

Abstract
Almost all invasive Neisseria meningitidis isolates express capsular polysaccharide. Antibody (Ab) is required for complement-dependent killing of meningococci. While alternative pathway evasion has received considerable attention, little is known about classical pathway (CP) inhibition by meningococci and forms the basis of this study. We engineered capsulated and unencapsulated isogenic mutant strains of groups A, B, C, W and Y meningococci to express similar amounts of the same factor H-binding protein (fHbp; a key component of group B meningococcal vaccines) molecule. Despite similar anti-fHbp mAb binding, significantly less C4b was deposited on all five encapsulated mutants compared to their unencapsulated counterparts (P<0.01), when purified C1 and C4 were used to deposit C4b. Reduced C4b deposition was the result of capsule-mediated inhibition of C1q engagement by Ab. C4b deposition correlated linearly with C1q engagement by anti-fHbp. While B, C, W and Y capsules limited CP-mediated killing by anti-fHbp, the unencapsulated group A mutant paradoxically was more resistant than its encapsulated counterpart. Strains varied considerably in their susceptibility to anti-fHbp and complement despite similar Ab binding, which may have implications for the activity of fHbp-based vaccines. Capsule also limited C4b deposition by anti-porin A mAbs. Capsule expression decreased binding of an anti-LOS IgM mAb (~1.2 to 2-fold reduction in fluorescence). Akin to observations with IgG, capsule also decreased IgM-mediated C4b deposition when IgM binding to the mutant strain pairs was normalized. In conclusion, we show that capsular polysaccharide, a critical meningococcal virulence factor, inhibits the CP of complement.
Keywords: Neisseria meningitidis, meningococci, capsular polysaccharide, complement, classical pathway, antibody, C4, C1q
Introduction
Complement-dependent bactericidal activity is an established correlate of protection against meningococcal disease. Intrathecal serotherapy with equine anti-meningococcal antiserum was shown to be beneficial in meningococcal meningitis as far back as the early 1900s by Flexner and Jobling (1, 2), which suggested a role for antibody (Ab) in host defenses against invasive disease. Elegant studies by Goldschneider and colleagues firmly established a role for Ab in protection against invasive meningococcal infection (3). They reported an inverse relationship between the incidence of meningococcal disease and age-specific serum bactericidal activity against prototypic groups A, B and C meningococcal strains (3). These findings were corroborated by a more recent study in the U.K (4).
While colonization of the human nasopharynx by Neisseria meningitidis is a relatively common event, invasive disease is rare (5). Previously ‘nonimmune’ individuals may be protected against invasive infection if colonization with Neisseria meningitidis elicits adequate titers of protective Ab (6). With a few exceptions (7–9), almost every strain of N. meningitidis recovered from the bloodstream or cerebrospinal fluid expresses a polysaccharide capsule. By contrast, unencapsulated (“capsule null”, or cnl) isolates comprise ~15–20 % of meningococci isolated from the nasopharynges of asymptomatic carriers (10, 11). Capsule appears to be critical for invasion of the bacteria into the bloodstream of individuals who lack adequate titers of specific protective Ab, where high levels of complement are encountered.
Over the past several years, much attention has focused on the role of membrane proteins in complement evasion, in particular the interactions between factor H (FH), an inhibitor of the alternative pathway of complement, and meningococcal factor H binding protein (fHbp (12); also referred to as LP2086 (13)) (14–16). fHbp is a component of a group B meningococcal vaccine candidate that has recently been licensed for clinical use in several countries (17). Other meningococcal surface molecules that inhibit the alternative pathway include Neisserial surface protein A (NspA) (18) and PorB2 (19), which directly bind to FH. Lipooligosaccharide (LOS) sialic acid, in concert with surface-bound C3 fragments increases FH recruitment (20). Capsular polysaccharide is critical for meningococci to evade killing by high levels of complement and loss of capsule expression (alone) is associated with a dramatic decrease in complement resistance (21, 22) and virulence in the infant rat and infant mouse models (23, 24).
The role of sialic acids, such as N-acetyl neuraminic acid, in regulating the alternative pathway of complement on host cells (25, 26) and on microbial surfaces (27, 28) including N. meningitidis (20, 29–32), is well described. Capsules expressed by four of the five most widely distributed meningococcal groups (groups B, C, W and Y) contain N-acetyl neuraminic acid and in light of the well-established link between sialic acids and the alternative pathway, studies pertaining to complement regulation by capsules have also focused on the alternative pathway. Given the importance of the classical pathway (CP) for complement-dependent killing of meningococci (22, 33) and the central role of capsule in serum resistance, we sought to determine how capsule modulated early events in Ab-dependent CP activation.
Materials and Methods
Bacterial strains and mutants
Isogenic mutant strains were derived from N. meningitidis strains A2594 (WUE2594), H44/76, C2120, W171 and Y2225. Their relevant characteristics are listed in Table 1. LOS sialylation was abrogated by deleting lst (encodes lipooligosaccharide sialyltransferase) as described previously (18). Note that group A isolates do not sialylate their LOS unless supplied with cytidinemonophospho-N-acetyl neuraminic acid (CMP-Neu5Ac) in growth media. To delete capsule from group A strain 2594, mynB (its newly proposed designation is csaB (34)) was deleted as described previously (18). The groups B, C W and Y isolates were rendered unencapsulated by deleting their respective polysialyltransferase siaD genes (19, 33) – these genes are now designated csb, csc, csw and csy, respectively (34). fHbp from A2594, C2120, W171 and Y2220 was replaced with fHbp from H44/76 (variant 1 fHbp) as follows. We previously constructed a mutant of group Y strain Y2220, called Y2220 siaDH44/76GNA1870+ (fHbp was previously called Genome-derived Neisseria Antigen (GNA) 1870 (12)) that expresses the full length H44/76 fHbp (14). This mutant contains the tetracycline-resistance marker immediately 3′ to fHbp. DNA extracted from Y2220 siaDH44/76GNA1870+ was used to transform encapsulated strains that lacked LOS sialylation (A2594, C2120 lst, W171 lst and Y2225 lst) and unencapsulated / LOS unsialylated mutants A2594 mynB, C2120 siaD lst, W171 siaD lst and Y2225 siaD lst. Tetracycline-resistant colonies were selected (the MIC of tetracycline for each strain was determined) and screened for binding to mAb JAR 1 that is specific for variant 1 fHbp expressed by H44/76 (35). The fHbp of clones that bound to mAb JAR 1 was sequenced to verify the presence of the entire H44/76 fHbp sequence. For simplicity, we will henceforth refer to the strains by their original serogroup (i.e., A, B, C, W or Y) followed by Cap+ or Cap− to denote encapsulated and unencapsulated mutants, respectively. All mutants used in this study lack LOS sialic acid and express the same variant 1 fHbp molecule.
Table 1.
Characteristics of wild-type N. meningitidis used to derive mutants for this study
| Strain | Group | Serotype (Class) |
SerosubtypeA | MLST | Origin (Year) |
Ref. |
|---|---|---|---|---|---|---|
| A2594 (WUE 2594) | A | 4 (PorB3) | 1–9 | ST-5 | Germany, 1991 | (63) |
| H44/76 | B | 15 (PorB3) | 1.7,16 | ST-32 | Norway, 1976 | (64) |
| C2120 | C | NTB (PorB2) | 1.5,2 | ST-11 | NAC | (65) |
| W171 | W | NT (PorB2) | 1.10 | ST-11/CC-11 | USA | (66) |
| Y2225 | Y | NTB (PorB2) | 1.2 | ST-11/CC-11 | Germany 1994 | (66) |
defined either by anti-PorA mAb reactivity or by variable region (VR) sequence typing
NT, Non-typeable
NA, not available
Complement proteins
Purified C1 complex (200 µg/ml), C1q (1 mg/ml) and C4 (4 mg/ml) were from Complement Technology, Inc (Tyler, TX). Protease inhibitors in purified C1 (EDTA, benzamidine and EACA) were removed by dialysis at 4 °C against PBS using an Amicon® Ultra-4 centrifugal Filter Unit (30 kD cutoff) prior to use.
IgG and IgM depleted serum
IgG and IgM were depleted from serum as described previously by passage over protein G sepharose and anti-human IgM agarose (36). Briefly, serum that was treated with EDTA (10 mM) to prevent complement activation and 1M NaCl to minimize C1q loss during immunodepletion was passed over immobilized protein G and anti-human IgM at 4 °C. The estimated amount of IgG and IgM in serum did not exceed the binding capacity of the columns. The fall-through was spin-concentrated and dialyzed against PBS with an Amicon® Ultra-15 (30 kD cutoff). Hemolytic activity was verified with the Total Hemolytic Complement kit (The Binding Site, Birmingham, U.K). IgG and IgM depletion was ascertained by dot blot assays on PVDF membranes as described previously (36). Serum was stored in single-use aliquots at −70 °C.
Antibodies and conjugates
Anti-variant 1 fHbp mAb JAR 1 (mouse IgG3) has been described previously (35). For simplicity, JAR 1 will henceforth be called anti-fHbp in this paper. Anti-PorA mAbs 1.9, 1.7, 1.5 and 1.2 that recognize PorA expressed by A2594, H44/76, C2120 and Y2225, respectively, were obtained from the National Institute for Biological Standards and Control (NIBSC), Hertfordshire, U.K. mAb P1.9 was supplied as ascites, while the other anti-PorA mAbs were provided as concentrated tissue culture supernatants. The mAb 3F11 hybridoma cell line was provided by Dr. Michael A. Apicella (University of Iowa); mAb 3F11 (mouse IgM) recognizes the lacto-N-neotetraose LOS species in the unsialylated state (37, 38). Polyclonal chicken anti-human C4 biotin used in flow cytometry assays was from Cedarlane Laboratories. Sheep anti-human C1q-FITC was from Biodesign (MP Biomedicals). Sheep polyclonal anti-human C4 used in western blotting assays was from Biodesign (MP Biomedicals). FITC-conjugated anti-mouse IgG, IgM and extravidin, and alkaline phosphatase conjugated anti-sheep IgG were from Sigma. All conjugates for flow cytometry were used at a dilution of 1:200 in PBS/1% BSA and anti-sheep IgG ALP was used at 1:1000 in PBS/0.05% Tween 20. Function blocking anti-factor Bb mAb (Quidel Corporation; Cat. No. A227) was added to serum at a concentration of 100 µg/ml to block alternative pathway activation (39, 40).
Flow cytometry
Flow cytometry to detect bound Ab, C4b or C1q was performed using previously described methods (18–20). To detect C4b deposition using pure complement components, ~107 bacteria in HBSS containing 1 mM Ca2+ and 1mM Mg2+ (HBSS++) were incubated with anti-fHbp (10 µg/ml) or the indicated dilutions of anti-PorA or mAb 3F11, followed by C4 (50 µg/ml) and C1 complex (7.5 µg/ml) in a final reaction volume of 50 µl. Control reactions that lacked anti-fHbp were included to compare C4b deposition that was mediated by Ab versus C4b deposition that may have occurred independently of Ab binding – for example, by direct binding of C1 to bacteria. Following incubation of the reaction mixture for 30 min at 37 °C, bacteria were washed and deposited C4b was measured using anti-C4 biotin and extravidin FITC. To measure C1q engagement by anti-fHbp, bacteria were incubated with anti-fHbp (10 µg/ml) and purified C1q (20 µg/ml) for 30 min at 37 °C, washed and bound C1q was detected with anti-C1q FITC. Data were collected on a BD FACSCalibur (Becton Dickinson) flow cytometer and analyzed with FlowJo software (Tree Star).
Western blotting
C4b deposited on bacteria was analyzed by western blotting as described previously (41, 42). Bacteria were incubated with anti-fHbp, C1 and C4 as described above, washed thrice and pellets were lysed with 4× LDS sample buffer containing 2-ME. The lysates were electrophoresed on 4–12% Bis-Tris gels and transferred to a PVDF membrane by western blotting. Blots were cut horizontally at the 50 kD marker; proteins that migrated above this were probed with sheep anti-human C4 to detect adducts of the C4b α' chain covalently linked to its bacterial targets, while proteins migrating faster than the 50kD marker were stained with coomassie blue, which served as a control to indicate equal loading of Cap+ and Cap− bacterial lysates.
Serum bactericidal assay
Serum bactericidal assays were performed as described previously (40). Bacteria that had been harvested from an overnight culture on chocolate agar plates were repassaged onto fresh chocolate agar and allowed to grow for 4–5 h at 37°C in an atmosphere containing 5% CO2 and then suspended in HBSS++. Approximately 2000 CFU of N. meningitidis with anti-fHbp (0.25 µg/ml) were incubated with IgG/IgM-depleted serum (5%) that contained 100 µg/ml of the anti-factor Bb mAb, in a final reaction volume of 75 µl. This concentration of mAb and complement was determined following preliminary titration experiments. Aliquots of 12.5-µl reaction mixtures were plated onto chocolate agar in duplicate at the beginning of the assay (t0) and again after incubation at 37°C for 30 min (t30). Survival was calculated as the number of viable colonies at t30 relative to t0.
Results
Capsule inhibits C4b deposition by anti-fHbp mAb JAR 1
To determine the effect of capsule on CP activation mediated by an anti-fHbp mAb, we replaced the fHbp molecule of the isogenic pairs of strains – A2594 and A2594 mynB, C2120 lst and C2120 siaD lst, W171 lst and W171 siaD lst and Y2225 lst and Y2225 siaD lst – with the variant 1 fHbp molecule of strain H44/76. We elected to use mutants lacking LOS sialic acid (lst mutants) because LOS is a target for C4b deposition on the pathogenic Neisseriae (41, 42) and LOS sialylation also limits C4b deposition on bacteria (42). Further, deleting capsule (siaD mutants) on groups B, C, W and Y isolates may ‘shunt’ cytidinemonophospho-N-acetyl neuraminic acid (CMP-NANA) from capsule biosynthesis on to LOS and result in a greater proportion of sialylated LOS on Cap− strains (22). As shown in Figure 1, all mutants bound similar amounts of anti-fHbp by flow cytometry.
Fig. 1.

Isogenic meningococcal mutants that differ in capsule expression express similar amounts of fHbp. Variant 1 fHbp expression levels were measured by flow cytometry with anti-fHbp mAb JAR 1. Each encapsulated (Cap+) strain and its isogenic unencapsulated (Cap−) mutant was incubated with anti-fHbp (10 µg/ml), followed by detection with anti-mouse IgG-FITC. X-axis, fluorescence (log10 scale); Y-axis, counts. One representative experiment of at least two repeats is shown.
Bacteria were then incubated with anti-fHbp (10 µg/ml), purified C1 complex (7.5 µg/ml) and pure C4 (50 µg/ml), and C4b deposited on the bacterial surface was measured by flow cytometry. Significantly less C4b was deposited on each of the Cap+ strains compared to its corresponding Cap− mutant (Fig. 2A; representative histogram tracings are shown in Supplemental Fig. S1). C4b deposition was dependent on the presence of specific Ab, as revealed by minimal C4b deposition when bacteria were incubated with pure C1 and C4 alone (Fig. 2B). Considerable variation in the amount of C4b deposited across the Cap− mutants was noted; the least amount of C4b was deposited on the group A Cap− mutant, intermediate amounts on the group B Cap-mutant, while high levels of C4b were deposited on the groups C, W and Y Cap− mutants. Bacteria were then incubated with anti-fHbp and IgG/IgM-depleted normal human serum and C4b deposition was measured. With the exception of the group A mutant pair where similar and low amounts of C4b was deposited, inhibition of C4b deposition by capsular polysaccharide mirrored findings with purified C1 and C4 (Figs. 2C). Similar to observations in Fig. 2B, minimal C4b was deposited when anti-fHbp was excluded from the reaction mixture (≤10% of the fluorescence seen when anti-fHbp was present in the reaction mixture; data not shown). We also performed western blotting analysis of bacteria that were incubated with anti-fHbp and purified C1 and C4 (Fig. 2D), which confirmed findings of the flow cytometry assays using a second independent method. It is worth noting that the targets for C4b deposited by anti-fHbp were LOS and opacity protein (Opa), as described previously in studies using normal human serum (41). Collectively, the data suggest that all five major meningococcal capsular polysaccharides interfere with C4b deposition mediated by an anti-fHbp mAb.
Fig. 2.

Capsule expression limits anti-fHbp−mediated C4b deposition. A. Bacteria were incubated with anti-fHbp (10 µg/ml), purified C1 complex (7.5 µg/ml) and pure C4 (50 µg/ml) and C4b deposited on bacteria (median fluorescence) was measured by flow cytometry. ‘Control’ represents the fluorescence of a reaction mixture that lacked complement components. The median fluorescence of each reaction was recorded and each bar represents the mean (SEM) of 3 experiments (Supplemental Figure S1 shows representative histograms). Y-axis, median fluorescence of C4 deposition. ***, P<0.001 (ANOVA). B. C4b deposition on meningococci is Ab-dependent. Bacteria were incubated with C1 and C4 in the absence of anti-fHbp and C4b deposition was measured by flow cytometry. Axes and controls are as described in A (note the magnified lower half of the Y-axis compared to panel A). C. Capsule inhibits anti-fHbp−mediated C4b deposition in the presence of human serum. Isogenic capsule mutants were incubated with anti-fHbp, followed by the addition of IgG and IgM depleted human serum at a final concentration 3.3%. Axes are as described above. **, P<0.01; *, P<0.05; ns, not significant (ANOVA). D. Western blotting confirms capsule-mediated inhibition of C4b deposition and identifies LOS and opacity proteins (Opa) as targets for C4b deposited by anti-fHbp. Bacteria were incubated with anti-fHbp, C1 and C4 as described in A. The ~87 kD α' chain of C4b binds to its bacterial targets covalently though ester or amide bonds. Electrophoresis under reducing conditions results in the ~87 kD α' migrating as a complex with its bacterial targets. Proteins were transferred to a PVDF membrane by western blotting to reveal C4b α'-bacterial target complexes. The locations of the ~90 kD and ~115 kD complexes of (LOS + C4b α'; labeled “α'+LOS (~90 kD)”) and (Opa + C4b α'; labeled “α'+Opa (~115 kD)”), respectively, are indicated; the identity of these bands was defined previously (41). The lane marked ‘C4’ contains pure C4 and the positions of the intact 95 kD α (labeled “α (95 kD)”) and 75 kD β (labeled “β (75 kD)”) chains are shown. Control lanes contained bacteria alone (labeled ‘W Cap− (alone)), or bacteria plus C1 and C4 with no added anti-fHbp. Proteins migrating below the 50 kD marker were stained with coomassie blue to ensure similar protein loading across the isogenic Cap+ and Cap− mutants.
Capsule interferes with C1q engagement by anti-fHbp
C4b deposition on a surface is the result of several steps. C1 complex binding by Ab induces a conformational change in C1q, which results in autoactivation of C1r. Activated C1r then cleaves and activates C1s and the latter cleaves a 9 kD fragment from the amino terminus of the α-chain of C4, called C4a, which exposes an internal thioester bond in C4. Nucleophilic attack of the exposed and metastable thioester in the C4b fragment by hydroxyl or amino groups leads to formation of covalent ester or amide bonds, respectively (43, 44).
The ability of anti-fHbp to engage C1q represents an early step in Ab-mediated CP activation and was studied next. As shown in Fig. 3A (Supplemental Fig. S2 shows representative histograms), capsule expression interfered with C1q binding by anti-fHbp. The amount of C1q bound also varied greatly across Cap− mutants and showed a statistically significant positive correlation with the amount of anti-fHbp -mediated C4b deposition (Fig. 3B). Collectively, these data show that despite similar anti-fHbp binding, the activity of anti-fHbp to activate the CP and deposit C4b varies across strains.
Fig. 3.

Capsule inhibits engagement of C1q by anti-fHbp. A. The Cap+ and Cap− isogenic mutants were incubated with anti-fHbp (10 µg/ml) followed by C1q (20 µg/ml); bound C1q was detected by flow cytometry. Reaction mixtures lacking anti-fHbp is shown by the hatched bars. ‘Control’ represents bacteria plus anti-C1q FITC. *** P<0.001; ** P<0.01; ns, not significant (ANOVA). Representative histograms are shown in Supplemental Figure S2. B. C4b deposition on Cap− mutants correlates directly with the amount of C1q engaged by anti-fHbp. C4b deposition (mean fluorescence values derived from Fig. 2A) was plotted as a function of C1q binding (mean fluorescence values derived from Fig. 3A). Correlation was determined by linear regression with Pearson’s coefficient.
Effects of capsular polysaccharide expression on CP-mediated killing of meningococci by anti-fHbp
We next determined the ability of anti-fHbp to mediate complement-dependent bactericidal activity through the classical pathway. Anti-factor Bb mAb (100 µg/ml) was added to IgG/IgM-depleted serum to block the alternative pathway of complement (40). As shown in Fig. 4, the groups B, C, W and Y encapsulated mutants were significantly more resistant to CP-mediated killing by the anti-fHbp mAb. Paradoxically, the Cap− group A mutant was more resistant than the isogenic Cap+ mutant. Controls where bacteria were incubated with complement (IgG/IgM depleted serum) alone resulted in 120 – 200% survival in every instance (data not shown).
Fig. 4.
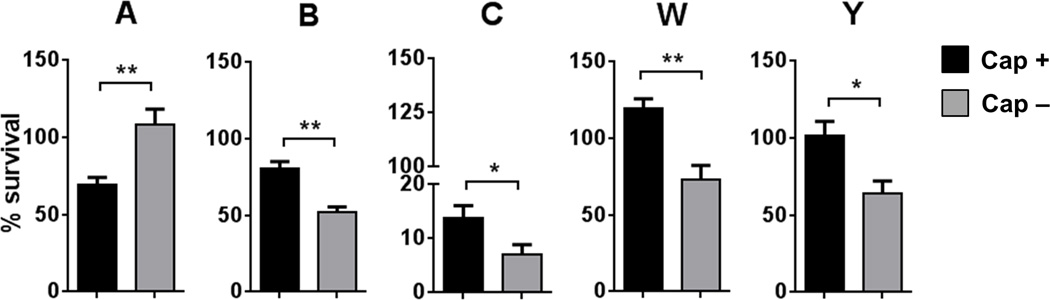
The effects of meningococcal capsular polysaccharide on CP-mediated killing by anti-fHbp. The five Cap− meningococcal mutants were incubated with anti-fHbp (0.25 µg/ml) followed by the addition of serum immunodepleted of IgG and IgM (5%) to which anti-Bb mAb (100 µg/ml) was added to block AP activation. Percentage survival of bacteria at 30 min (mean (SEM) of 5 separate experiments) is shown on the Y-axis. Note the segmented Y-axis for the group C strain. *, P<0.05; **, P<0.01 compared to the corresponding isogenic mutant (two-tailed t test).
Capsule inhibits C4b deposition by anti-PorA mAbs
To ensure that the effects of capsular polysaccharide on CP activation was not specific for anti-fHbp Ab, we next measured C4b deposition mediated by anti-PorA mAbs. mAbs that were available against four of the strains used in this study (groups A, B, C and Y) were tested for their ability to deposit C4b. While capsule expression did not affect binding of the mAbs to the bacterial surface (Fig. 5A), decreased C4b deposition was noted on the Cap+ mutants (Fig. 5B). Taken together, these data suggest that capsule limits C4b deposition by specific IgG mAbs.
Fig. 5.

Capsule limits C4b deposition mediated by anti-PorA mAbs. A. Cap+ and Cap− isogenic mutants bind similar amounts of anti-PorA mAb as measured by flow cytometry. X-axis, reciprocal dilution of ascites (P1.9) or tissue culture supernatants (P1.7, P1.5 and P1.2); Y-axis, median fluorescence. B. Capsule limits C4b deposition mediated by anti-Por mAbs. Cap+ and Cap− isogenic mutants were incubated with equal amounts of their respective anti-PorA mAbs, followed by the addition of purified C1 complex and C4 as described in Fig. 2, and C4b deposition was measured by flow cytometry. Representative histogram tracings of one experiment (of two reproducible repeats) are shown. X-axis, fluorescence (log10 scale); Y-axis, counts. C4b deposition on Cap+ and Cap− strains are shown by the grey shaded and solid black line histograms, respectively. Controls, where complement proteins were excluded, are shown by the dashed line. The numbers alongside each histogram represents the median fluorescence of the entire bacterial population and the shading and outline corresponds to that of the histogram.
Capsule inhibits CP activation by IgM directed against LOS
IgM is very efficient in fixing complement because a single IgM molecule can engage C1. However, IgM (Mr ~900 kD) is considerably larger than IgG (Mr ~150 kD) and the effects of capsule on binding and activity of IgM directed against noncapsular membrane antigens is not clear and was investigated next.
Anti-LOS IgM mAb directed against the unsialylated lacto-N-neotetraose structure that was elaborated by all strains used in this study was tested for its ability to bind to the Cap+ and Cap− mutants. At all dilutions tested, we noted a trend toward lower binding of mAb 3F11 to the Cap+ mutants (Fig. 6A). The differences were the least with the group A mutant pair (~1.2-fold), and the highest (~2-fold) with the group B strain pair.
Fig. 6.

Capsule limits binding of and C4b deposition by anti-LOS IgM mAb 3F11. A. Binding of mAb 3F11 to Cap+ and Cap− isogenic meningococcal mutants. Bacteria were incubated with varying dilutions (X-axis) of tissue culture supernatant containing mAb 3F11 and 3F11 binding was measured by flow cytometry (shown as median fluorescence on the log2 Y-axis; mean of 3 experiments). Binding to Cap+ is shown by the solid line and binding to Cap− by the broken line. B. All 5 capsular groups limit anti-LOS IgM-mediated C4 deposition. Cap+ (black bars) and Cap− (grey bars) isogenic mutants were incubated with low (1/32 and 1/64 for Cap+ and Cap− mutants, respectively) or high (1/8 and 1/16 for Cap+ and Cap− mutants, respectively) concentrations of 3F11 supernatants. Two-fold higher concentrations of 3F11 were used on Cap+ mutants as a conservative estimate to normalize for differences in 3F11 binding as shown in panel A. C4 deposition was measured by FACS following the addition of purified C1 complex and C4. Median fluorescence of C4b binding was recorded and each bar shows the mean (range) of two independent experiments. Supplemental Figure S3 shows representative histograms of C4b deposition.
To determine whether capsule interfered with C4b deposition by IgM mAb 3F11, we first ‘normalized’ the amount of 3F11 binding to Cap+ and Cap− mutants. Based on results above, a 2-fold higher concentration of 3F11 supernatant was used with each Cap+ mutant compared to the Cap− mutant and represented a conservative estimate to normalize binding. Two doses of mAb 3F11 were tested; the ‘high dose’ compared 1:8 and 1:16 dilutions, and the low dose compared 1:16 and 1:32 dilutions of 3F11 supernatant across the Cap+ and Cap− mutants, respectively. Again, in every instance, capsule expression was associated with lower C4b deposition (Fig. 6B).
Discussion
A novel finding in this study was that all five major meningococcal capsules interfered with engagement of pure C1q by IgG Fc, which led to decreased C4b deposition and inhibition of CP activation. We confirmed reduced C4b deposition using specific Abs directed against distinct antigens on five meningococcal groups. These observations may explain, at least in part, how capsular polysaccharides contribute to inhibition of the classical pathway. We emphasize that attenuation of the CP by capsule is stoichiometric and can be overcome as the titers of Ab and complement exceed a threshold, as occurs following immunization.
In a previous study we showed that the five meningococcal capsular groups interacted with the alternative pathway of complement in different ways (33). Groups B and C capsules inhibited the human alternative pathway, the group A capsule had no effect, while the groups W and Y capsules enhanced alternative pathway activation. Paradoxical enhancement of alternative pathway activation on the groups W and Y strains would theoretically place these strains at a disadvantage were complement activation to proceed in an unimpeded manner. However, the site of complement activation may be a critical factor in determining whether C5b-9 is generated and if the generated C5b-9 is bactericidal. Smooth LPS expressed by Klebsiella pneumoniae results in C3b deposited at locations too distant from the membrane to permit generated C5b-9 to elicit bactericidal activity (45). Elegant studies by Joiner and colleagues showed that similar amounts of C3 and C9 were deposited on Escherichia coli O111B4 incubated with complement alone compared to E. coli presensitized with immune Ab plus complement, but bacterial killing was observed only in the presence of Ab or F(ab')2 (46). These data suggest that the location of complement activation plays a critical role in bacterial killing. The AP alone does not kill wild-type N. meningitidis, including groups W and Y isolates where capsule activates the AP and serves as a site for C3 fragment deposition (33). We speculate that the distal locations of C3b on capsule may not result in C5b-9 formation that inserts into the bacterial membrane to effectively facilitate killing. Further, Rawal and Pangburn have shown that AP C5 convertase (C3bC3b,Bb) has a 6- to 9-fold lower catalytic rate than CP C5 convertase (C4bC3b,2a) (47), which may explain at least in part the ineffectiveness of the AP alone in mediating killing of Neisseriae. The ability of groups W and Y capsules to regulate the CP that is triggered by Ab bound to membrane antigens provides them a means to reduce CP-mediated complement activation that may otherwise have resulted in effective membrane attack complex insertion and bacterial death.
None of the capsule groups interfered with binding of specific IgG Ab, which is consistent with prior data, where similar amounts of anti-fHbp bound to strains that expressed different amounts of capsule (48). McNeil et al tested diverse wild-type encapsulated strains and we have confirmed and extended their observations using isogenic mutant strains that differed only in capsule expression. While capsule did not affect the binding of specific high-affinity IgG mAbs to their targets (in this instance, fHbp and PorA), it is not clear whether capsule would limit binding of lower affinity IgG Abs, for example, ‘natural’ Abs present in unimmunized individuals. By contrast, capsule expression decreased binding of an anti-LOS IgM mAb and the extent of inhibition varied across strains. Capsule likely poses an impediment to the bulky (~900 kD) pentameric IgM molecule from binding to its LOS epitope. A previous study showed that group B capsule expression reduced the binding of another large protein, C4b binding protein (C4BP; Mr ~470 kD) to bacteria by about 50% (49).
In a similar manner, capsule may also decrease engagement of the relatively large C1q molecule (~460 kD) by anti-fHbp Fc. C1q is formed by 18 polypeptide chains (6 A, B and C chains) that are organized into 6 subunits, each comprising an A, B and C chain. Engagement of C1q by IgG is a complex event. Recent data have shown that the IgG bound to surfaces form ordered Ab hexamers through noncovalent interactions of Fc, which results in binding of activated C1 (50). Further, the interaction between the globular head of C1q and IgG interaction is ionic in nature (51) and interactions between charged amino acids contribute to C1q-IgG binding (52–54). How capsules interfere with C1q engagement by Ab remains unclear. Based on published literature, possibilities include restriction of IgG hexamer formation and/or interference of ionic interactions between C1q and IgG by the negatively charged capsules.
Another interesting finding was that the major targets for C4b deposition when Ab was specifically directed against fHbp were LOS and Opa, similar to that we have described previously when normal human serum that contained ‘natural’ Ab was used to activate complement and deposit C4b (41). We have shown previously that groups W and Y capsules themselves serve as targets for C3 fragments (33). Akin to C3b, C4b (derived from the C4B isoform of C4) also forms covalent ester binds with electron-donating –OH groups (43, 44). Whether –OH groups of glycans in capsular polysaccharides also serve as acceptors for C4b when complement is activated in close proximity, for example by anti-capsular Ab, merits further study.
Uria et al showed that group C strains of N. meningitidis that expressed high levels of capsule because of the presence of an insertion sequence (IS1301) between the capsule biosynthesis and transport operons were resistant to killing by vaccine-elicited anti-capsular Ab (55). This finding appeared seemingly counterintuitive because of greater expression of the target antigen would have permitted higher levels of Ab binding and more killing. As stated above, group C capsule regulates the alternative pathway and up-regulation of capsule could dampen alternative pathway amplification initiated by the CP. Other possibilities such as activation of complement relatively distal to the membrane, which prevents insertion of ‘bactericidal’ membrane attack complex, may have also contributed to serum resistance. These capsule ‘hyperproducers’ were also resistant to killing by an anti-PorA mAb P1.5 (55); although the reason cited was inhibition of the alternative pathway, an additional but not mutually exclusive explanation is capsule-mediated CP inhibition as we have reported here. Whether complement activation by anti-capsular Ab and Ab directed against membrane antigens differ quantitatively or qualitatively is an important consideration from the perspective of vaccine development.
An additional important finding that emerged from this study was the wide variation in the amount of C4b deposited by anti-fHbp across the five unencapsulated meningococcal mutants, despite similar amounts of Ab binding. Levels of NspA expression (20) and different PorB alleles (19) are two variables that modulate alternative pathway activity, which necessitated the use of a complement source devoid of alternative pathway activity (achieved in this instance by blocking factor B function) to interpret results of serum bactericidal assays as they related to CP activation. With the exception of the group A mutants, capsule expression decreased CP-dependent killing by anti-fHbp. The reason for the greater (and unexpected) resistance to killing of the Cap− group A mutant relative to its Cap+ counterpart (Fig. 4) remains unclear, but it is worth noting that inhibition of C4b by group A capsule that was seen in the presence of anti-fHbp, pure C1 and pure C4 (Fig. 2A) was not seen in the presence of anti-fHbp and IgG/IgM depleted serum (Fig. 2C). Complement deposition and bactericidal activity in the context of serum requires participation of several proteins of the complement cascade and is modulated by complement inhibitors, which underscores the importance of corroborating data obtained using pure complement components with serum. Variation in the amount of C4b deposited and CP-mediated killing across strains despite similar amounts of anti-fHbp binding has important implications for the activity and evaluation of efficacy of fHbp-based vaccines. As an example, these data suggest that binding of anti-fHbp Ab alone may not predict the amount of CP activation and/or complement-dependent bacterial killing on different strains. The factor(s) that contribute to variations in C4b deposition among the strains remain to be elucidated. An example of surface variation that lead to differences in complement activation was described by Brown et al, where C2 cleavage varied markedly across human, guinea pig and sheep erythrocytes that were all sensitized with similar amounts of C1 and C4 (56).
Direct binding of C1 to several bacterial species can trigger CP activation independently of Ab (57–62). However, in this study only a low amount of C4b was deposited on meningococci that were incubated with C1 and C4 in the absence of Ab (Fig. 2B). These data are consistent with a requirement for Ab to kill meningococci (complement alone does not kill the bacteria), which forms the basis of evaluation of Ab responses to immunization by the serum bactericidal assay to predict protection against invasive disease (3).
In conclusion, we have shown that meningococcal capsular polysaccharides attenuate the CP of complement. These data highlight how this key virulence factor can modulate multiple facets of the complement cascade to subvert host immune responses.
Supplementary Material
Acknowledgements
We thank Dr. Ulrich Vogel, Universität Würzburg, Würzburg, Germany for providing the following strains: A2594 and its mynB mutant, H44/76, C2120, W171 and Y2225 and their lst and siaD lst mutants. We thank Dr. Dan M. Granoff (Childrens Hospital Oakland Research Institute) for providing mAb JAR 1 and Dr. Sunita Gulati (University of Massachusetts Medical School) for providing mAb 3F11 supernatants.
This work was supported by National Institutes of Health grants AI054544, AI084048 and AI32725.
Abbreviations used in this article
- LOS
lipooligosaccharide
- NHS
normal human serum
- HBSS
Hanks Balanced Salt Solution
- lst
lipooligosaccharide sialyltransferase
References
- 1.Flexner S. The Results of the Serum Treatment in Thirteen Hundred Cases of Epidemic Meningitis. J Exp Med. 1913;17:553–576. doi: 10.1084/jem.17.5.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flexner S, Jobling JW. An Analysis of Four Hundred Cases of Epidemic Meningitis Treated with the Anti-Meningitis Serum. J Exp Med. 1908;10:690–733. doi: 10.1084/jem.10.5.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldschneider I, Gotschlich EC, Artenstein MS. Human immunity to the meningococcus. I. The role of humoral antibodies. J Exp Med. 1969;129:1307–1326. doi: 10.1084/jem.129.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones GR, Williams JN, Christodoulides M, Jolley K, Heckels JE. Lack of immunity in university students before an outbreak of serogroup C meningococcal infection. J Infect Dis. 2000;181:1172–1175. doi: 10.1086/315352. [DOI] [PubMed] [Google Scholar]
- 5.Yazdankhah SP, Caugant DA. Neisseria meningitidis: an overview of the carriage state. J Med Microbiol. 2004;53:821–832. doi: 10.1099/jmm.0.45529-0. [DOI] [PubMed] [Google Scholar]
- 6.Goldschneider I, Gotschlich EC, Artenstein MS. Human immunity to the meningococcus. II. Development of natural immunity. J Exp Med. 1969;129:1327–1348. doi: 10.1084/jem.129.6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Findlow H, Vogel U, Mueller JE, Curry A, Njanpop-Lafourcade BM, Claus H, Gray SJ, Yaro S, Traore Y, Sangare L, Nicolas P, Gessner BD, Borrow R. Three cases of invasive meningococcal disease caused by a capsule null locus strain circulating among healthy carriers in Burkina Faso. J Infect Dis. 2007;195:1071–1077. doi: 10.1086/512084. [DOI] [PubMed] [Google Scholar]
- 8.Hoang LM, Thomas E, Tyler S, Pollard AJ, Stephens G, Gustafson L, McNabb A, Pocock I, Tsang R, Tan R. Rapid and fatal meningococcal disease due to a strain of Neisseria meningitidis containing the capsule null locus. Clin Infect Dis. 2005;40:e38–e42. doi: 10.1086/427875. [DOI] [PubMed] [Google Scholar]
- 9.Vogel U, Claus H, von Muller L, Bunjes D, Elias J, Frosch M. Bacteremia in an immunocompromised patient caused by a commensal Neisseria meningitidis strain harboring the capsule null locus (cnl) J Clin Microbiol. 2004;42:2898–2901. doi: 10.1128/JCM.42.7.2898-2901.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caugant DA, Maiden MC. Meningococcal carriage and disease--population biology and evolution. Vaccine. 2009;27(Suppl 2):B64–B70. doi: 10.1016/j.vaccine.2009.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claus H, Maiden MC, Wilson DJ, McCarthy ND, Jolley KA, Urwin R, Hessler F, Frosch M, Vogel U. Genetic analysis of meningococci carried by children and young adults. J Infect Dis. 2005;191:1263–1271. doi: 10.1086/428590. [DOI] [PubMed] [Google Scholar]
- 12.Masignani V, Comanducci M, Giuliani MM, Bambini S, Adu-Bobie J, Arico B, Brunelli B, Pieri A, Santini L, Savino S, Serruto D, Litt D, Kroll S, Welsch JA, Granoff DM, Rappuoli R, Pizza M. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med. 2003;197:789–799. doi: 10.1084/jem.20021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fletcher LD, Bernfield L, Barniak V, Farley JE, Howell A, Knauf M, Ooi P, Smith RP, Weise P, Wetherell M, Xie X, Zagursky R, Zhang Y, Zlotnick GW. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect Immun. 2004;72:2088–2100. doi: 10.1128/IAI.72.4.2088-2100.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Madico G, Welsch JA, Lewis LA, McNaughton A, Perlman DH, Costello CE, Ngampasutadol J, Vogel U, Granoff DM, Ram S. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J Immunol. 2006;177:501–510. doi: 10.4049/jimmunol.177.1.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider MC, Prosser BE, Caesar JJ, Kugelberg E, Li S, Zhang Q, Quoraishi S, Lovett JE, Deane JE, Sim RB, Roversi P, Johnson S, Tang CM, Lea SM. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature. 2009 doi: 10.1038/nature07769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seib KL, Serruto D, Oriente F, Delany I, Adu-Bobie J, Veggi D, Arico B, Rappuoli R, Pizza M. Factor H-binding protein is important for meningococcal survival in human whole blood and serum and in the presence of the antimicrobial peptide LL-37. Infect Immun. 2009;77:292–299. doi: 10.1128/IAI.01071-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorringe AR, Pajon R. Bexsero: a multicomponent vaccine for prevention of meningococcal disease. Hum Vaccin Immunother. 2012;8:174–183. doi: 10.4161/hv.18500. [DOI] [PubMed] [Google Scholar]
- 18.Lewis LA, Ngampasutadol J, Wallace R, Reid JE, Vogel U, Ram S. The Meningococcal Vaccine Candidate Neisserial Surface Protein A (NspA) Binds to Factor H and Enhances Meningococcal Resistance to Complement. PLoS Pathog. 2010;6:e1001027. doi: 10.1371/journal.ppat.1001027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis LA, Vu DM, Vasudhev S, Shaughnessy J, Granoff DM, Ram S. Factor H-Dependent Alternative Pathway Inhibition Mediated by Porin B Contributes to Virulence of Neisseria meningitidis. MBio. 2013;4:e00339–e00313. doi: 10.1128/mBio.00339-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis LA, Carter M, Ram S. The relative roles of factor H binding protein, neisserial surface protein A, and lipooligosaccharide sialylation in regulation of the alternative pathway of complement on meningococci. J Immunol. 2012;188:5063–5072. doi: 10.4049/jimmunol.1103748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammerschmidt S, Birkholz C, Zahringer U, Robertson BD, van Putten J, Ebeling O, Frosch M. Contribution of genes from the capsule gene complex (cps) to lipooligosaccharide biosynthesis and serum resistance in Neisseria meningitidis. Mol Microbiol. 1994;11:885–896. doi: 10.1111/j.1365-2958.1994.tb00367.x. [DOI] [PubMed] [Google Scholar]
- 22.Kahler CM, Martin LE, Shih GC, Rahman MM, Carlson RW, Stephens DS. The (alpha2-->8)-linked polysialic acid capsule and lipooligosaccharide structure both contribute to the ability of serogroup B Neisseria meningitidis to resist the bactericidal activity of normal human serum. Infect Immun. 1998;66:5939–5947. doi: 10.1128/iai.66.12.5939-5947.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackinnon FG, Borrow R, Gorringe AR, Fox AJ, Jones DM, Robinson A. Demonstration of lipooligosaccharide immunotype and capsule as virulence factors for Neisseria meningitidis using an infant mouse intranasal infection model. Microb Pathog. 1993;15:359–366. doi: 10.1006/mpat.1993.1085. [DOI] [PubMed] [Google Scholar]
- 24.Vogel U, Hammerschmidt S, Frosch M. Sialic acids of both the capsule and the sialylated lipooligosaccharide of Neisseria meningitis serogroup B are prerequisites for virulence of meningococci in the infant rat. Med Microbiol Immunol (Berl) 1996;185:81–87. doi: 10.1007/s004300050018. [DOI] [PubMed] [Google Scholar]
- 25.Fearon DT. Regulation by membrane sialic acid of beta1H-dependent decay- dissociation of amplification C3 convertase of the alternative complement pathway. Proc Natl Acad Sci U S A. 1978;75:1971–1975. doi: 10.1073/pnas.75.4.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc. Natl. Acad. Sci. U S A. 1990;87:3982–3986. doi: 10.1073/pnas.87.10.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Severi E, Hood DW, Thomas GH. Sialic acid utilization by bacterial pathogens. Microbiology. 2007;153:2817–2822. doi: 10.1099/mic.0.2007/009480-0. [DOI] [PubMed] [Google Scholar]
- 28.Varki A, Gagneux P. Multifarious roles of sialic acids in immunity. Ann N Y Acad Sci. 2012;1253:16–36. doi: 10.1111/j.1749-6632.2012.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarvis GA. Analysis of C3 deposition and degradation on Neisseria meningitidis and Neisseria gonorrhoeae. Infect Immun. 1994;62:1755–1760. doi: 10.1128/iai.62.5.1755-1760.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarvis GA, Vedros NA. Sialic acid of group B Neisseria meningitidis regulates alternative complement pathway activation. Infect Immun. 1987;55:174–180. doi: 10.1128/iai.55.1.174-180.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogel U, Claus H, Heinze G, Frosch M. Functional characterization of an isogenic meningococcal alpha-2,3- sialyltransferase mutant: the role of lipooligosaccharide sialylation for serum resistance in serogroup B meningococci. Med Microbiol Immunol (Berl) 1997;186:159–166. doi: 10.1007/s004300050059. [DOI] [PubMed] [Google Scholar]
- 32.Vogel U, Claus H, Heinze G, Frosch M. Role of lipopolysaccharide sialylation in serum resistance of serogroup B and C meningococcal disease isolates. Infect Immun. 1999;67:954–957. doi: 10.1128/iai.67.2.954-957.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ram S, Lewis LA, Agarwal S. Meningococcal group W-135 and Y capsular polysaccharides paradoxically enhance activation of the alternative pathway of complement. J Biol Chem. 2011;286:8297–8307. doi: 10.1074/jbc.M110.184838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harrison OB, Claus H, Jiang Y, Bennett JS, Bratcher HB, Jolley KA, Corton C, Care R, Poolman JT, Zollinger WD, Frasch CE, Stephens DS, Feavers I, Frosch M, Parkhill J, Vogel U, Quail MA, Bentley SD, Maiden MC. Description and nomenclature of Neisseria meningitidis capsule locus. Emerg Infect Dis. 2013;19:566–573. doi: 10.3201/eid1904.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welsch JA, Rossi R, Comanducci M, Granoff DM. Protective activity of monoclonal antibodies to genome-derived neisserial antigen 1870, a Neisseria meningitidis candidate vaccine. J Immunol. 2004;172:5606–5615. doi: 10.4049/jimmunol.172.9.5606. [DOI] [PubMed] [Google Scholar]
- 36.Ray TD, Lewis LA, Gulati S, Rice PA, Ram S. Novel blocking human IgG directed against the pentapeptide repeat motifs of Neisseria meningitidis Lip/H.8 and Laz lipoproteins. J Immunol. 2011;186:4881–4894. doi: 10.4049/jimmunol.1003623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Apicella MA, Bennett KM, Hermerath CA, Roberts DE. Monoclonal antibody analysis of lipopolysaccharide from Neisseria gonorrhoeae and Neisseria meningitidis. Infect Immun. 1981;34:751–756. doi: 10.1128/iai.34.3.751-756.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apicella MA, Shero M, Jarvis GA, Griffiss JM, Mandrell RE, Schneider H. Phenotypic variation in epitope expression of the Neisseria gonorrhoeae lipooligosaccharide. Infect Immun. 1987;55:1755–1761. doi: 10.1128/iai.55.8.1755-1761.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giuntini S, Reason DC, Granoff DM. The Combined Roles of Human IgG Subclass, Alternative Complement Pathway Activation, and Epitope Density on Bactericidal Activity of Antibodies to Meningococcal Factor H Binding Protein. Infect Immun. 2011 doi: 10.1128/IAI.05956-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gulati S, Agarwal S, Vasudhev S, Rice PA, Ram S. Properdin is critical for antibody-dependent bactericidal activity against Neisseria gonorrhoeae that recruit C4b-binding protein. J Immunol. 2012;188:3416–3425. doi: 10.4049/jimmunol.1102746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis LA, Ram S, Prasad A, Gulati S, Getzlaff S, Blom AM, Vogel U, Rice PA. Defining targets for complement components C4b and C3b on the pathogenic neisseriae. Infect Immun. 2008;76:339–350. doi: 10.1128/IAI.00613-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ram S, Cox AD, Wright JC, Vogel U, Getzlaff S, Boden R, Li J, Plested JS, Meri S, Gulati S, Stein DC, Richards JC, Moxon ER, Rice PA. Neisserial lipooligosaccharide is a target for complement component C4b: Inner core phosphoethanolamine residues define C4b linkage specificity. J Biol Chem. 2003;278:50853–50862. doi: 10.1074/jbc.M308364200. [DOI] [PubMed] [Google Scholar]
- 43.Carroll MC, Fathallah DM, Bergamaschini L, Alicot EM, Isenman DE. Substitution of a single amino acid (aspartic acid for histidine) converts the functional activity of human complement C4B to C4A. Proc Natl Acad Sci U S A. 1990;87:6868–6872. doi: 10.1073/pnas.87.17.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Isenman DE, Young JR. Covalent binding properties of the C4A and C4B isotypes of the fourth component of human complement on several C1-bearing cell surfaces. J Immunol. 1986;136:2542–2550. [PubMed] [Google Scholar]
- 45.Merino S, Camprubi S, Alberti S, Benedi VJ, Tomas JM. Mechanisms of Klebsiella pneumoniae resistance to complement-mediated killing. Infect Immun. 1992;60:2529–2535. doi: 10.1128/iai.60.6.2529-2535.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joiner KA, Goldman RC, Hammer CH, Leive L, Frank MM. Studies on the mechanism of bacterial resistance to complement-mediated killing. V. IgG and F(ab')2 mediate killing of E. coli 0111B4 by the alternative complement pathway without increasing C5b-9 deposition. J Immunol. 1983;131:2563–2569. [PubMed] [Google Scholar]
- 47.Rawal N, Pangburn MK. Formation of high affinity C5 convertase of the classical pathway of complement. J Biol Chem. 2003;278:38476–38483. doi: 10.1074/jbc.M307017200. [DOI] [PubMed] [Google Scholar]
- 48.McNeil LK, Murphy E, Zhao XJ, Guttmann S, Harris S, Scott A, Tan C, Mack M, Dasilva I, Alexander K, Jiang HQ, Zhu D, Mininni T, Zlotnick GW, Hoiseth SK, Jones TR, Pride M, Jansen KU, Anderson A. Detection of LP2086 on the cell surface of Neisseria meningitidis and its accessibility in the presence of serogroup B capsular polysaccharide. Vaccine. 2009 doi: 10.1016/j.vaccine.2009.01.075. [DOI] [PubMed] [Google Scholar]
- 49.Jarva H, Ram S, Vogel U, Blom AM, Meri S. Binding of the complement inhibitor C4bp to serogroup B Neisseria meningitidis. J Immunol. 2005;174:6299–6307. doi: 10.4049/jimmunol.174.10.6299. [DOI] [PubMed] [Google Scholar]
- 50.Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJ, van de Winkel JG, Wilson IA, Koster AJ, Taylor RP, Saphire EO, Burton DR, Schuurman J, Gros P, Parren PW. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–1263. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burton DR, Boyd J, Brampton AD, Easterbrook-Smith SB, Emanuel EJ, Novotny J, Rademacher TW, van Schravendijk MR, Sternberg MJ, Dwek RA. The Clq receptor site on immunoglobulin G. Nature. 1980;288:338–344. doi: 10.1038/288338a0. [DOI] [PubMed] [Google Scholar]
- 52.Duncan AR, Winter G. The binding site for C1q on IgG. Nature. 1988;332:738–740. doi: 10.1038/332738a0. [DOI] [PubMed] [Google Scholar]
- 53.Kojouharova MS, Gadjeva MG, Tsacheva IG, Zlatarova A, Roumenina LT, Tchorbadjieva MI, Atanasov BP, Waters P, Urban BC, Sim RB, Reid KB, Kishore U. Mutational analyses of the recombinant globular regions of human C1q A, B, and C chains suggest an essential role for arginine and histidine residues in the C1q-IgG interaction. J Immunol. 2004;172:4351–4358. doi: 10.4049/jimmunol.172.7.4351. [DOI] [PubMed] [Google Scholar]
- 54.Roumenina LT, Ruseva MM, Zlatarova A, Ghai R, Kolev M, Olova N, Gadjeva M, Agrawal A, Bottazzi B, Mantovani A, Reid KB, Kishore U, Kojouharova MS. Interaction of C1q with IgG1, C-reactive protein and pentraxin 3: mutational studies using recombinant globular head modules of human C1q A, B, and C chains. Biochemistry. 2006;45:4093–4104. doi: 10.1021/bi052646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uria MJ, Zhang Q, Li Y, Chan A, Exley RM, Gollan B, Chan H, Feavers I, Yarwood A, Abad R, Borrow R, Fleck RA, Mulloy B, Vazquez JA, Tang CM. A generic mechanism in Neisseria meningitidis for enhanced resistance against bactericidal antibodies. J Exp Med. 2008;205:1423–1434. doi: 10.1084/jem.20072577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown EJ, Ramsey J, Hammer CH, Frank MM. Surface modulation of classical pathway activation: C2 and C3 convertase formation and regulation on sheep, guinea pig, and human erythrocytes. J Immunol. 1983;131:403–408. [PubMed] [Google Scholar]
- 57.Alberti S, Marques G, Camprubi S, Merino S, Tomas JM, Vivanco F, Benedi VJ. C1q binding and activation of the complement classical pathway by Klebsiella pneumoniae outer membrane proteins. Infect Immun. 1993;61:852–860. doi: 10.1128/iai.61.3.852-860.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Betz SJ, Isliker H. Antibody-independent interactions between Escherichia coli J5 and human complement components. J Immunol. 1981;127:1748–1754. [PubMed] [Google Scholar]
- 59.Bredt W, Wellek B, Brunner H, Loos M. Interactions between mycoplasma pneumoniae and the first components of complement. Infect Immun. 1977;15:7–12. doi: 10.1128/iai.15.1.7-12.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clas F, Loos M. Antibody-independent binding of the first component of complement (C1) and its subcomponent C1q to the S and R forms of Salmonella minnesota. Infect Immun. 1981;31:1138–1144. doi: 10.1128/iai.31.3.1138-1144.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mintz CS, Arnold PI, Johnson W, Schultz DR. Antibody-independent binding of complement component C1q by Legionella pneumophila. Infect Immun. 1995;63:4939–4943. doi: 10.1128/iai.63.12.4939-4943.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tenner AJ, Ziccardi RJ, Cooper NR. Antibody-independent C1 activation by E. coli. J Immunol. 1984;133:886–891. [PubMed] [Google Scholar]
- 63.Schoen C, Weber-Lehmann J, Blom J, Joseph B, Goesmann A, Strittmatter A, Frosch M. Whole-genome sequence of the transformable Neisseria meningitidis serogroup A strain WUE2594. J Bacteriol. 2011;193:2064–2065. doi: 10.1128/JB.00084-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frasch CE, Zollinger WD, Poolman JT. Serotype antigens of Neisseria meningitidis and a proposed scheme for designation of serotypes. Rev Infect Dis. 1985;7:504–510. doi: 10.1093/clinids/7.4.504. [DOI] [PubMed] [Google Scholar]
- 65.Vogel U, Morelli G, Zurth K, Claus H, Kriener E, Achtman M, Frosch M. Necessity of molecular techniques to distinguish between Neisseria meningitidis strains isolated from patients with meningococcal disease and from their healthy contacts. J Clin Microbiol. 1998;36:2465–2470. doi: 10.1128/jcm.36.9.2465-2470.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Claus H, Borrow R, Achtman M, Morelli G, Kantelberg C, Longworth E, Frosch M, Vogel U. Genetics of capsule O-acetylation in serogroup C, W-135 and Y meningococci. Mol Microbiol. 2004;51:227–239. doi: 10.1046/j.1365-2958.2003.03819.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.