

Abstract
Rationale
The E3 ubiquitin ligase IDOL triggers lysosomal degradation of the LDL receptor. The tissue-specific effects of the IDOL pathway on plasma cholesterol and atherosclerosis have not been examined.
Objective
Given that the liver is the primary determinant of plasma cholesterol levels, we sought to examine the consequence of effect of chronic liver-specific expression of a dominant active form of IDOL in mice.
Methods and Results
We expressed a degradation-resistant, dominant-active form of IDOL (sIDOL) in C57Bl/6J mice from the liver-specific albumin promoter (L-sIDOL transgenics). L-sIDOL mice were fed a Western diet for 20 or 30 weeks and then analyzed for plasma lipid levels and atherosclerotic lesion formation. L-sIDOL mice showed dramatic reductions in hepatic LDLR protein and increased plasma LDL cholesterol levels on both chow and Western diets. Moreover, L-sIDOL mice developed marked atherosclerotic lesions when fed a Western diet. Lesion formation in L-sIDOL mice was more robust than in ApoE*3 Leiden mice and did not require the addition of cholate to the diet. Western diet-fed L-sIDOL mice had elevated expression of LXR target genes and pro-inflammatory genes in their aortas.
Conclusions
Liver-specific expression of dominant-active IDOL is associated with hypercholesterolemia and a marked elevation in atherosclerotic lesions. Our results show that increased activity of the IDOL pathway in the liver can override other LDLR regulatory pathways leading to cardiovascular disease. L-sIDOL mice are a robust, dominantly-inherited, diet-inducible model for the study of atherosclerosis.
Keywords: Atherosclerosis, lipoprotein metabolism, mouse model, E3 ubiquitin ligase, LDL cholesterol, nuclear receptor
INTRODUCTION
The E3 ligase inducible degrader of the LDL receptor (IDOL) ubiquitinates and facilitates lysosomal degradation of the LDL receptor (LDLR).1 IDOL expression is controlled by the sterol-responsive LXR nuclear receptors. In response to elevated cellular cholesterol levels, LXRs induce IDOL expression, thereby causing degradation of the LDLR and inhibiting additional cholesterol uptake by the cell. The LXR-IDOL pathway provides a complementary mechanism to the SREBPs for feedback regulation of LDL uptake by sterols. We have identified the molecular basis for IDOL target recognition and shown that loss of IDOL expression in cells alters LDLR protein levels and cholesterol uptake.2, 3 However, the tissue-specific effects of the IDOL pathway on plasma cholesterol levels and cardiovascular disease are unknown. The ability of IDOL to affect atherosclerosis development in mice has not been addressed previously.
Human genetic studies support a link between the activity of the LXR-IDOL pathway and plasma lipid levels. Genome-wide association studies have identified non-coding variants within IDOL that are associated with LDL cholesterol levels.4 In addition, a coding single nucleotide polymorphism (SNP) in IDOL, N342S, was associated with lower plasma cholesterol levels in Mexicans.5 Most recently, a complete loss-of-function IDOL allele was reported to be associated with low LDL levels.6
The importance of the LDLR in the regulation of plasma cholesterol levels is well established. Loss-of-function LDLR mutations in humans reduce hepatic LDL clearance, elevate plasma LDL levels and accelerate atherosclerosis.7 Moreover, the LDLR KO mouse is one of the most widely used models of hypercholesterolemia and atherosclerosis. The relative contribution of LDLR deletion in specific tissues to hypercholesterolemia and atherosclerosis in these mice has not been defined. As the liver is the key organ for the uptake of LDL cholesterol, we sought to examine the consequence of chronic liver-specific expression of a dominant active form of IDOL in mice. Our results show that chronic stable expression of IDOL effectively reduces hepatic LDLR protein levels and raises plasma LDL cholesterol levels, and that this effect is durable over the life of the animal. Liver-selective IDOL transgenic mice are a robust, dominantly-inherited, diet-inducible model for the study of atherosclerosis.
METHODS
An expanded methods section is available in the Online Data Supplement.
Mice
Briefly, mice were generated expressing a dominant active form of IDOL (K293R, K309R, K310R, K320R) known as super IDOL (sIDOL) under the direction of the albumin enhancer/promoter. At 6 weeks of age L-sIDOL mice and their wild type littermates were fed a western diet (WD; 21% fat 0.21% cholesterol; Research Diets) or maintained on a chow diet for 20 or 30 weeks as indicated. LDLR KO mice and ApoE*3 Leiden mice were placed on a WD from 6 weeks of age for 20 weeks. All mice were maintained on a 12-hour light/dark cycle and had access to food and water ad libitum. Mice were fasted for 6 hours prior to sacrifice. Animal studies were carried out in accordance with the Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals and the UCLA Animal Research Committee guidelines. Lesion area was quantitated by en face and aortic root analysis.
RESULTS
Generation of L-sIDOL transgenic mice
The E3 ubiquitin ligase IDOL is a potent post-transcriptional modifier of LDLR protein levels. In addition to targeting the LDLR for lysosomal degradation, IDOL also targets itself for proteasomal degradation through autoubiquitination. We previously identified lysine residues in the IDOL FERM domain critical for IDOL autoubiquitination and stability. An IDOL mutant lacking these residues (K293R, K309R, K310R, K320R) is unable to undergo autoubiquitination but maintains its ability to degrade the LDLR.2 We generated transgenic mice expressing this degradation-resistant, dominant-active form of human IDOL (sIDOL) in C57Bl/6J mice from the liver-specific albumin promoter (L-sIDOL transgenic mice; Fig. 1A). Realtime PCR analysis confirmed that transgene expression was selective for liver (Fig. 1B). Two independent lines of L-sIDOL mice exhibited a marked reduction in hepatic LDLR protein expression, confirming increased IDOL activity (Fig. 1C). We chose to perform subsequent studies on line #1, as this line was associated with slightly more hepatic LDLR degradation and higher plasma cholesterol levels than line #2 (Fig. 1C, D). As expected, there was no change in LDLR mRNA, consistent with the role of IDOL as a post-translational modifier of LDLR protein levels (Fig. 1E).
Figure 1. Generation of L-sIDOL transgenic mice.

(A) Schematic of the construct used to generate L-sIDOL mice demonstrating albumin (Alb) promoter, super IDOL and bovine growth hormone poly A tail (bGH polyA); (B) Expression of human IDOL mRNA in tissues normalised to Rplp0; (C) Immunoblot of hepatic LDLR and actin expression in wild type (WT) and L-sIDOL mice at 6 weeks of age on chow diet; (D) Plasma cholesterol levels of mice on chow diet at 6 weeks of age; (E) Hepatic mRNA expression of mouse LDLR normalised to Rplp0; Data is expressed as mean ± SEM. ***p<0.001 vs matched wild-type.
L-sIDOL mice and their littermate controls were fed chow or Western diet (WD) for 20 or 30 weeks. There was no difference in body or liver weight between wild-type (WT) mice and L-sIDOL mice within their respective age or diet groups (Table 1 and Online Table I). WD feeding was associated with increased weight gain across all groups at both time points. Male L-sIDOL mice showed a small reduction in hepatic fat mass as assessed by ex vivo magnetic resonance imaging, consistent with reduced hepatic lipid uptake (p<0.05; Fig. 2A). L-sIDOL expression did not cause overt hepatotoxicity, as plasma ALT was not different from WT controls on chow or WD (Fig. 2B). There was no significant difference in mouse IDOL or ABCA1 mRNA expression in the liver between WT and L-sIDOL transgenic mice across all groups (Fig. 2C). Thus, the sIDOL transgene does not appear to provoke a compensatory response in the activity of the endogenous LXR pathway in the liver.
Table 1.
Male mouse metabolic data.
| Chow 20 weeks | WD 20 weeks | Chow 30 weeks | WD 30 weeks | WD 20 weeks | |||||
|---|---|---|---|---|---|---|---|---|---|
| WT | L-sIDOL | WT | L-sIDOL | WT | L-sIDOL | WT | L-sIDOL | LDLR KO† | |
| n | 5 | 6 | 7 | 10 | 5 | 7 | 7 | 8 | 7 |
| Body Weight (g) | 28.6±1.4 | 28.1±0.8 | 47.1±0.7 | 45.8±1.8 | 31.6±1.0 | 31.5±1.3 | 48.1±1.2 | 47.2±1.1 | 44.2±1.9 |
| Liver Weight (g) | 1.1±0.0 | 1.0±0.1 | 4.3±0.3 | 3.9±0.5 | 1.4±0.1 | 1.3±0.1 | 4.0±0.2 | 3.7±0.2 | 2.7±0.4 |
| LW/BW (%) | 3.9±0.1 | 3.6±0.2 | 9.2±0.5 | 8.3±0.7 | 4.4±0.1 | 4.2±0.1 | 8.3±0.4 | 7.8±0.4 | 6.0±0.5 |
| TGs (mmol/L) | 0.79±0.04 | 0.46±0.04*** | 0.67±0.04 | 1.00±0.06** | 0.60±0.05 | 0.50±0.03 | 0.29±0.03 | 0.54±0.04*** | 6.35±1.09 |
BW – body weight; LW – liver weight; TGs - triglycerides; L-sIDOL – Albumin driven super IDOL transgenic mice; WT – wild type.
p<0.01,
p<0.001 vs matched wild type.
not included in statistical analysis.
Figure 2. Effect of L-sIDOL expression on hepatic function and gene expression.

(A) Hepatic fat mass (as assessed by magnetic resonance imaging) in male mice fed a western diet for duration as indicated; (B) Plasma alanine aminotransferase (ALT) levels in male mice on diets for durations as indicated; (C) Hepatic mRNA expression of mouse IDOL and ABCAI in mice on diet as indicated. Data normalised to Rplp0 and to chow fed WT mice at each time point for males and females; Data is expressed as mean ± SEM. *p<0.05 vs matched wild-type.
Stable expression of IDOL in mouse liver raises plasma LDL cholesterol levels
Plasma cholesterol levels were markedly elevated in L-sIDOL mice, not only in association with a WD, but even on a chow diet (Fig. 3A, B). This effect was even more profound after 30 weeks (p<0.001). However, L-sIDOL mice did not develop hypercholesterolemia to the level seen in LDLR KO mice on WD, likely reflecting either residual hepatic LDLR expression or preserved peripheral LDLR expression, in L-sIDOL mice (p<0.001; Fig. 7C). HPLC profiling of plasma lipid distribution revealed that the increase in cholesterol could be attributed primarily to an increase in the LDL fraction (Fig. 3D, G). Consistent with the HPLC data, we observed increased expression of ApoB protein levels in both chow and WD-fed L-sIDOL mice by immunoblotting (Fig. 3C, F). In contrast to LDL cholesterol, we observed little difference in HDL cholesterol fractions as assessed by HPLC profiling (Fig. 3D, G), and there was no difference ApoA-I protein expression (Fig. 3C, F). Interestingly, chow-fed male L-sIDOL mice showed a reduction in the VLDL triglyceride levels compared to WT, but this difference was not observed in females or when the mice were fed WD (Fig. 3E, H; Table 1, Supplemental Table 1). Similar to LDLR KO mice, L-sIDOL mice exhibited elevated plasma triglyceride levels in the VLDL and LDL fractions compared to WT littermates when maintained on WD (Table 1, Online Table I, Fig. 3H), although these values were not elevated to the levels seen in LDLR KO mice (Table 1).
Figure 3. Altered plasma lipid and apolipoprotein profiles in L-sIDOL mice.

(A, B) Plasma cholesterol levels in male and female mice on diets for durations as indicated; (C, F) Immunoblot of apolipoprotein B (ApoB) and apolipoprotein A-I (ApoA-I) in fractionated plasma for mice on 20 weeks diet as indicated; (D, G) HPLC chromatogram of plasma cholesterol after 20 weeks on chow or western diet (WD); (E, H) HPLC chromatogram of plasma triglycerides after 20 weeks on chow or western diet (WD); Data is expressed as mean ± SEM (A, B). *p<0.05, **p<0.01, ***p<0.001 vs matched wild-type.
Figure 7. Comparison of atherosclerotic mouse models.

(A) Representative pictures of en face assessment of atherosclerotic lesions L-sIDOL and LDLR KO mice fed a western diet; (B) Quantitation of en face aortic lesions in L-sIDOL and LDLR KO mice; (C) Plasma cholesterol levels of L-sIDOL and LDLR KO mice; Quantitation of (D) aortic root lesions and (E) en face aortic root lesions, in WT, L-sIDOL and ApoE*3 Leiden mice following 20 weeks of western diet; data for WT and L-sIDOL mice are the same data shown in Figure 4B; (F) HPLC chromatogram of plasma cholesterol following 20 weeks on western diet; data for WT and L-sIDOL mice are the same data shown in Figure 3G. ** p<0.01; ***p<0.001.
Stable expression of IDOL in mouse liver promotes atherosclerosis
To determine the impact of chronic expression of IDOL and the consequent downregulation of hepatic LDLR protein on the development of cardiovascular disease, we analysed atherosclerosis with both en face and aortic root section approaches. En face analysis after 20 weeks on WD revealed substantial lesion development in the aortas of L-sIDOL mice, whereas WT controls developed no detectable lesions at this time point (Fig. 4A, B). The extent of lesions in L-sIDOL mice was further enhanced when they were maintained on WD for 30 weeks. Interestingly, lesion formation in L-sIDOL mice was highly diet-dependent, as only minimal lesions were seen on chow diet (Fig. 4A, B).
Figure 4. En face atherosclerosis analysis in L-sIDOL mice.

(A) Representative pictures of en face assessment of atherosclerotic lesions in chow and western diet-fed male wild type (WT) and L-sIDOL mice after 20 weeks and 30 weeks of diet; (B) Quantitation of en face aortic lesions area in mice as indicated; **p<0.01 vs matched wild-type.
We observed similar effects of L-sIDOL expression on atherosclerosis development in the aortic root (Fig. 5A). L-sIDOL mice had a marked increased in lesion area compared to the negligible lesion development in WT mice (p<0.001). H & E suggested that some lesions were beginning to develop an acellular core (Fig. 5B, top). Trichrome staining further revealed collagen deposition in lesions of L-sIDOL mice (Fig. 5B, bottom). Immunohistochemical staining for CD68 confirmed that the lesions formed in L-sIDOL aortas were macrophage-rich (Fig. 5B, middle bottom). Overall, the architecture of the L-sIDOL lesions was consistent with early lesions observed in LDLR KO mice.
Figure 5. L-sIDOL mice develop atherosclerosis in the aortic root.

(A) Quantitation of aortic root lesions in male mice following 30 weeks of western diet; (B) Representative pictures of aortic root lesions in WT and L-sIDOL mice stained with haemotoxylin and eosin (H&E; top), Oil red O (top middle), CD68 (bottom middle) and trichrome (bottom) at magnifications as indicated.
We next assessed gene expression in the aortas of WT and L-sIDOL mice maintained on WD. Real-time PCR analysis of mRNA expression in whole aorta revealed an upregulation of the LXR target genes IDOL and AIM, consistent with increased sterols within the vessel wall of the lesion-rich transgenic mice (Fig. 6). We also observed an upregulation in inflammatory genes, including the p65 subunit of NK-κB, relA, as well as the proatherogenic vascular cell adhesion marker 1, VCAM-1 (Fig. 6). Increased scavenger receptor A (SR-A) was also observed, consistent with increased macrophage content (Fig. 6).
Figure 6. L-sIDOL mice aortic mRNA expression.
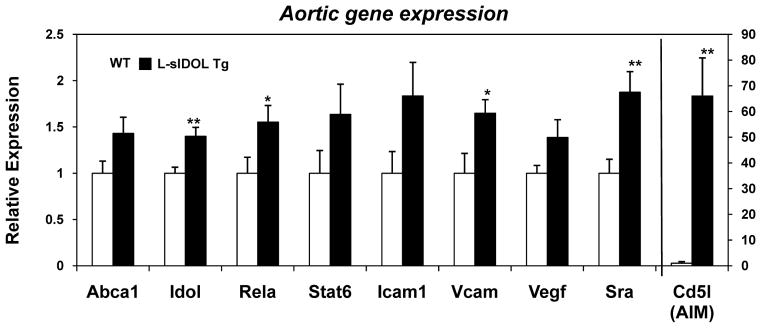
(A) mRNA expression of genes in aortas of male mice after 20 weeks WD, normalised to Rplp0; Data expressed as fold increase over wild type and represented as mean ± SEM. *p<0.05, **p<0.01 vs wild-type.
As the L-sIDOL mice represent a new monoallelic mouse model of atherogenesis, we compared plasma lipid levels and lesion development between L-sIDOL mice and two widely-used mouse models, LDLR KO mice and ApoE*3 Leiden mice. In comparison to LDLR KO mice, the phenotype of L-sIDOL mice is less severe, both in terms of the degree of plasma LDL cholesterol elevation and the extent of lesion development by en face and root section analysis (Fig. 7A–C). The higher LDL cholesterol levels in LDLR KO mice, despite the severely reduced hepatic LDLR protein levels in L-sIDOL mice, are suggestive of a significant role for extra-hepatic LDLR in LDL clearance in this setting. We also compared L-sIDOL mice to female ApoE*3 Leiden mice, as lesion development is reported to be greatest in females in this strain due to higher cholesterol and triglyceride levels than in males.19 When both L-sIDOL and ApoE*3 Leiden mice were fed WD for 20 weeks, L-sIDOL transgenic mice developed substantially more lesions as assessed by both en face and aortic root section analysis (Fig. 7D, E). Plasma total and LDL cholesterol levels were comparable between L-sIDOL and ApoE*3 Leiden mice on WD (L-sIDOL: 11.87 +/− 1.16 mmol/L (n=10), E3L: 13.28 +/− 0.63 mmol/L (n=8); Fig. 7F). Thus, L-sIDOL mice fed WD developed more severe atherosclerosis despite similar plasma cholesterol levels. Consistent with the ApoE mutation, however, ApoE*3 Leiden mice had higher levels of VLDL cholesterol compared to L-sIDOL mice.
In summary, chronic stable expression of IDOL in mouse liver is sufficient to durably increase plasma LDL cholesterol levels and promote substantial atherosclerotic lesion formation. Atherosclerosis development in L-sIDOL mice is robust for a transgenic mouse on the C57BL/6J background in the absence of a concurrent genetic lesion in LDLR or ApoE. Furthermore, lesion formation in this model does not require the addition of cholate to the diet.
DISCUSSION
We have shown here that chronically increased hepatic IDOL activity results not only in elevated plasma cholesterol levels, but also marked atherosclerotic lesion deposition on a WD. Our data indicate that dominant activation of the IDOL pathway in the mouse liver can override other LDLR regulatory pathways leading to cardiovascular disease.
Prior work has established that susceptibility to atherosclerosis differs markedly between mouse strains.20 In C57Bl/6 mice, there appears to be a threshold above which atherosclerosis development is observed. In our study, feeding C57Bl/6J mice WD raised plasma cholesterol levels to a measureable degree, but we observed virtually no lesion development. Introduction of the L-sIDOL transgene further increased plasma cholesterol levels in response to WD feeding and this resulted in substantial lesion formation. Interestingly, although the plasma cholesterol levels in L-sIDOL mice on WD are below those of LDLR KO mice, they are clearly above the threshold for lesion development, since we observe robust atherosclerosis in the aortic arch. The lower cholesterol levels seen in L-sIDOL mice compared to LDLR KO mice may be due to low level residual hepatic LDLR expression in L-sIDOL transgenic mice; however, it is also possible that extrahepatic LDLR expression contributes meaningfully to LDL clearance in a setting in which hepatic LDLR expression is lost. No tissue-specific models of the LDLR exist, so this has not been directly addressed; however some studies have suggested a role for intestinal LDLR in systemic cholesterol regulation.21
There are several advantages to a monoallelic transgenic mouse model for the study of atherosclerosis. Most importantly, the breeding strategy required to generate a compound mutant is greatly simplified, saving both time and money. L-sIDOL mice represent an attractive new model to test the effect of other transgenic or knockout alleles on atherosclerosis. Lesion deposition in L-sIDOL mice is highly dependent upon diet, with few lesions being observed in chow-fed mice. In this respect, L-sIDOL mice are similar to LDLR KO mice but different from ApoE KO mice.22 The fact that lesion deposition is driven by diet makes L-sIDOL mice an attractive model system. It is also notable that L-sIDOL mice develop substantial atherosclerotic lesion formation on standard WD formulation (41% calories from fat, 0.2% cholesterol). Unlike what has been reported for some other monoallelic atherosclerosis models, L-sIDOL mice do not require high levels of dietary cholesterol or the addition of cholate to induce lesion formation.23 Also, since the LDLR is still genetically present in L-sIDOL mice, pathways that affect LDLR expression or function could potentially still be studied with this model.
A further advantage of the L-sIDOL mice is the greater similarity of the lipid profile to humans as compared to other mouse models. In contrast to some other transgenic models of hypercholesterolemia, L-sIDOL mice carry the majority of their cholesterol in the LDL fraction, and thus represent a more humanized model. L-sIDOL mice are distinct from ApoE KO mice and ApoE*3 Leiden mice, which carry much of their cholesterol in the VLDL fraction, a profile that is not seen in humans.19, 24 ApoE*3 Leiden mice have been bred with CETP transgenic mice to exhibit or a more “humanized” lipid profile;25 however, a complex breeding strategy is required to introduce the transgenic alleles into a transgenic or knockout line of interest. We observed substantially greater lesion formation in L-sIDOL mice in a direct comparison to ApoE*3 Leiden mice after 20 weeks of WD feeding. Most published studies using ApoE*3 Leiden mice have employed more extreme diets, typically containing 0.5–1.0% w/w cholesterol. Many studies also included 0.1–0.5% cholate to increase the absorption of fat and cholesterol.26 A smaller number of studies have utilized diets comparable to the WD used here.25, 27
The majority of lesions observed in the aortic root of L-sIDOL mice are early lesions comprised primarily of foam-cells and smooth muscle cells, with some, such as that shown in Fig. 5, beginning to develop an acellular core. We have not observed the highly advanced lesions that can be observed with the LDLR KO or ApoE KO models. Thus, at the present time our model, like the ApoE*3 Leiden model, appears best suited to the study of early lesion formation. It remains possible that more extreme dietary feeding protocols might further enhance lesion severity and complexity in L-sIDOL mice, and we are actively testing alternate regimens. Westerterp et al. demonstrated that while ApoE*3 Leiden mice can develop complex lesions, the vast majority of lesions observed were early lesions comprised of fatty streaks and foam cells.25, 27 ApoE*3 Leiden mice must be crossed with hCETP transgenic mice in order to develop severe lesions on a WD.25, 27 Interestingly, although it is difficult to compare lesion quantitations performed in different laboratories, prior studies have estimated the total atherosclerotic burden in the aortic root of ApoE*3 Leiden mice to be ~50,000μm2 after 19 weeks on a WD.25, 27 These findings are in excellent agreement with our quantification of the ApoE*3 Leiden mice on 20 weeks of WD, which showed a mean total lesion area of ~47,000μm2.
In summary, hepatic expression of dominant active IDOL results in diet-induced hypercholesterolemia and atherosclerotic lesion deposition. These data establish that increased activity of the IDOL pathway in the liver can override other LDLR regulatory mechanisms and result in cardiovascular disease. In addition, we anticipate that L-sIDOL mice will prove to be a valuable tool for the study of atherosclerosis development in transgenic and knockout models.
Supplementary Material
Novelty and Significance.
What Is Known?
The low density lipoprotein receptor (LDLR) plays an important role in the regulation of plasma cholesterol levels and the LDLR null mouse is a common model of atherosclerosis.
Inducible degrader of the LDLR, IDOL, is an E3 ligase that targets the LDLR for ubiquitination and subsequent degradation and can modulate plasma cholesterol levels in humans.
What new information does this article contribute?
Chronic hepatic expression of a dominant-active form of IDOL, (L-sIDOL) is associated with marked hypercholesterolemia and atherosclerosis in a diet-inducible manner.
L-sIDOL mice are a novel mouse model to study atherosclerosis with potential advantages over other models related to their lipid profile, breeding strategy and susceptibility to lesions on diet
Current mouse models of atherosclerosis have limitations, including the use of extreme diets to generate atherosclerotic lesions, non-“human-like” lipid profiles or the need for complex breeding strategies to study a gene of interest. The LDLR is well established to play a role in hypercholesterolaemia and the LDLR null mouse is one of the most common models of atherosclerosis, but the relative contribution of specific tissues to this phenotype are currently unknown. We aimed to determine the effect of chronic hepatic LDLR degradation by a dominant-active form of the E3 ligase IDOL in mice. L-sIDOL mice exhibited degradation of hepatic LDLR protein and marked hypercholesterolaemia. Lesion deposition was observed in the aortic arch and aortic root in association with a western diet. Our data demonstrate that the hepatic IDOL-LDLR pathway can override other LDLR pathways, leading to vascular disease. In summary, L-sIDOL mice are a robust, dominantly-inherited, diet-inducible model of atherosclerosis. Advantages over other models include their “human-like” lipid profile, their susceptibility to develop lesions without being placed on an extreme dietary regimen and a simplified breeding strategy to test a transgenic or knockout alleles of interest, saving both time and money.
Acknowledgments
SOURCES OF FUNDING
ACC was funded by a National Heart Foundation of Australia Overseas Fellowship (O 08M 3934). P.T. is an Investigator of the Howard Hughes Medical Institute. This work was supported by NIH grant R01 HL066088.
Nonstandard Abbreviations and Acronyms
- AIM
Apoptosis inhibitor expressed in macrophages
- ABCAI
ATP-binding cassette transporter AI
- ALT
Alanine aminotransferase
- ApoE
Apolipoprotein E
- BSA
Bovine serum albumin
- CCD
Couple-charged device
- CD
Cluster of differentiation
- EDTA
Ethylenediaminetetraacetic acid
- HDL
High-density lipoprotein
- HPLC
High-performance liquid chromatography
- ICAM
Intracellular adhesion molecule
- IDOL
Inducible degrader of the LDLR
- KO
Knockout
- LDLR
Low-density lipoprotein receptor
- L-sIDOL
Liver-specific super IDOL transgene
- LXR
Liver X receptor
- OCT
Optimal cutting temperature
- PBS
Phosphate buffered saline
- PCR
Polymerase chain reaction
- RelA
v-rel avian reticuloendotheliosis viral oncogene homolog A (p65)
- SNP
Single nucleotide polymorphism
- SR-A
Scavenger receptor A
- SREBP
Sterol regulatory element-binding protein
- STAT
Signal transducer and activator of transcription
- Tg
Transgenic
- VCAM
Vascular cell adhesion molecule
- VLDL
Very low-density lipoprotein
- VEGF
Vascular endothelial growth factor
- WD
Western Diet
Footnotes
In May 2014, the average time from submission to first decision for all original research papers submitted to Circulation Research was 14.87 days.
DISCLOSURES
None.
References
- 1.Zelcer N, Hong C, Boyadjian R, Tontonoz P. Lxr regulates cholesterol uptake through idol-dependent ubiquitination of the ldl receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calkin AC, Goult BT, Zhang L, Fairall L, Hong C, Schwabe JW, Tontonoz P. Ferm-dependent e3 ligase recognition is a conserved mechanism for targeted degradation of lipoprotein receptors. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:20107–20112. doi: 10.1073/pnas.1111589108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scotti E, Hong C, Yoshinaga Y, Tu Y, Hu Y, Zelcer N, Boyadjian R, de Jong PJ, Young SG, Fong LG, Tontonoz P. Targeted disruption of the idol gene alters cellular regulation of the low-density lipoprotein receptor by sterols and liver x receptor agonists. Mol Cell Biol. 2011;31:1885–1893. doi: 10.1128/MCB.01469-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chasman DI, Pare G, Mora S, Hopewell JC, Peloso G, Clarke R, Cupples LA, Hamsten A, Kathiresan S, Malarstig A, Ordovas JM, Ripatti S, Parker AN, Miletich JP, Ridker PM. Forty-three loci associated with plasma lipoprotein size, concentration, and cholesterol content in genome-wide analysis. PLoS Genet. 2009;5:e1000730. doi: 10.1371/journal.pgen.1000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissglas-Volkov D, Calkin AC, Tusie-Luna T, Sinsheimer JS, Zelcer N, Riba L, Tino AM, Ordonez-Sanchez ML, Cruz-Bautista I, Aguilar-Salinas CA, Tontonoz P, Pajukanta P. The n342s mylip polymorphism is associated with high total cholesterol and increased ldl receptor degradation in humans. The Journal of clinical investigation. 2011;121:3062–3071. doi: 10.1172/JCI45504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorrentino V, Fouchier SW, Motazacker MM, Nelson JK, Defesche JC, Dallinga-Thie GM, Kastelein JJ, Kees Hovingh G, Zelcer N. Identification of a loss-of-function inducible degrader of the low-density lipoprotein receptor variant in individuals with low circulating low-density lipoprotein. Eur Heart J. 2013;34:1292–1297. doi: 10.1093/eurheartj/ehs472. [DOI] [PubMed] [Google Scholar]
- 7.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 8.Pinkert CA, Ornitz DM, Brinster RL, Palmiter RD. An albumin enhancer located 10 kb upstream functions along with its promoter to direct efficient, liver-specific expression in transgenic mice. Genes Dev. 1987;1:268–276. doi: 10.1101/gad.1.3.268. [DOI] [PubMed] [Google Scholar]
- 9.van den Maagdenberg AM, Hofker MH, Krimpenfort PJ, de Bruijn I, van Vlijmen B, van der Boom H, Havekes LM, Frants RR. Transgenic mice carrying the apolipoprotein e3-leiden gene exhibit hyperlipoproteinemia. The Journal of biological chemistry. 1993;268:10540–10545. [PubMed] [Google Scholar]
- 10.Lee RG, Kelley KL, Sawyer JK, Farese RV, Jr, Parks JS, Rudel LL. Plasma cholesteryl esters provided by lecithin: Cholesterol acyltransferase and acyl-coenzyme a:Cholesterol acyltransferase 2 have opposite atherosclerotic potential. Circ Res. 2004;95:998–1004. doi: 10.1161/01.RES.0000147558.15554.67. [DOI] [PubMed] [Google Scholar]
- 11.Tangirala RK, Rubin EM, Palinski W. Quantitation of atherosclerosis in murine models: Correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in ldl receptor-deficient and apolipoprotein e-deficient mice. Journal of lipid research. 1995;36:2320–2328. [PubMed] [Google Scholar]
- 12.Qiao JH, Xie PZ, Fishbein MC, Kreuzer J, Drake TA, Demer LL, Lusis AJ. Pathology of atheromatous lesions in inbred and genetically engineered mice. Genetic determination of arterial calcification. Arteriosclerosis and thrombosis: a journal of vascular biology/American Heart Association. 1994;14:1480–1497. doi: 10.1161/01.atv.14.9.1480. [DOI] [PubMed] [Google Scholar]
- 13.Mehrabian M, Allayee H, Wong J, Shi W, Wang XP, Shaposhnik Z, Funk CD, Lusis AJ. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91:120–126. doi: 10.1161/01.res.0000028008.99774.7f. [DOI] [PubMed] [Google Scholar]
- 14.Hong C, Bradley MN, Rong X, Wang X, Wagner A, Grijalva V, Castellani LW, Salazar J, Realegeno S, Boyadjian R, Fogelman AM, Van Lenten BJ, Reddy ST, Lusis AJ, Tangirala RK, Tontonoz P. Lxralpha is uniquely required for maximal reverse cholesterol transport and atheroprotection in apoe-deficient mice. Journal of lipid research. 2012;53:1126–1133. doi: 10.1194/jlr.M022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradley MN, Hong C, Chen M, Joseph SB, Wilpitz DC, Wang X, Lusis AJ, Collins A, Hseuh WA, Collins JL, Tangirala RK, Tontonoz P. Ligand activation of lxr beta reverses atherosclerosis and cellular cholesterol overload in mice lacking lxr alpha and apoe. The Journal of clinical investigation. 2007;117:2337–2346. doi: 10.1172/JCI31909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P. Synthetic lxr ligand inhibits the development of atherosclerosis in mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masson P. Some histological methods; trichrome stainings and their preliminary technique. J Tech Methods. 1929;12:75–90. [Google Scholar]
- 18.Orozco LD, Kapturczak MH, Barajas B, Wang X, Weinstein MM, Wong J, Deshane J, Bolisetty S, Shaposhnik Z, Shih DM, Agarwal A, Lusis AJ, Araujo JA. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ Res. 2007;100:1703–1711. doi: 10.1161/CIRCRESAHA.107.151720. [DOI] [PubMed] [Google Scholar]
- 19.van Vlijmen BJ, van’t Hof HB, Mol MJ, van der Boom H, van der Zee A, Frants RR, Hofker MH, Havekes LM. Modulation of very low density lipoprotein production and clearance contributes to age- and gender- dependent hyperlipoproteinemia in apolipoprotein e3-leiden transgenic mice. The Journal of clinical investigation. 1996;97:1184–1192. doi: 10.1172/JCI118532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 21.Le May C, Berger JM, Lespine A, Pillot B, Prieur X, Letessier E, Hussain MM, Collet X, Cariou B, Costet P. Transintestinal cholesterol excretion is an active metabolic process modulated by pcsk9 and statin involving abcb1. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1484–1493. doi: 10.1161/ATVBAHA.112.300263. [DOI] [PubMed] [Google Scholar]
- 22.Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. The Journal of clinical investigation. 1994;93:1885–1893. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell-Huynh DA, Farese RV, Jr, Johnson DF, Flynn LM, Pierotti V, Newland DL, Linton MF, Sanan DA, Young SG. Transgenic mice expressing high levels of human apolipoprotein b develop severe atherosclerotic lesions in response to a high-fat diet. The Journal of clinical investigation. 1995;95:2246–2257. doi: 10.1172/JCI117915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein e. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 25.Westerterp M, van der Hoogt CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, Havekes LM, Rensen PC. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in apoe*3-leiden mice. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2552–2559. doi: 10.1161/01.ATV.0000243925.65265.3c. [DOI] [PubMed] [Google Scholar]
- 26.Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 27.den Boer MA, Westerterp M, de Vries-van der Weij J, Wang Y, Hu L, Espirito Santo SM, Kooistra T, Reiss P, Romijn JA, Havekes LM, Rensen PC. Ritonavir protects against the development of atherosclerosis in apoe*3-leiden mice. Atherosclerosis. 2010;210:381–387. doi: 10.1016/j.atherosclerosis.2009.11.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.