOXO3-induced Bim expression induces apoptosis in paclitaxel-treated breast cancer cell lines [32], sulindac sulfide (a non-steroidal anti-inflammatory drug) induces thyroid cell apoptosis through FOXO3-induced Bim1 and GADD45 expression [33], and Bim and Puma were abundantly increased after inhibition of HER2 in a mouse breast cancer model [34]. On the other hand, FOXO3a also contributed to drug resistance through gene expression and cell survival pathways. Hui et al. reported that FOXO3a induces PIK3CA (the catalytic subunit of Class 1A PI3K) expression, thereby increasing drug resistance via the PI3K-AKT pathway in leukemia cells [35]. Recent studies identified IGF-1R, IGFBP1, INSR, and HER3 as FOXO3a target genes that, when expressed, result in enhancement of the PI3K-AKT feedback pathway that contributes to survival mechanisms [22,35-37]. We also observed the elevated HER3 protein level by lapatinib single treatment, and it is inhibited by combination treatment with entinostat. The possible explanation for this effect is epigenetic modulation by entinostat. Previous studies have suggested that entinostat downregulates HER2 and HER3 expression by induction of repressor microRNAs [38], and HDAC inhibitors repress the transcription of highly expressed genes as well as high-copy-number genes in HER2+ breast cancer genomes [39]. However, further mechanistic study is necessary to elucidate how entinostat regulates transcriptional activity of FOXO3a in conjunction with lapatinib treatment.
The rescue effect produced by Bim1 siRNA on cell survival was significantly increased under combination drug treatment at high-dose concentrations, but cell viability did not fully recover. These data imply that there may be other mechanisms that independently contribute to the anti-tumor effect of the entinostat/lapatinib combination. Our data indicate that entinostat additionally induces growth inhibition itself through induction of cell cycle arrest genes (p21Waf, GADD45), thereby blocking cell cycle progression, represented by the observed G1 arrest (BT474) or G2 arrest (SUM190). Previous studies have shown that enhanced p21Waf levels induced by an HDAC inhibitor promoted proteasomal degradation of cyclin B1 and resulted in G2/M arrest [40] and that GADD45 promotes G2/M arrest via nuclear export and kinase activity of Cdc2 [41]. We observed that single treatment with entinostat appeared more effective in inhibiting growth than single treatment with lapatinib in soft agar culture. We presume that this phenomenon is caused by p21Waf and cyclin D1 expression level. Gua et al. reported that increased p21Waf resulted in markedly reduced colony formation ability [42]. We also observed a lower cyclin D1 expression level for entinostat than for lapatinib in both the SUM190 and BT474 cell lines (data not shown).
Differing from the in vitro proliferation assay data, we observed a differential in vivo response to the entinostat/lapatinib combination in BT474 and SUM190 xenograft models. Bim1 expression was not strongly expressed in SUM190 xenograft tissue samples with low-dose drug treatment. It is speculated that PIK3CA mutation (H1047R) status may be correlated with the different in vitro and in vivo results. The SUM190 cell line has an endogenous PIK3CA-H1047R mutation that confers resistance to HER2 targeted drugs in HER2+ breast cancer cell lines [43]. Furthermore, ectopic overexpression of PIK3CA-H1047R in BT474 cells leads to resistance to lapatinib [44]. The tumor microenvironment may also correlate with differential drug efficacy. When cells were tested in a proliferation assay in a Type I collagen-coated flask, SUM190 cells showed elevated growth rate (30-45%) and high IC50 value of lapatinib (unpublished data). Therefore, we speculated that SUM190 cells showed growth inhibition rather than cell death under low-dose conditions both in vitro and in vivo.
Acquired resistance to HER2-targeted drugs may be caused by multiple mechanisms, such as genetic modifications, post-translational modification, activation of bypass pathways, hypoxia, or EMT [45-47]. Thus, to overcome this resistance, multiple means of reversing resistance mechanisms must be accomplished simultaneously. Previous studies have identified that entinostat sensitizes TRAIL-resistant breast cancer cells by upregulation of E-cadherin and downregulation of N-cadherin, Snail, Slug, and ZEB1 [48]; overcomes trastuzumab resistance by disrupting HER2/HER3 interaction and inactivating PI3K/Akt signaling [29]; restores responsiveness in the setting of letrozole resistance by reducing expression of HER2 and HPS90 [18]; inhibits HIF-1a expression and angiogenesis [49]; and reverses EMT to MET [50,51]. This evidence indicates that entinostat can modulate epigenetic change as well as post-translational modifications that caused re-sensitization of resistant cells to the HER2-targeted drug. We also observed blockade of ErbB1/2/3 and Akt signaling by entinostat treatment. Indeed, our unpublished data showed that entinostat induced pro-apoptotic proteins and resulted in inhibition of MCL-1, which confers multi-drug resistance [52]. Therefore, further studies are warranted to investigate in vitro mechanisms and in vivo experiments of entinostat-induced resensitization to lapatinib and trastuzumab in resistant cells.
In summary, we show a novel synergistic mechanism of the enhanced anti-tumor effect of the entinostat and lapatinib combination over that of either single agent in HER2+ breast cancer cells through apoptosis regulated by FOXO3-mediated Bim1 expression. Taken together, our results provide a strong rationale for clinical investigation targeting HER2+ breast cancer with lapatinib, entinostat and trastuzumab. Recently, based on these findings, we have started a phase I study of entinostat in combination with lapatinib and trastuzumab in patients with HER2+ metastatic breast cancer in whom trastuzumab has failed (NCT01434303).
Supplementary Material
Acknowledgments
This study was supported by the Morgan Welch Inflammatory Breast Cancer Research Program (NTU), the State of Texas Rare and Aggressive Breast Cancer Research Program, and the National Institutes of Health/National Cancer Institute [through CA123318 (NTU) and MD Anderson's Cancer Center Support Grant, P30CA016672]. We thank Dr. Mien-Chie Hung (MD Anderson Cancer Center) for providing AKT-CA plasmid constructs. We thank Sunita Patterson (Department of Scientific Publications, MD Anderson) for editorial assistance and the Flow Cytometry and Cellular Imaging Facility at MD Anderson for assistance with cell-cycle analysis.
Footnotes
Electronic supplementary material: This article contains supplementary material information.
Conflicts of interest: Peter Ordentlich is an employee of Syndax Pharmaceuticals. All other authors have no conflicts of interest to disclose.
Contributor Information
Jangsoon Lee, Section of Translational Breast Cancer Research, Morgan Welch Inflammatory Breast Cancer Research Program and Clinic, Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Chandra Bartholomeusz, Section of Translational Breast Cancer Research, Morgan Welch Inflammatory Breast Cancer Research Program and Clinic, Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Oula Mansour, Section of Translational Breast Cancer Research, Morgan Welch Inflammatory Breast Cancer Research Program and Clinic, Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Juliane Humphries, Section of Translational Breast Cancer Research, Morgan Welch Inflammatory Breast Cancer Research Program and Clinic, Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Gabriel N. Hortobagyi, Section of Translational Breast Cancer Research, Morgan Welch Inflammatory Breast Cancer Research Program and Clinic, Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA
Peter Ordentlich, Syndax Pharmaceuticals, Inc., Waltham, Massachusetts, USA.
References
- 1.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, Nielsen TO, Moorman PG, Earp HS, Millikan RC. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 2.Stern HM. Improving Treatment of HER2-Positive Cancers: Opportunities and Challenges. Sci Transl Med. 2012;4(127):127rv–122. doi: 10.1126/scitranslmed.3001539. [DOI] [PubMed] [Google Scholar]
- 3.Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001;61(12):4744–4749. [PubMed] [Google Scholar]
- 4.Baselga J, Albanell J, Molina MA, Arribas J. Mechanism of action of trastuzumab and scientific update. Semin Oncol. 2001;28(5 Suppl 16):4–11. doi: 10.1016/s0093-7754(01)90276-3. [DOI] [PubMed] [Google Scholar]
- 5.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 6.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 7.Spector NL, Xia W, Burris H, 3rd, Hurwitz H, Dees EC, Dowlati A, O'Neil B, Overmoyer B, Marcom PK, Blackwell KL, Smith DA, Koch KM, Stead A, Mangum S, Ellis MJ, Liu L, Man AK, Bremer TM, Harris J, Bacus S. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol. 2005;23(11):2502–2512. doi: 10.1200/JCO.2005.12.157. [DOI] [PubMed] [Google Scholar]
- 8.Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21(41):6255–6263. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- 9.Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11(24):3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- 10.Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18(6):977–984. doi: 10.1093/annonc/mdl475. [DOI] [PubMed] [Google Scholar]
- 11.Kim H, Kim SN, Park YS, Kim NH, Han JW, Lee HY, Kim YK. HDAC inhibitors downregulate MRP2 expression in multidrug resistant cancer cells: implication for chemosensitization. Int J Oncol. 2011;38(3):807–812. doi: 10.3892/ijo.2010.879. [DOI] [PubMed] [Google Scholar]
- 12.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 13.Heightman TD. Therapeutic prospects for epigenetic modulation. Expert Opin Ther Targets. 2011;15(6):729–740. doi: 10.1517/14728222.2011.561786. [DOI] [PubMed] [Google Scholar]
- 14.Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A. 1999;96(8):4592–4597. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh TR, Shankar S, Srivastava RK. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene. 2005;24(29):4609–4623. doi: 10.1038/sj.onc.1208585. [DOI] [PubMed] [Google Scholar]
- 16.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee MJ, Trepel JB. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31(17):2128–2135. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabnis GJ, Goloubeva O, Chumsri S, Nguyen N, Sukumar S, Brodie AM. Functional activation of the estrogen receptor-alpha and aromatase by the HDAC inhibitor entinostat sensitizes ER-negative tumors to letrozole. Cancer Res. 2011;71(5):1893–1903. doi: 10.1158/0008-5472.CAN-10-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabnis GJ, Goloubeva OG, Kazi AA, Shah P, Brodie AH. HDAC inhibitor entinostat restores responsiveness of letrozole-resistant MCF-7Ca xenografts to aromatase inhibitors through modulation of Her-2. Mol Cancer Ther. 2013;12(12):2804–2816. doi: 10.1158/1535-7163.MCT-13-0345. 10.1158/1535-7163.MCT-13-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov. 2004;3(6):509–520. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- 20.Ohrt C, Willingmyre GD, Lee P, Knirsch C, Milhous W. Assessment of azithromycin in combination with other antimalarial drugs against Plasmodium falciparum in vitro. Antimicrob Agents Chemother. 2002;46(8):2518–2524. doi: 10.1128/AAC.46.8.2518-2524.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loupakis F, Vasile E, Santini D, Masi G, Falcone A, Graziano F. EGF-receptor targeting with monoclonal antibodies in colorectal carcinomas: rationale for a pharmacogenomic approach. Pharmacogenomics. 2008;9(1):55–69. doi: 10.2217/14622416.9.1.55. [DOI] [PubMed] [Google Scholar]
- 22.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, Ferraro F, Mercier F, Singh H, Brumme KM, Acharya SS, Scholl C, Tothova Z, Attar EC, Frohling S, DePinho RA, Armstrong SA, Gilliland DG, Scadden DT. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146(5):697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santo EE, Stroeken P, Sluis PV, Koster J, Versteeg R, Westerhout EM. FOXO3a is a major target of inactivation by PI3K/AKT signaling in aggressive neuroblastoma. Cancer Res. 2013;73(7):2189–2198. doi: 10.1158/0008-5472.CAN-12-3767. [DOI] [PubMed] [Google Scholar]
- 25.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 26.Wang L, Zhang Q, Zhang J, Sun S, Guo H, Jia Z, Wang B, Shao Z, Wang Z, Hu X. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer. 2011;11:248. doi: 10.1186/1471-2407-11-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurokawa H, Arteaga CL. ErbB (HER) receptors can abrogate antiestrogen action in human breast cancer by multiple signaling mechanisms. Clin Cancer Res. 2003;9(1 Pt 2):511S–515S. [PubMed] [Google Scholar]
- 28.Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, Schmidt M, Mills GB, Mendelsohn J, Fan Z. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003;22(21):3205–3212. doi: 10.1038/sj.onc.1206394. [DOI] [PubMed] [Google Scholar]
- 29.Huang X, Wang S, Lee CK, Yang X, Liu B. HDAC inhibitor SNDX-275 enhances efficacy of trastuzumab in erbB2-overexpressing breast cancer cells and exhibits potential to overcome trastuzumab resistance. Cancer Lett. 2011;307(1):72–79. doi: 10.1016/j.canlet.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 30.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 31.Hagenbuchner J, Ausserlechner MJ. Mitochondria and FOXO3: breath or die. Front Physiol. 2013;4:147. doi: 10.3389/fphys.2013.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, Lam EW. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278(50):49795–49805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- 33.Weidinger C, Krause K, Mueller K, Klagge A, Fuhrer D. FOXO3 is inhibited by oncogenic PI3K/Akt signaling but can be reactivated by the NSAID sulindac sulfide. J Clin Endocrinol Metab. 2011;96(9):E1361–1371. doi: 10.1210/jc.2010-2453. [DOI] [PubMed] [Google Scholar]
- 34.Bean GR, Ganesan YT, Dong Y, Takeda S, Liu H, Chan PM, Huang Y, Chodosh LA, Zambetti GP, Hsieh JJ, Cheng EH. PUMA and BIM are required for oncogene inactivation-induced apoptosis. Sci Signal. 2013;6(268):ra20. doi: 10.1126/scisignal.2003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hui RC, Gomes AR, Constantinidou D, Costa JR, Karadedou CT, Fernandez de Mattos S, Wymann MP, Brosens JJ, Schulze A, Lam EW. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol Cell Biol. 2008;28(19):5886–5898. doi: 10.1128/MCB.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puig O, Tjian R. Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev. 2005;19(20):2435–2446. doi: 10.1101/gad.1340505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sanchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, Manning HC, Chang J, Arteaga CL. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci U S A. 2011;108(12):5021–5026. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, Huang J, Lyu H, Lee CK, Tan J, Wang J, Liu B. Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis. 2013;4:e556. doi: 10.1038/cddis.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim YJ, Greer CB, Cecchini KR, Harris LN, Tuck DP, Kim TH. HDAC inhibitors induce transcriptional repression of high copy number genes in breast cancer through elongation blockade. Oncogene. 2013;32(23):2828–2835. doi: 10.1038/onc.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mateen S, Raina K, Jain AK, Agarwal C, Chan D, Agarwal R. Epigenetic modifications and p21-cyclin B1 nexus in anticancer effect of histone deacetylase inhibitors in combination with silibinin on non-small cell lung cancer cells. Epigenetics. 2012;7(10):1161–1172. doi: 10.4161/epi.22070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maeda T, Hanna AN, Sim AB, Chua PP, Chong MT, Tron VA. GADD45 regulates G2/M arrest, DNA repair, and cell death in keratinocytes following ultraviolet exposure. J Invest Dermatol. 2002;119(1):22–26. doi: 10.1046/j.1523-1747.2002.01781.x. [DOI] [PubMed] [Google Scholar]
- 42.Guo Y, Kyprianou N. Overexpression of transforming growth factor (TGF) beta1 type II receptor restores TGF-beta1 sensitivity and signaling in human prostate cancer cells. Cell Growth Differ. 1998;9(2):185–193. [PubMed] [Google Scholar]
- 43.Kataoka Y, Mukohara T, Shimada H, Saijo N, Hirai M, Minami H. Association between gain-of-function mutations in PIK3CA and resistance to HER2-targeted agents in HER2-amplified breast cancer cell lines. Ann Oncol. 2010;21(2):255–262. doi: 10.1093/annonc/mdp304. [DOI] [PubMed] [Google Scholar]
- 44.Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, Baselga J. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–9230. doi: 10.1158/0008-5472.CAN-08-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tortora G. Mechanisms of resistance to HER2 target therapy. Journal of the National Cancer Institute Monographs. 2011;2011(43):95–98. doi: 10.1093/jncimonographs/lgr026. [DOI] [PubMed] [Google Scholar]
- 46.Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69(17):6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 47.Wetterskog D, Shiu KK, Chong I, Meijer T, Mackay A, Lambros M, Cunningham D, Reis-Filho JS, Lord CJ, Ashworth A. Identification of novel determinants of resistance to lapatinib in ERBB2-amplified cancers. Oncogene. 2014;33(8):966–976. doi: 10.1038/onc.2013.41. [DOI] [PubMed] [Google Scholar]
- 48.Srivastava RK, Kurzrock R, Shankar S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol Cancer Ther. 2010;9(12):3254–3266. doi: 10.1158/1535-7163.MCT-10-0582. 1535-7163.MCT-10-0582. [DOI] [PubMed] [Google Scholar]
- 49.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. Journal of cellular biochemistry. 2009;107(4):600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagathihalli NS, Massion PP, Gonzalez AL, Lu P, Datta PK. Smoking induces epithelial-to-mesenchymal transition in non-small cell lung cancer through HDAC-mediated downregulation of E-cadherin. Mol Cancer Ther. 2012;11(11):2362–2372. doi: 10.1158/1535-7163.MCT-12-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah P, Gau Y, Sabnis G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res Treat. 2014;143(1):99–111. doi: 10.1007/s10549-013-2784-7. [DOI] [PubMed] [Google Scholar]
- 52.Hermanson DL, Das SG, Li Y, Xing C. Overexpression of Mcl-1 confers multidrug resistance, whereas topoisomerase IIbeta downregulation introduces mitoxantrone-specific drug resistance in acute myeloid leukemia. Mol Pharmacol. 2013;84(2):236–243. doi: 10.1124/mol.113.086140. mol.113.086140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.