

Abstract
Highly chemoselective intramolecular amination of propargylic C(sp3)–H bonds has been demonstrated for N-bishomopropargylic sulfamoyl azides via Co(II)-based metalloradical catalysis. Supported by D2h-symmetric amidoporphyrin ligand 3,5-DitBu-IbuPhyrin, the Co(II)-catalyzed C–H amination process can proceed effectively under neutral and nonoxidative conditions without the need of any additives, generating N2 as the only byproduct. The metalloradical amination is suitable to both secondary and tertiary propargylic C–H substrates with an unusually high degree of functional group tolerance, providing a direct method for high-yielding synthesis of functionalized propargylamine derivatives.
Keywords: propargylic C–H amination, metalloradical catalysis, cobalt porphyrin, sulfamoyl azide, chemoselectivity
Significant efforts have been devoted to develop synthetic methods for propargylamines as they serve as versatile intermediates in organic synthesis,[1] as well as important structural elements in natural and synthetic products with interesting biological activities.[2] Traditionally, propargylamines have been prepared through addition of metal alkynylides to imines. Since this traditional method typically requires the stoichiometric amounts of metal alkynylides, which are known to be highly moisture sensitive,[3] it would reduce the degree of functional group tolerance and has largely restricted the applications. Consequently, there has been continued interest in developing new methods for the synthesis of propargylamines under mild conditions with a high degree of functional group tolerance. Among different approaches that have been developed recently, transition metal-catalyzed three-component one-pot coupling of an aldehyde, an alkyne and an amine represents one of the most general and atom-economic methods. This so-called “A3-coupling” provides a catalytic method for efficient synthesis of propargylamines under mild conditions with H2O as the only byproduct.[4,5] Since “A3-coupling” is mainly suitable for aldehydes as one of the coupling partners, its application has been limited to the preparation of propargylamines bearing tertiary carbon center at the propargylic position.[6]
Selective amination of omnipresent C–H bonds via metal-mediated nitrene insertion represents a powerful approach for direct introduction of valuable amino functionalities into molecules.[7] This direct transformation has the potential to serve as an efficient alternative to traditional approaches for amine synthesis that are based on functional group transformations. Its realization may have far-reaching impact for amine synthesis and their practical applications in different fields. Accordingly, the direct synthesis of propargylamines based on metal-catalyzed amination of propargylic C–H bonds could become an alternative approach to traditional methods. In addition to tertiary carbon-containing propargylamines, catalytic propargylic C–H amination would also allow for preparation of propargylamines bearing a quaternary carbon center at the propargylic position. While metal-catalyzed amination has been successfully demonstrated with several different types of C–H substrates,[7] few catalytic systems are known for chemoselective amination of propargylic C(sp3)–H bonds.[8a,9] Due to the electrophilic nature of the key metallonitrene intermediates, its addition to more electron-rich C≡C π bonds would be typically preferred over amination of the propargylic C–H σ bonds for propargylic C–H substrates.[8] Through decreasing the electrophilicity of the corresponding Rh2-nitrene intermediates by replacing sulfamates with carbamates, Schomaker and coworkers reported recently that intramolecular propargylic C–H amination of homopropargylic carbamates could be successfully catalyzed by Rh2(esp)2 in combination with PhI(OAc)2 and MgO, generating 5-membered propargylamine derivatives in good yields.[9a] However, due to the competitive electrophilic addition of Rh2-nitrene intermediate to the electron-rich C≡C π bonds under these catalytic conditions,[8a] the intramolecular propargylic C–H amination of sulfamates gave the corresponding 6-membered propargylamines in only moderate yields.[8a,9a] It should be noted that the ring size of the resulting heterocycles from intramolecular C–H amination is typically governed by the substrate geometry and tether length of the substrates.[7a]
Cobalt(II) complexes of porphyrins, [Co(Por)], which exist as stable metalloradicals, have emerged as a new class of catalysts that have proven to be effective to activate azides as nitrene sources for amination of various types of C–H bonds, including challenging primary and electron-deficient C–H bonds.[10,11] Different from electrophilic metallonitrene intermediates associated with Rh2-catalyzed systems, the Co(II)-based metalloradical amination has been demonstrated to proceed via a stepwise radical mechanism.[12] Consequently, it has been shown that the reactivity and selectivity profile of the Co(II)-based amination system is governed by the bond-dissociation energy (BDE) rather than electron density of the reacting C–H bonds.[11a–c] This concept of metalloradical catalysis (MRC) has been successfully applied to address chemoselectivity issues in intramolecular allylic C–H amination over competitive C=C aziridination.[11b] Considering the fact that the BDE of secondary propargylic C–H bonds is similar to that of secondary allylic C–H bonds (~83 kcal/mol),[13] we anticipated the possibility of chemoselective amination of propargylic C–H bonds via Co(II)-based MRC if the radical addition of the Co(III)-nitrene radical to C≡C triple bonds could be disfavored. Herein, we report that [Co(Por)] are effective catalysts for intramolecular amination of sulfamoyl azides with complete control of chemoselectivity toward propargylic C–H bonds without direct involvement of the normally more reactive C≡C π bonds and other common C–H bonds (Scheme 1).
Scheme 1.
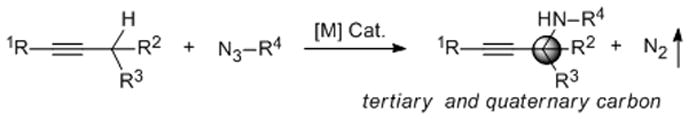
Synthesis of Propargylamines via Catalytic C–H Amination.
At the onset of our study, N-bishomopropargylic sulfamoyl azide 1a[14,15] was used as the initial substrate for intramolecular C–H amination by [Co(Por)] (Scheme 2). We were delighted to find that intramolecular amination of the secondary propargylic C–H bonds positioned α- to the unprotected terminal alkyne in 1a could be catalyzed by [Co(TPP)] (2 mol % at 40 °C), forming the corresponding propargylamine 2a in a moderate yield (42%). The catalytic reaction exhibited complete chemoselectivity toward the propargylic C–H amination, without observation of the competitive homopropargylic C–H amination as well as the C≡C addition reaction. When [Co(P1)], in which the D2h-symmetric porphyrin ligand 3,5-DitBu-IbuPhyrin P1 is functionalized with amide functionalities as hydrogen-bonding donors at the ortho-positions of the meso-phenyl groups,[16] was used as the catalyst, the yield for chemoselective formation of 2a was dramatically improved to be near quantitative. This represents another demonstration of hydrogen-bonding acceleration in Co(II)-based MRC.[11a–e] It is worth emphasizing the operational simplicity and cleanness of the Co(II)/azide-based system, which gave rise to a catalytic process where the desired product 2a existed as a sole compound after completion of the reaction.
Scheme 2.
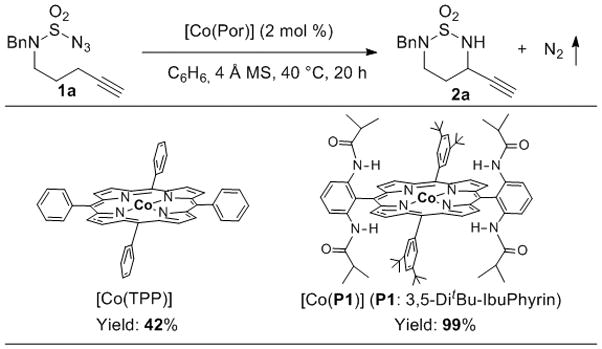
Ligand Effect on [Co(Por)]-Catalyzed Intramolecular Propargylic C–H Amination of Sulfamoyl Azide.
Under the optimized condition (2 mol % of [Co(P1)] in benzene at 40 °C for 20 h), the [Co(P1)]-catalyzed intramolecular propargylic amination was shown to be applicable to sulfamoyl azides 1 having varied N-substitutions (Table 1). For example, the substrates containing electron-donating N-alkyl substituents, whose corresponding sulfamides would be degraded under the Rh2(esp)2/PhI(OAc)2/MgO catalytic system,[17] have proven to be suitable substrates for the Co(II)-based metalloradical system, as shown by the chemoselective C–H amination reactions of sulfamoyl azides with N-benzyl (entry 1), N-methyl (entry 2), N-ethyl (entry 3), N-isopropyl (entry 4), N-allyl (entry 14) and N-butyl (entry 16) substituents. Additionally, the propargylic C–H bonds in sulfamoyl azides with electron-withdrawing N-substituents could also be successfully aminated with complete chemoselectivity, as exemplified by the high-yielding formation of the six-membered propargylamine 2e from the corresponding N-Boc-protected azide on 1.0 mmol scale (entry 5). This result indicates that the radical reactivity of the corresponding Co(III)-nitrene radical intermediate remained as the dominating factor despite its increased electrophilicity owing to the presence of the electron-withdrawing N-substituent.
Table 1.
Synthesis of Functionalized Propargylamine Derivatives from Chemoselective Propargylic C–H Amination of Sulfamoyl Azides Catalyzed by [Co(P1)]a,b
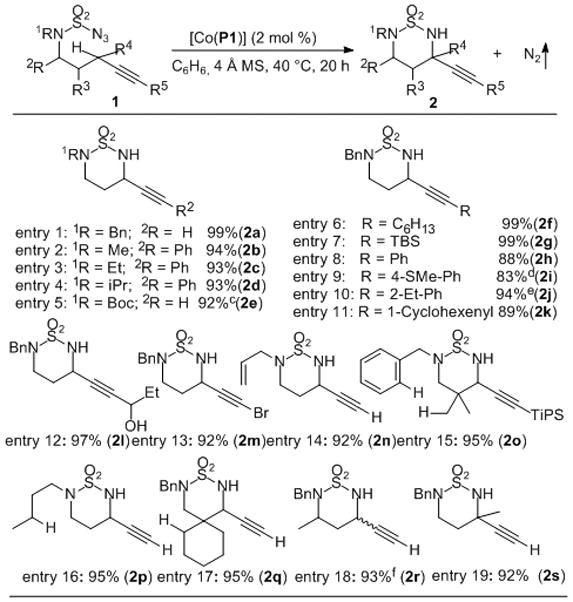
|
Performed in C6H6 at 40 °C for 20 h using 2 mol % [Co(P1)] under N2 in the presence of 4 Å MS; [azide 1] = 0.10 M.
Isolated yields.
5 mol % [Co(P1)], azide 1e = 1.0 mmol.
80 °C for 3 h.
Isolated yields through repeating the reaction three times without isolating the catalyst.
dr: 68/32.
Moreover, the Co(II)-catalyzed chemoselective C–H amination could be applied to a wide range of sulfamoyl azides bearing different alkyne elements (Table 1). In addition to the azides with terminal alkynes (entries 1, 5, 14 and 16–19), the catalytic system could also be suitable for azides derived from internal alkynes with diverse substituents, as demonstrated by the high-yielding amination reactions of propargylic C–H bonds of alkynes substituted with aryl (entries 2–4 and 8–10), alkyl (entries 6 and 12), and silyl (entries 7 and 15) group. It should be noted that the sulfide group in azide 1i (entry 9) was well tolerated due to the non-oxidative condition. Furthermore, intramolecular propargylic C–H amination of enyne-based sulfamoyl azide 1k could also be chemoselectively catalyzed by [Co(P1)], forming the propargylamine 2k without affecting the conjugated enyne functionality (entry 11). The high degree of functional group tolerance and chemoselectivity of the Co(II)-based metalloradical system was further demonstrated with the amination of sulfamoyl azide 1l containing unprotected propargylic secondary alcohol (entry 12). Remarkably, the catalytic reaction afforded the desired propargylic amine 2l in an excellent yield without any side-reactions from the propargylic alcohol unit. It is noted that the intramolecular C–H amination of the corresponding carbamate with the similar propargylic alcohol unit was shown to be problematic for the Rh2-based catalytic system.[9] Surprisingly, the Co(II)-catalyzed system could be even extended to 1-bromoalkyne-derived sulfamoyl azide 1m, forming the corresponding amination product 2m in a high yield with no complication from the bromoethynyl functional group (entry 13). The outstanding chemoselectivity toward propargylic C–H amination was further highlighted by the catalytic reactions of the sulfamoyl azide 1n with N-allyl substituent (entry 14). Despite the existence of both alkene and alkyne functionalities that are normally prone toward electrophilic addition, the metalloradical catalyst [Co(P1)] could chemoselectively aminate the propargylic C–H bond without affecting the potentially reactive C=C and C≡C π bonds. These multi-functionalized propargylic amine products such as 2k–n may serve as intermediates for synthesis of other useful amine derivatives. For example, propargylic amine 2l may allow for access to the corresponding allenic amine, which is difficult to prepare.[9a] In addition to chemoselectivity, our preliminary results also demonstrated the possibility of controlling enantioselectivity of the C–H amination process through the employment of Co(II) complexes of D2-symmetric chiral amidoporphyrins [Co(D2-Por*)] as chiral metalloradical catalysts (see Table S1 in Supporting Information for details).[18] Furthermore, to enhance the practicality of the catalytic system, it was shown that the amination reaction of azide 1j (entry 10) could be successfully repeated three times without isolating the catalyst [Co(P1)], affording the desired product 2j in similarly high yields (see Table S2 in Supporting Information for details).
Bond-dissociation energy (BDE) was recognized as a fundamentally important factor in metalloradical amination for controlling and differentiating reactivity and selectivity of various C–H bonds.[11] This governing principle was also well demonstrated in the current catalytic system by [Co(P1)] for regioselective amination of propargylic C–H bonds of sulfamoyl azides (Table 1). Previous report indicated that the normal aliphatic C–H bonds, including primary, secondary and tertiary C(sp3)–H bonds, could also be effectively aminated by [Co(P1)] under the similar conditions.[11a] Due to the relatively lower bond-dissociation energy of propargylic C–H bonds (BDE of propargylic C–H bonds: ~85 kcal/mol; BDE of aliphatic C–H bonds: ~98 kcal/mol),[13] we showed that the intramolecular amination of propargylic C–H bonds could be selectively catalyzed by [Co(P1)] in the presence of different types of normal aliphatic C–H bonds (Table 1). Using the N-benzyl-N-bishomopropargyl sulfamoyl azide 1o as an example, the Co(II)-based metalloradical system could achieve highly selective 1,6-amination of the propargylic C–H bonds in 1o without amination of the much stronger primary C(sp3)–H bonds and aromatic C(sp2)–H bonds located at the equal positions (entry 15). Similarly, highly regioselective propargylic amination could be accomplished in the presence of acyclic and cyclic secondary C(sp3)–H bonds as shown with the catalytic reactions of azides 1p (entry 16) and 1q (entry 17), respectively.
In addition to the above examples of various sulfamoyl azides with secondary propargylic C–H bonds for formation of tertiary carbon-containing propargylamines, the Co(II)-catalyzed 1,6-C–H amination process worked equally well with tertiary propargylic C–H substrates, as illustrated by the effective amination reaction of sulfamoyl azide 1s, which gave the corresponding propargylamine 2s containing a synthetically challenging quaternary carbon center in 92% yield (entry 19).[19]
The demonstrated intramolecular propargylic C–H amination by the Co(II)-based MRC paves a practical route to access various unsymmetric cyclic sulfamide derivatives 2. In view of the impressively diverse array of biological activities of cyclic sulfamide-containing compounds reported in numerous recent patents,[20] the products 2, bearing other functionalities in addition to the highly versatile alkyne functional groups, may serve as valuable synthetic intermediates for applications in biology and medicine. Furthermore, the neutral and non-oxidative reaction conditions of the Co(II)-based catalytic system may allow for direct use of substrates that have pharmaceutical relevance and often contain various functionalities. For example, the [Co(P1)] catalyst could be successfully utilized for intramolecular amination of deoxyuridine-based substrate 1t, providing the new deoxyuridine derivative containing cyclic sulfamide 2t in 90% yield (Scheme 3). When the reaction was scaled up to 0.65 mmol and at a lower catalyst loading (0.5 mol %), a similarly high yield (88%) was obtained in 70 h. Given that the variants of deoxyuridine, such as idoxuridine and trifluridine, have been used as antiviral drugs,[21] this type of modifications of deoxyuridine with cyclic sulfamide units may offer an attractive opportunity for obtaining new compounds having interesting biological activities. Such a high degree of functional group tolerance exhibited by Co(II)-based MRC should further enhance the practicality of this catalytic system for synthetic applications.
Scheme 3.
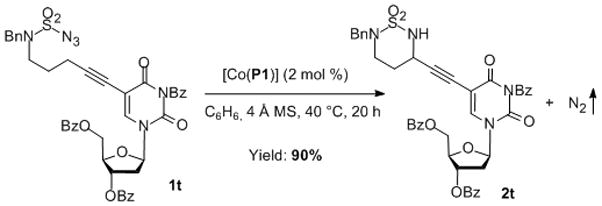
Application of Catalytic Propargylic C–H Amination for Deoxyuridine-Based Sulfamoyl Azide.
As another exploration of their synthetic applications, the 6-membered amination products 2 could serve as convenient precursors for the synthetically valuable 1,3-diamines. For example, the -SO2- bridging unit in cyclic sulfamide 2e could be conveniently opened on 0.9 mmol scale by alcohols such as 2,2,2-trichloroethanol in the presence of DMAP,[17] providing the corresponding alkyne-containing 1,3-diamine 3e in a high yield (Scheme 4). Since the two amine units in 3e are differentially protected by Boc and Tces groups, it should be easily transformed to various 1,3-diamine derivatives by utilizing the diverse transformations known for alkynes.
Scheme 4.
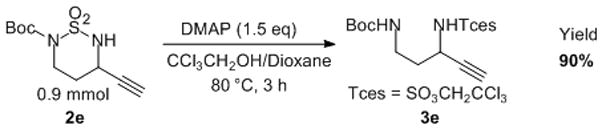
Ring-Opening Reaction for Generation of 1,3-Propargylic Diamine with Different Protecting Groups.
In summary, we have demonstrated for the first time a chemoselective catalytic system via Co(II)-based metalloradical catalysis for intramolecular amination of propargylic C(sp3)–H bonds of N-bishomopropargylic sulfamoyl azides. The metalloradical catalyst [Co(P1)] has proven to be highly effective for chemoselective amination of different types of propargylic C–H bonds with excellent regioselectivity, affording 6-membered cyclic sulfamides with unaffected alkyne substituents in excellent yields.[22] In addition to the alkyne functionality, the Co(II)-based metalloradical amination, which operates under neutral and nonoxidative conditions, has been shown to tolerate a range of other functional groups, including alkenes and unprotected alcohols. The resulting tertiary and quaternary carbon-containing propargylamine products from this new catalytic process may find a myriad of synthetic applications.
Supplementary Material
Footnotes
We are grateful for financial support by NSF (CHE-1152767) and NIH (R01-GM098777)
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
Contributor Information
Prof. Dr. Hongjian Lu, Email: hongjianlu@nju.edu.cn, Department of Chemistry, University of South Florida, Tampa, Florida 33620-5250, United States. Institute of Chemistry and BioMedical Sciences, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, China
Dr. Chaoqun Li, Department of Chemistry, University of South Florida, Tampa, Florida 33620-5250, United States
Dr. Huiling Jiang, Department of Chemistry, University of South Florida, Tampa, Florida 33620-5250, United States
Christopher L. Lizardi, Department of Chemistry, University of South Florida, Tampa, Florida 33620-5250, United States
Prof. Dr. X. Peter Zhang, Email: xpzhang@usf.edu, Department of Chemistry, University of South Florida, Tampa, Florida 33620-5250, United States
References
- 1.Selected examples: Nilsson B, Vargas HM, Ringdahl B, Hacksell U. J Med Chem. 1992;35:285–294. doi: 10.1021/jm00080a013.Jenmalm A, Berts W, Li YL, Luthman K, Csoeregh I, Hacksell U. J Org Chem. 1994;59:1139–1148.Miura M, Enna M, Okuro K, Nomura M. J Org Chem. 1995;60:4999–5004.Davidson MH, McDonald FE. Org Lett. 2004;6:1601–1603. doi: 10.1021/ol049630m.Trost BM, Chung CK, Pinkerton AB. Angew Chem. 2004;116:4427–4429.Angew Chem Int Ed. 2004;43:4327–4329. doi: 10.1002/anie.200460058.Nakamura H, Kamakura T, Ishikura M, Biellmann JF. J Am Chem Soc. 2004;126:5958–5959. doi: 10.1021/ja039175+.Detz RJ, Abiri Z, le Griel R, Hiemstra H, van Maarseveen JH. Chem Eur J. 2011;17:5921–5930. doi: 10.1002/chem.201003727.Ding CH, Hou XL. Chem Rev. 2011;111:1914–1937. doi: 10.1021/cr100284m.Aubert C, Buisine O, Malacria M. Chem Rev. 2002;102:813–834. doi: 10.1021/cr980054f.Naota T, Takaya H, Murahashi SI. Chem Rev. 1998;98:2599–2660. doi: 10.1021/cr9403695.
- 2.a) Konishi M, Ohkuma H, Tsuno T, Oki T, Vanduyne GD, Clardy J. J Am Chem Soc. 1990;112:3715–3716. [Google Scholar]; b) Yu PH, Davis BA, Boulton AA. J Med Chem. 1992;35:3705–3713. doi: 10.1021/jm00098a017. [DOI] [PubMed] [Google Scholar]; c) Huffman MA, Yasuda N, Decamp AE, Grabowski EJJ. J Org Chem. 1995;60:1590–1594. [Google Scholar]; d) Enders D, Reinhold U. Tetrahedron-Asymmetry. 1997;8:1895–1946. [Google Scholar]; e) Kauffman GS, Harris GD, Dorow RL, Stone BRP, Parsons RL, Pesti JA, Magnus NA, Fortunak JM, Confalone PN, Nugent WA. Org Lett. 2000;2:3119–3121. doi: 10.1021/ol006321x. [DOI] [PubMed] [Google Scholar]
- 3.a) Aubrecht KB, Winemiller MD, Collum DB. J Am Chem Soc. 2000;122:11084–11089. [Google Scholar]; b) Tuulmets A, Pallin V, Tammiku-Taul J, Burk P, Raie K. J Phys Org Chem. 2002;15:701–705. [Google Scholar]; c) Murai T, Ohta Y, Mutoh Y. Tetrahedron Lett. 2005;46:3637–3640. [Google Scholar]; d) Ma Y, Lobkovsky E, Collum DB. J Org Chem. 2005;70:2335–2337. doi: 10.1021/jo0480895. [DOI] [PubMed] [Google Scholar]; e) Magueur G, Crousse B, Bonnet-Delpon D. Tetrahedron Lett. 2005;46:2219–2221. [Google Scholar]; f) Ding CH, Chen DD, Luo ZB, Dai LX, Hou XL. Synlett. 2006:1272–1274. [Google Scholar]; g) Badorrey R, Diaz-de-Villegas MD, Diez R, Galvez JA. Eur J Org Chem. 2007:2114–2120. [Google Scholar]; h) Wee AGH, Zhang B. Tetrahedron Lett. 2007;48:4135–4138. [Google Scholar]
- 4.For recent reviews on “A3-coupling”, see: Peshkov VA, Pereshivko OP, Van der Eycken EV. Chem Soc Rev. 2012;41:3790–3807. doi: 10.1039/c2cs15356d.Yoo WJ, Zhao L, Li CJ. Aldrichimica Acta. 2011;44:43–51.Li CJ. Acc Chem Res. 2010;43:581–590. doi: 10.1021/ar9002587.Kouznetsov VV, Méndez LYV. Synthesis. 2008;4:491–506.Zani L, Bolm C. Chem Commun. 2006:4263–4275. doi: 10.1039/b607986p.Wei CM, Li ZG, Li CJ. Synlett. 2004:1472–1483.
- 5.For synthesis of propargylamines through alkynylation of C–H bonds adjacent to nitrogen atoms of existing amines, see: Li ZP, Li CJ. Org Lett. 2004;6:4997–4999. doi: 10.1021/ol047814v.Li ZP, Li CJ. J Am Chem Soc. 2004;126:11810–11811. doi: 10.1021/ja0460763.Niu M, Yin Z, Fu H, Jiang Y, Zhao Y. J Org Chem. 2008;73:3961–3963. doi: 10.1021/jo800279j.Detz RJ, Abiri Z, le Griel R, Hiemstra H, van Maarseveen JH. Chem-Eur J. 2011;17:5921–5930. doi: 10.1002/chem.201003727.Sugiishi T, Nakamura H. J Am Chem Soc. 2012;134:2504–2507. doi: 10.1021/ja211092q.
- 6.For examples of “A3-coupling” using activated acyclic ketones as one of the coupling partners to form quaternary carbon-containing propargylamines, see: Pereshivko OP, Peshkov VA, Van der Eycken EV. Org Lett. 2010;12:2638–2641. doi: 10.1021/ol1008312.Pierce CJ, Larsen CH. Green Chem. 2012;14:2672–2676.; For examples of “A3-coupling” using unactivated ketones to form quaternary carbon-containing propargylamines, see: Cheng M, Zhang Q, Hu XY, Li BG, Ji JX, Chan ASC. Adv Synth Catal. 2011;353:1274–1278.Pierce CJ, Nguyen M, Larsen CH. Angew Chem. 2012;124:12455–12458. doi: 10.1002/anie.201206674.Angew Chem Int Ed. 2012;51:12289–12292. doi: 10.1002/anie.201206674.Tang X, Kuang J, Ma S. Chem Commun. 2013;49:8976–8978. doi: 10.1039/c3cc45301d.
- 7.Selected reviews: Roizen JL, Harvey ME, Du Bois J. Acc Chem Res. 2012;45:911–922. doi: 10.1021/ar200318q.Ramirez TA, Zhao B, Shi Y. Chem Soc Rev. 2012;41:931–942. doi: 10.1039/c1cs15104e.Collet F, Lescot C, Dauban P. Chem Soc Rev. 2011;40:1926–1936. doi: 10.1039/c0cs00095g.Du Bois J. Org Proc Res Dev. 2011;15:758–762. doi: 10.1021/op200046v.Zalatan DN, Du Bois J. Top Curr Chem. 2010;292:347–378. doi: 10.1007/128_2009_19.Collet F, Dodd RH, Dauban P. Chem Commun. 2009:5061–5074. doi: 10.1039/b905820f.Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485.Davies HML. Angew Chem. 2006;118:6574–6577.Angew Chem Int Ed. 2006;45:6422–6425. doi: 10.1002/anie.200601814.Davies HML, Long MS. Angew Chem. 2005;117:3584–3586.Angew Chem Int Ed. 2005;44:3518–3520. doi: 10.1002/anie.200500554.Muller P, Fruit C. Chem Rev. 2003;103:2905–2919. doi: 10.1021/cr020043t.
- 8.a) Thornton AR, Blakey SB. J Am Chem Soc. 2008;130:5020–5021. doi: 10.1021/ja7111788. [DOI] [PubMed] [Google Scholar]; b) Thornton AR, Martin VI, Blakey SB. J Am Chem Soc. 2009;131:2434–2435. doi: 10.1021/ja809078d. [DOI] [PubMed] [Google Scholar]; c) Mace N, Thornton AR, Blakey SB. Angew Chem. 2013;125:5948–5951. doi: 10.1002/anie.201301087. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:5836–5839. doi: 10.1002/anie.201301087. [DOI] [PubMed] [Google Scholar]
- 9.For Rh2-catalyzed intramolecular propargylic C–H amination with homopropargylic carbamates as the substrates, see: Grigg RD, Rigoli JW, Pearce SD, Schomaker JM. Org Lett. 2012;14:280–283. doi: 10.1021/ol203055v.; For two examples of Rh2-catalyzed intermolecular propargylic C–H amination using 1-aryl-1-butynes as the substrates and N-mesyloxycarbamate as the nitrene source, see: Lebel H, Trudel C, Spitz C. Chem Commun. 2012;48:7799–7801. doi: 10.1039/c2cc33689h.
- 10.For reviews on the use of azides as nitrene sources for metal-catalyzed nitrene transfers, Lu HJ, Zhang XP. Chem Soc Rev. 2011;40:1899–1909. doi: 10.1039/c0cs00070a.Che CM, Lo VKY, Zhou CY, Huang JS. Chem Soc Rev. 2011;40:1950–1975. doi: 10.1039/c0cs00142b.Driver TG. Org Biomol Chem. 2010;8:3831–3846. doi: 10.1039/c005219c.Cenini S, Gallo E, Caselli A, Ragaini F, Fantauzzi S, Piangiolino C. Coord Chem Rev. 2006;250:1234–1253.Katsuki T. Chem Lett. 2005;34:1304–1309.
- 11.Selected examples: Lu HJ, Jiang HL, Wojtas L, Zhang XP. Angew Chem. 2010;122:10390–10394.Angew Chem Int Ed. 2010;49:10192–10196. doi: 10.1002/anie.201005552.Lu HJ, Jiang HL, Hu Y, Wojtas L, Zhang XP. Chem Sci. 2011;2:2361–2366.Lu HJ, Hu Y, Jiang H, Wojtas L, Zhang XP. Org Lett. 2012;14:5158–5161. doi: 10.1021/ol302511f.Ruppel JV, Kamble RM, Zhang XP. Org Lett. 2007;9:4889–4892. doi: 10.1021/ol702265h.Lu HJ, Tao JR, Jones JE, Wojtas L, Zhang XP. Org Lett. 2010;12:1248–1251. doi: 10.1021/ol100110z.Lu HJ, Subbarayan V, Tao JR, Zhang XP. Organometallics. 2010;29:389–393.Ragaini F, Penoni A, Gallo E, Tollari S, Gotti CL, Lapadula M, Mangioni E, Cenini S. Chem Eur J. 2003;9:249–259. doi: 10.1002/chem.200390018.Cenini S, Gallo E, Penoni A, Ragaini F, Tollari S. Chem Commun. 2000:2265–2266.
- 12.For detailed studies on the radical mechanism of [Co(Por)]-catalyzed C–H amination, including EPR observation of Co(III)-nitrene radical intermediates, see: Lyaskovskyy V, Suarez AIO, Lu HJ, Jiang HL, Zhang XP, de Bruin B. J Am Chem Soc. 2011;133:12264–12273. doi: 10.1021/ja204800a.; For related DFT studies on the radical mechanism of [Co(Por)]-catalyzed olefin aziridination, see: Olivos Suarez AI, Jiang HJ, Zhang XP, de Bruin B. Dalton Trans. 2011;40:5697–5705. doi: 10.1039/c1dt10027k.Hopmann KH, Ghosh A. ACS Catalysis. 2011;1:597–600.; For detailed studies on the similar radical mechanism involving Co(III)-carbene radical intermediates, see: Dzik WI, Xu X, Zhang XP, Reek JNH, de Bruin B. J Am Chem Soc. 2010;132:10891–10902. doi: 10.1021/ja103768r.Belof JL, Cioce CR, Xu X, Zhang XP, Space B, Woodcock HL. Organometallics. 2011;30:2739–2746. doi: 10.1021/om2001348.Lu HJ, Dzik WI, Xu X, Wojtas L, de Bruin B, Zhang XP. J Am Chem Soc. 2011;133:8518–8521. doi: 10.1021/ja203434c.
- 13.Luo YR. Comprehensive handbook of chemical bond energies. CRC; Boca Raton, FL: 2007. [Google Scholar]
- 14.a) Sulfamoyl azides were reported to be chemically stable, even under strong acidic and basic conditions (see ref. 15). Our DSC experiments indicated that these sulfamoyl azides were thermally stable without decomposition up to at least 100 °C; see Supporting Information for a representative DSC plot of azide 1a; b) N,N-Disubstituted sulfamoyl azides 1 were prepared from secondary amines by a one-step procedure (see Supporting Information). Attempts to prepare unprotected N-monosubstituted sulfamoyl azides from primary amines were unsuccessful, possibly due to high reactivity of the N-monosubstituted sulfamoyl azides with amines (see ref. 15b).
- 15.a) Griffith J. J Chem Soc C-Organic. 1971:3191–3195. [Google Scholar]; b) Matier WL, Comer WT, Deitchma D. J Med Chem. 1972;15:538–541. doi: 10.1021/jm00275a025. [DOI] [PubMed] [Google Scholar]; c) Goddard-Borger ED, Stick RV. Org Lett. 2007;9:3797–3800. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 16.Ruppel JV, Jones JE, Huff CA, Kamble RM, Chen Y, Zhang XP. Org Lett. 2008;10:1995–1998. doi: 10.1021/ol800588p. [DOI] [PubMed] [Google Scholar]
- 17.Kurokawa T, Kim M, Du Bois J. Angew Chem. 2009;121:2815–2817. doi: 10.1002/anie.200806192. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:2777–2779. doi: 10.1002/anie.200806192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.With the Co(II) complex of 3,5-DitBu-QingPhyrin, [Co(P4)], as the catalyst, asymmetric intramolecular C–H amination of bishomopropargylic azide 1j could afford the corresponding propargylic amine 2j in 92% yield with 38% ee (see Table S1 in Supporting Information).
- 19.For one of the most versatile methods for preparation of quaternary amines, see: Xu H, Chowdhury S, Ellman JA. Nat Protocol. 2013;8:2271–2280. doi: 10.1038/nprot.2013.134.
- 20.For selected reviews, see: Reitz AB, Smith GR, Parker MH. Expert Opin Ther Patents. 2009;19:1449–1453. doi: 10.1517/13543770903185920.Winum JY, Scozzafava A, Montero JL, Supuran CT. Expert Opin Ther Patents. 2006;16:27–47. doi: 10.1517/13543776.16.1.27.
- 21.a) Kaufman HE, Varnell ED, Thompson HW. Arch Ophth. 1998;116:777–780. doi: 10.1001/archopht.116.6.777. [DOI] [PubMed] [Google Scholar]; b) Romanowski EG, Bartels SP, Gordon YJ. Invest Ophth Vis Sci. 1999;40:378–384. [PubMed] [Google Scholar]; c) Bernard PH, Mounier M, Dupuy P. J Eur Acad Derm Ven. 2003;17:246–246. doi: 10.1046/j.1468-3083.2003.00577_14.x. [DOI] [PubMed] [Google Scholar]; d) Seth AK, Misra A, Umrigar D. Pharm Dev Technol. 2004;9:277–289. doi: 10.1081/pdt-200031432. [DOI] [PubMed] [Google Scholar]
- 22.Efforts are on the way to expand the application of the Co(II)-based amination system for formation of 5- and 7-membered cyclic sulfamides as well as sulfamates.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.