

Abstract
The onset of autoantibodies in systemic autoimmunity can be the result of a breakdown in tolerance at multiple checkpoints. Genetic, hormonal, and immunological factors can combine with environmental influences to accelerate the onset of disease and aggravate disease outcome. Here, we describe a novel mechanism relating to the regulatory role of Neutrophil Gelatinase Associated Lipocalin (NGAL) in modulating the levels of autoantibodies in pristane induced lupus. Following a single injection of pristane intraperitoneally, NGAL expression was induced in both the serum and spleen. Furthermore, NGAL deficient mice were more susceptible to the induction of pristane stimulated autoimmunity, and displayed higher numbers of autoantibody secreting cells and increased expression of activation induced cytidine deaminase (AID) and other inflammatory mediators in the spleen. In contrast, kidney damage was milder in NGAL deficient mice, indicating that NGAL was detrimental in autoantibody mediated kidney disease. These studies indicate that NGAL plays differential roles in different tissues in the context of lupus, and suggest a previously unrecognized role for NGAL in adaptive immunity.
Keywords: NGAL, lipocalin-2, autoantibodies, SLE, pristane
1. Introduction
Neutrophil gelatinase associated lipocalin (NGAL)/lipocalin-2 is a 25kD member of the lipocalin superfamily. NGAL is expressed by different tissues (including spleen, lung, kidney, liver, brain, heart, and testis) under healthy conditions in humans and mice [1,2]. During ischemic or inflammatory conditions, NGAL is upregulated in various cell types including neutrophils, hepatocytes, alveolar epithelial cells, and renal resident cells [3-5]. Recent studies have suggested that serum NGAL levels are useful as a biomarker in a broad variety of pathological conditions, including breast cancer, pancreatic cancer, acute and chronic kidney diseases, and systemic lupus erythematosus (SLE) [6,7].
Numerous studies have established that NGAL is involved in innate immune responses during infection due to its siderophore scavenging ability, with NGAL deficient mice exhibiting increased sensitivity to bacterial infection [3,8,9]. NGAL expression is also associated with cell metastasis and breast cancer progression via induction of angiogenesis [10,11]. Recently, in the context of autoimmune disease, NGAL was determined to be instrumental in the pathogenesis of experimental autoimmune encephalitis (EAE) [12]. Moreover, apoptotic effects of NGAL have been reported in various cell types [13,14] including human hematopoietic cells such as the progenitors of red blood cells (RBC) and macrophages [15].
Previously, we reported that NGAL is upregulated in glomerular mesangial cells and in murine kidneys following exposure to pathogenic anti-DNA antibodies in vitro and in vivo, respectively [4]. The MRL-lpr/lpr mouse strain carries a mutant Fas gene, leading to defective apoptosis, persistence of autoreactive lymphocytes, and eventually spontaneous development of circulating autoantibodies and organ involvement (including nephritis) very similar to human disease. MRL-lpr/lpr lupus mice exhibited significant upregulation of NGAL in kidneys with increasing disease severity [4]. In human disease, we found that NGAL was upregulated in the serum and urine of patients with lupus nephritis as compared to normal controls [16]. Furthermore, we determined that NGAL is detrimental in antibody-mediated nephritis, as NGAL−/− mice were protected against renal injury induced by challenge with nephrotoxic serum [17].
In the current study, our goal was to establish the role of NGAL in the adaptive immune response and in triggering humoral autoimmunity in a widely used murine model, viz. pristane induced lupus [18-22]. Pristane (a naturally occurring hydrocarbon oil) is an isoprenoid alkane, which induces a lupus like syndrome in several non-autoimmune prone mouse strains [23]. Injection of pristane intraperitoneally stimulates the formation of lupus associated autoantibodies to multiple nuclear antigens as well as polyclonal hypergammaglobulinemia [24], and promotes the development of lipogranulomas, or ectopic lymphoid tissues, which develop distinct T cell/dendritic cell and B cell zones [25].
We found that NGAL regulates the onset of autoantibodies to nuclear antigens in pristane induced lupus. Post pristane challenge, NGAL deficient mice had increased levels of serum autoantibodies as well as elevated numbers of autoantibody secreting cells in the spleen. Furthermore, expression of inflammatory mediators in the spleen and polyclonal hypergammaglobulinemia were NGAL dependent. In contrast, renal injury was significantly attenuated in NGAL−/− as compared with NGAL sufficient mice. Taken together, our results demonstrate that NGAL regulates the onset of autoantibodies and exerts differential responses in various tissues in the context of murine lupus.
2. Materials and methods
2.1. Mice
Six to eight week old C57BL/6 (B6) female mice were purchased from Jackson Laboratories (Bar Harbor, Maine). Mice were housed 3-5 per cage in the animal facility of the Albert Einstein College of Medicine (Bronx, NY), and acclimatized in the facility for 2 weeks prior to experiments. B6.NGAL−/− mice were generously provided by Drs. Thorsten Berger and Tak M. Mak, The Campbell Family Institute for Breast Cancer Research and the Ontario Cancer Institute, University Health Network (Toronto, CA), and bred at the Einstein Institute for Animal Studies. Generation of NGAL−/− mice on the B6 background has been described; these mice appear normal and display no gross phenotype, as previously reported [8,17,26]. We confirmed that the D1Mit105 lupus susceptibility locus on chromosome 1 in the B6.NGAL−/− strain was of B6 (non-risk), not 129/Sv origin (data not shown). The housing conditions were controlled, with a temperature of 21-23°C and a 12:12 hours light:dark cycle. All animal study protocols were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine, Bronx, New York.
2.2. Treatment with pristane/TMPD (2,6,10,14-Tetramethylpentadecane)
NGAL sufficient (wild type) B6 and B6.NGAL−/− female mice were treated with single injection of pristane (Sigma-Aldrich, St. Louis, MO) 0.5 ml intraperitoneally (i.p.) per mouse at the age of 8-10 weeks. Control groups of mice of the above strains were injected with the same volume of PBS. Serum was collected before and 4 months after the pristane/PBS injection. The time point of 4 months was chosen based upon the kinetics of autoantibody development following exposure to pristane [24,25].
2.3. ELISA for serum antibodies
Serum antibodies against dsDNA, ssDNA, histone, and chromatin were measured as described previously [4,27-30]. In brief, dsDNA was generated from salmon sperm DNA (Invitrogen, Grand Island, NY) after S1 nuclease (Promega, Madison, WI) digestion. ssDNA was generated by heating the DNA at 95°C and immediately cooling on ice. Ninety-six well microplates (NUNC Maxisorp, Thermo scientific, Pittsburgh, PA) were coated with a solution of 1 μg/ml of poly-L-lysine for 1 hour at room temperature (RT), followed by washing and coating with dsDNA or ssDNA at 1 μg/ml overnight at 4°C. The plates were washed and blocked with blocking buffer (2% BSA in PBS) followed by washing and incubation with diluted (1:200 in blocking buffer) serum samples and standards for 2 hours at 37°C. Alkaline-phosphataseconjugated anti-mouse IgG, IgG2a, IgG2b, IgG3, and IgG1 antibodies (Southern Biotech, Birmingham, Alabama) were used for the detection of bound antibodies in serum, followed by addition of phosphatase substrate (Sigma-Aldrich) for the color development which was read at 405 nm. The anti-IgG2a antibody reagent was used to provide a relative measure of IgG2c levels in the different experimental groups. For the anti-histone and anti-chromatin IgG ELISA, the plates were coated with 10 μg/ml of histone and chromatin, respectively, overnight at 4°C, followed by the same protocol as above. Serum from 20 week old female MRL/lpr mice was used as a positive control in each assay.
For analysis of total and subclass-specific IgG, 96 well plates were coated overnight with goat anti-mouse IgG, IgG1, IgG2a, IgG2b, and IgG3 antibodies (all from Southern Biotech), and the ELISA continued using the same protocol described above. Monoclonal antibody standards run on each plate were used to calculate antibody concentrations.
2.4. Detection of antinuclear antibodies (ANA) with Hep-2 slides
The titer and pattern of ANA was determined by incubating serum samples on Hep-2 slides (Bio-RAD, Hercules, CA). Diluted serum samples (1:50) were incubated on the slides for 1 hour, followed by washing and incubation with FITC-labeled anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) for 20-30 minutes. Serum from 6-10 week old B6 mice was used as a negative control. The staining pattern was captured using fluorescence microscopy at 40x magnification. The fluorescent patterns of ANA were classified as homogenous nuclear and nucleolar, as described previously [18,31]. Four random fields were analyzed for scoring of each sample.
2.5. Immunoprecipitation
Serum autoantibodies to cellular proteins were analyzed by immunoprecipitating [35S] labeled proteins from K562 cell extracts using 5 μl serum per sample followed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and autoradiography, as described previously [24].
2.6. Splenocyte stimulation and cytokine measurement
Splenocytes were prepared from B6 and B6.NGAL−/− mice of 8-10 weeks of age, as described [32]. In brief, mice were anesthetized, sacrificed and spleens were isolated. The spleens were chopped into fine pieces in ice cold RPMI (Invitrogen) medium containing 10% heat inactivated fetal bovine serum (FBS) (Invitrogen) and 1% penicillin-streptomycin (PS), and then passed through 70 μm filters by gentle mincing. The cell suspension was then washed twice with the same media, and treated with a RBC lysis solution containing 0.14 M NH4CL, KHCO3 and EDTA, pH 7 for 5-10 minutes at RT. The cells were washed 3 times with RPMI medium and then resuspended in the same medium. Sterile 96 well or 24 well microplates (Costar, St. Louis, MO) were used for culturing the cells at density of 2 × 105 or 1 × 106 per well respectively. Plates were maintained in an incubator at 37°C/5% CO2 for stimulation experiments and ELISPOT assays. For the stimulation experiments, after 24 hours of incubation the cells were treated with different pristane concentrations for 24 hours. ELISA assays were performed to quantify NGAL concentrations in cell culture supernatants.
2.7. Isolation and stimulation of peritoneal macrophages
Ten week old B6 wild type female mice were injected with sterile 3.8% thioglycollate (protected from light and aged for 1 month) at a volume of 0.5 ml i.p. Three days later, the mice were sacrificed and the peritoneal cavity gently flushed with 3 ml of PBS containing 5 mM EDTA. The lavage fluid was collected and centrifuged at 200 g for 5 minutes. The resulting cell pellet was washed twice with PBS containing EDTA, and filtered through a 40 μm filter to prepare a single cell suspension. The cells were suspended in RBC lysis buffer and kept at RT for 5 min with intermittent mixing of the suspension. The cell suspension was then centrifuged and washed twice as above. A total of 500,000 cells were plated in 24 well plates and cultured in RPMI+10% FCS+1% PS+GM-CSF for 24 hours. The cells were then stimulated with pristane at different concentrations (determined empirically based upon the amount of pristane injected into the peritoneum in this model), and the supernatants collected after 24 hours. Desired concentrations of pristane were prepared by mixing with the media by sufficient vortexing, so that the oil globules were divided into small size particles.
2.8. Serum IL-12 and NGAL concentrations
Serum IL-12p40 was analyzed using an ELISA kit (BD Biosciences, San Jose, CA), following the manufacturer’s instructions. Serum NGAL was analyzed using the mouse NGAL ELISA duoset (R & D systems, Minneapolis, MN), using the manufacturer’s protocol with minor modifications. In brief, 96 well microplates were coated overnight with unconjugated anti-mouse NGAL antibody at a concentration of 4 μg/ml in PBS. The plate was washed and blocked with reagent diluent for 1 hour, followed by 3 washes. Samples and standards were then incubated for 2 hours followed by 3 washes, and the biotinylated detection antibody added and incubated for 2 hours. The plate was washed again, followed by streptavidin-AP for 20 minutes and phosphatase substrate for color development.
2.9. ELISPOT
Multiscreen 96 well filter plates (Millipore, Billerica, MA) were coated with poly-L-lysine followed by dsDNA, histone, or Sm/RNP (ImmunoVision, Springdale, AR) overnight at 4°C, washed once, and blocked with 200 μl of RPMI complete medium with 10% FCS incubated at RT for 2 hours. The blocking solution was discarded, and spleen cells were added at a density of 2×105 per well and incubated at 37°C/5% CO2 for 72 hours. The cells were washed twice with 200 μl per well of deionized water and three times with the wash buffer (PBS containing 0.05% Tween-20). An alkaline phosphatase conjugated anti-mouse IgG secondary antibody was added and incubated for 2 hours at RT. The plates were washed again 4 times with wash buffer and two times with PBS. Substrate solution was added (BCIP-p-toluidine 1 μg/ml in AMP buffer) and incubated in the dark at RT for 5-10 minutes while monitoring spot development. The reaction was stopped by washing the wells with deionized water. The plates were then air dried at RT overnight in the dark, and the spots were quantified by an ELISPOT plate reader (Autoimmun Diagnostika, Strassberg, Germany).
2.10. Real time PCR
RNA was extracted from spleen and kidney tissues using the RNeasy minikit (Qiagen, Valencia, CA). For cDNA synthesis, reverse transcription was performed from 2 μg of total RNA using the SuperScript II system from Invitrogen. Real time PCR was performed in duplicate or triplicate using the SYBR green PCR mastermix (Power SyBr, Applied Biosystems, Warrington, UK) and the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems), using the ΔCt method as described previously [17]. GAPDH was used as a housekeeping gene for normalization of the CT values for the gene of interest.
2.11. Histological analysis of kidney
Renal histology of kidneys from PBS and pristane challenged B6 and B6.NGAL−/− mice was evaluated by an experienced renal pathologist who was blinded to the mouse strains and treatment groups. Sections were stained with hematoxylin & eosin (H&E) and periodic acid Schiff (PAS), and glomeruli were evaluated for the presence of mesangial proliferation, endocapillary proliferation, immune complex deposition and tubulointerstitial lesions, as described previously [33,34]. Between 50 and 100 randomly chosen glomeruli were scored for each mouse. A score from 0-4 was given for each parameter above, based on the percentage of glomeruli displaying that lesion (0=not observed, 1=1-25%, 2=26-50%, 3=51-75%, 4>75%). Tubulointerstitial lesions including interstitial inflammation, tubulointerstitial fibrosis and acute tubular injury were also assessed and scored based on the percentage of cortical tissue displaying the pathologic lesion, using the 0-4 scale detailed above.
2.12 Immunohistochemical analysis of kidney
For analysis of neutrophils by immunohistochemistry, the sections were deparaffinized as described above and antigen retrieval was performed using citrate buffer (pH 6) at 95°C for 20 minutes. The sections were incubated with peroxide block (DAKO, Carpinteria, CA), followed by the blocking procedure described above. The sections were then incubated with rat anti-mouse Ly6B antibody (AbD Serotec, Raleigh, NC) for 2 hours at RT, washed, and incubated with biotin conjugated goat anti-rat IgG for 1 hour at RT. Sections were washed, incubated with streptavidin-HRP (Thermo Scientific) for 20 min, and washed again. The DAB substrate (DAKO) was used for color development for 1-2 min. Slides were rinsed with water for 5 min, followed by staining with Mayer’s Hematoxylin (Sigma-Aldrich) for another 2 minutes. Slides were rinsed in water, air dried, and mounted with Permount (Fisher, Pittsburgh, PA). Images were captured using light microscopy (Zeiss Axiscope) at 40x magnification.
2.13. In vitro stimulation of mesangial cells and tubular cells
Immortalized mesangial and tubular cells originally derived from MRL-lpr/lpr mice were used for the stimulation experiments. In brief, cells in 2 ml of DMEM media containing 10% FCS and 1% PS were plated at a density of 105 per well in 6 well plates. Upon reaching 70% confluence, cells were switched to serum free medium and stimulated with LPS (Invivogen, San Diego, CA) at 5 μg/ml or a combination of TNFα and IFNγ each at 5 ng/ml (eBioscience, San Diego, CA), for 6, 12, and 24 hours. RNA was isolated using the Qiagen RNeasy minikit, and real time PCR for NGAL was performed as above. Fold changes in mRNA levels were expressed as a ratio to basal mRNA levels (just prior to stimulation). In addition, supernatants were collected for analysis of secreted cytokines and the cells harvested for analysis of apoptosis. Apoptosis was analyzed by flow cytometry using Annexin V and 7-AAD (BD Biosciences).
In other experiments, mesangial cells were stimulated for 24 hours with recombinant NGAL in the presence and absence of anti-NGAL antibody or control mouse IgG (Sigma), and the amount of MCP-1 in the supernatant analyzed by ELISA (BD Biosciences).
The anti-NGAL antibody producing clone 6B9C3F12 (IgG1) was generated by immunization of a B6 mouse with murine NGAL, and isolation of NGAL-specific hybridomas (as initially determined by ELISA) using standard hybridoma technology. The 6B9C3F12 hybridoma was chosen based upon its affinity for NGAL, and its effectiveness in blocking NGAL effects in in-vitro studies. Furthermore, the specificity of this particular clone was confirmed by Western blot, where 6B9C3F12 displayed a similar binding pattern to purified NGAL as the polyclonal goat anti-mouse NGAL antibody from R&D systems (Minneapolis, MN). Clonality of 6B9C3F12 was verified, and the cell line was grown in chemically defined, protein free, serum free media containing 1% PS, in a two compartment bioreactor (Celline, Integra, Hudson, NH). The antibody was purified from the supernatant using Protein-G columns (Hi-Trap, GE healthcare), followed by extensive dialysis and filter sterilization.
2.14. Statistical analysis
Differences between groups were compared by two tailed unpaired t-test unless otherwise indicated. The results for each experiment were assessed for normality, and non-parametric and non-normally distributed data analyzed using Mann Whitney’s test. Graphing and statistical analyses were performed using GraphPad Prism (version 4), with p values ≤0.05 considered significant.
3. Results
3.1. Serum total IgG, IgG isotypes, and IgM are differentially regulated by NGAL
Pristane induces polyclonal hypergammaglobulinemia when given as a single intraperitoneal injection [23,35]. Hence, we analyzed the levels of total IgG and specific isotypes in serum of treated mice. Pristane challenged B6 mice had significantly higher levels of total IgG, IgG1 and IgM as compared to PBS treated B6 mice (Fig. 1A, D, F). Surprisingly, the pristane challenged B6.NGAL−/− mice had significantly higher levels of total IgG, IgG2b, and IgG2c as compared to the B6 mice (Fig. 1A, B, C). Furthermore, there was a trend towards increased IgG1 (p=0.09) in the pristane challenged B6.NGAL−/− group as compared to the B6 mice (Fig. 1D). Total IgM was upregulated in both the pristane challenged groups, although the increase in the pristane challenged B6 group was significantly higher than in the B6.NGAL−/− group (Fig. 1F). There was no significant difference in total IgG, IgG isotypes, and IgM titers between the PBS treated B6 and B6.NGAL−/− groups (Fig. 1).
Fig. 1.

NGAL regulates hypergammaglobulinemia. A-F. Serum levels of IgG, IgG2b, IgG2c, IgG1, IgG3 and IgM from B6 and B6.NGAL−/− mice, 4 months after pristane or PBS challenge, were analyzed by ELISA (n=4-5 per PBS group, n=10-11 per pristane group). The mice which received pristane will be referred to as B6 pristane and B6.NGAL−/− pristane, whereas mice which received PBS will be referred to as B6 PBS and B6.NGAL−/− PBS hereafter. Data are expressed as mean ± SEM and analyzed by the Mann Whitney test. Data are representative of results from three independent cohorts, *p< 0.05, **p<0.01, ***p<0.001.
3.2. NGAL deficiency leads to elevation of autoantibodies post pristane challenge
Pristane induces generation of autoantibodies in different strains of non-autoimmune prone mice such as BALB/c, SJL, and B6 [23-25,31]. Therefore, we analyzed the levels of autoantibodies in the serum for several nuclear antigens. A significant increase in the levels of ssDNA specific IgG, IgG2c, and IgG1 as well as dsDNA specific IgG, IgG2c, and IgG1 was seen in the pristane challenged B6 group as compared to the PBS treated B6 group (Fig. 2A, C, D and Fig. 3A, C, D). The pristane challenged B6 group also had significantly higher levels of chromatin and histone IgG as compared to PBS treated B6 group (Fig. 4).
Fig. 2.

Serum anti-ssDNA autoantibodies. Serum samples from B6 and B6.NGAL−/− mice, 4 months after pristane or PBS challenge, were analyzed. A-E. Anti-ssDNA IgG, IgG2b, IgG2c, IgG1, and IgG3 were measured by ELISA (n=4-5 per PBS group, n=10-11 per pristane group). Data are expressed as mean ± SEM and analyzed by the Mann Whitney test. Data are representative of results from three independent cohorts, *p< 0.05, **p<0.01, ***p<0.001.
Fig. 3.

Serum anti-dsDNA autoantibodies. Serum samples from B6 and B6.NGAL−/− mice, 4 months after pristane or PBS challenge, were analyzed. A-E. Anti-dsDNA IgG, IgG2b, IgG2c, IgG1, and IgG3 were measured by ELISA (n=4-5 per PBS group, n=10-11 per pristane group). Data are expressed as mean ± SEM and analyzed by the Mann Whitney test. Data are representative of results from three independent cohorts, *p< 0.05, **p<0.01, ***p<0.001.
Fig. 4.
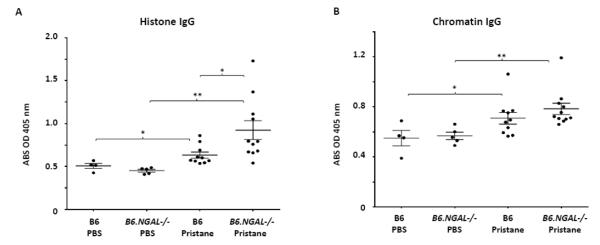
Serum anti-histone IgG and anti-chromatin IgG. Serum samples from B6 and B6.NGAL−/− mice, 4 months after pristane or PBS challenge, were analyzed. A. Anti-histone IgG, B. Anti-chromatin IgG, were analyzed by ELISA (n=4-5 per PBS group, n=10-11 per pristane group). Data are expressed as mean ± SEM and analyzed by the Mann Whitney test. Data are representative of results from three independent cohorts, *p< 0.05, **p<0.01.
Surprisingly, we found that the levels of anti-ssDNA and dsDNA IgG2b and IgG2c were significantly higher in pristane challenged B6.NGAL−/− mice as compared to B6 mice (Fig. 2B, C and 3B, C). In contrast, anti-ssDNA and dsDNA IgG1 antibodies (Fig. 2D and Fig. 3D) were significantly lower in the pristane challenged B6.NGAL−/− as compared to B6 mice. IgG3 anti-ssDNA (Fig. 2E) and anti-dsDNA antibodies (Fig. 3E) also trended higher in pristane challenged B6.NGAL−/− mice. Anti-histone IgG was significantly higher (Fig. 4A) in pristane challenged B6.NGAL−/− as compared to the B6 mice; however, anti-chromatin IgG (Fig. 4B) levels did not show any significant difference.
All the types of antinuclear antibodies were at similar levels in both the PBS challenged B6 and B6.NGAL−/− groups except for IgG1 anti-ssDNA and anti-dsDNA antibodies, which were significantly lower in the PBS treated B6.NGAL−/− mice (Fig. 2D, and Fig. 3D).
3.3. Elevated levels of anti-nuclear antibodies in pristane challenged NGAL deficient mice
To confirm the ELISA results showing differences in autoantibodies between the groups, we studied serum anti-nuclear antibodies (ANA) by immunofluorescence on fixed Hep-2 cells. Pristane immunized mice from several non-autoimmune strains (including B6) can display both homogenous and nucleolar ANA staining patterns (18,31,36-38). We found that the pristane challenged B6.NGAL−/− mice had higher levels of ANA than B6 mice, as observed by a significant increase in homogenous nuclear staining and a trend towards increased nucleolar nuclear pattern staining, as analyzed by immunofluorescence (Fig. 5A). PBS challenged B6 and B6.NGAL−/− groups were anti-nuclear antibody negative.
Fig. 5.


NGAL regulates the levels of anti-nuclear antibodies. A. Serum samples from B6 and B6.NGAL−/− mice, 4 months after pristane or PBS challenge, were analyzed for the presence of ANA using Hep-2 slides. Immunofluorescent staining patterns were compared between the groups using the Mann-Whitney test. The analysis was performed on randomly selected mice, n=3-4 per PBS group, n=5-7 per pristane group. Data are expressed as mean ± SEM, **p<0.01. B. Autoantibodies to Sm/RNP are upregulated in the serum of pristane challenged B6.NGAL−/− mice. 35S-labeled K562 cell extract was immunoprecipitated with sera from B6 and B6.NGAL−/− mice, 3-6 months after pristane or PBS challenge. Positions of the U1 snRNP-specific A and C proteins, and the Sm proteins B’/B, D, E/F, and G, are indicated by arrows. Positions of molecular weight standards in kilodaltons are shown on the left. Data are representative of results from two independent cohorts (n=3 per PBS group, n=14-16 per pristane group).
3.4. Upregulation of anti-Sm/RNP antibodies in response to pristane in the absence of NGAL
Analysis of autoantibodies using immunoprecipitation displayed a prominent anti-Sm and anti-RNP autoantibody response in the serum of pristane challenged B6.NGAL−/− mice (50%; 7/14) as compared to B6 mice (13%; 2/16) between 3-6 months of age (Fig. 5B). In contrast, serum from PBS treated B6 and B6.NGAL−/− mice were negative for anti-Sm/RNP antibodies (Fig. 5B).
3.5. NGAL deficiency increases nuclear antigen specific antibody secreting cells in spleen
As NGAL promotes apoptosis of hematopoietic cells, we investigated whether NGAL affects the numbers of autoantibody producing cells in the spleen. Autoantibody secreting cells (ASCs) specific for nuclear antigens such as histone, dsDNA and Sm/RNP were elevated in the pristane challenged as compared to the PBS treated groups. We observed a significant increase in the absolute number of anti-histone IgG producing cells in pristane challenged B6.NGAL−/− mice as compared to B6 mice (Fig. 6A), whereas anti-dsDNA and anti-Sm/RNP IgG producing cells showed a similar trend (Fig. 6B, C).
Fig. 6.

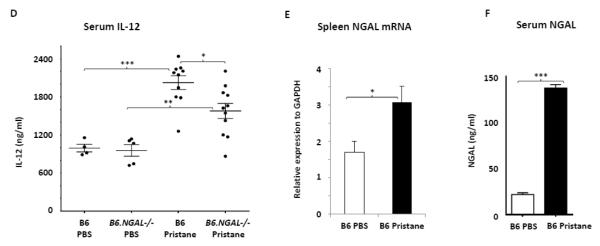
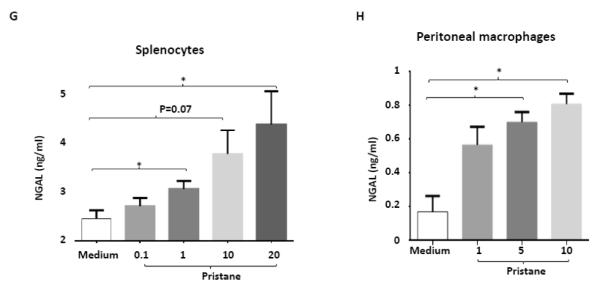
A-C. Autoantibody secreting cells specific to histone, dsDNA, and Sm/RNP in spleens from B6 and B6.NGAL−/− mice, 4 months after pristane or PBS challenge, were measured by ELISPOT in triplicate and analyzed by unpaired t-test. Data are expressed as mean ± SEM, n=3 mice randomly selected from each of the pristane and PBS groups, *p<0.05, **p<0.01, ***p<0.001. D. IL-12 is induced after pristane challenge. Serum IL-12 was analyzed at 4 months after pristane or PBS challenge in duplicate (n=4-5 per PBS group, n=10-11 per pristane group). Data are expressed as mean ± SEM and analyzed by the Mann Whitney test, *p< 0.05, **p<0.01, ***p<0.001. E. NGAL mRNA levels in spleen analyzed by real time PCR in B6 Pristane and B6 PBS groups of mice (n = 4 per PBS group and n=10 per pristane group). Relative expression of NGAL mRNA to GAPDH is plotted (Y-axis scale is x 10−4). Data are representative of results from two independent cohorts, performed in duplicate. Data are expressed as mean ± SEM and analyzed by unpaired t-test, *p<0.05. F. Serum NGAL was measured by ELISA at 4 months post pristane or PBS treatment in B6 mice (n = 4 per PBS group and n=10 per pristane group). Data are representative of results from two independent cohorts performed in triplicate. Data are expressed as mean ± SEM and analyzed by unpaired t-test, ***p<0.001. G. Splenocytes isolated from B6 mice were stimulated in vitro with either pristane or medium at concentrations of 0.1, 1, 10, and 20 ng/ml for 24 hours, and the supernatants analyzed for NGAL by ELISA. Data are representative of results from two independent experiments, performed in duplicate. Data are expressed as mean ± SEM and analyzed by unpaired t-test, *p< 0.05. H. Peritoneal macrophages were isolated from B6 mice 72 hours after i.p. injection of thioglycollate, and stimulated for 24 hrs with pristane at concentrations of 1, 5, and 10 μg/ml. Data are representative of results from two independent experiments, performed in duplicate. Data are expressed as mean ± SDEV and analyzed by unpaired t-test, *p< 0.05.
3.6. Serum IL-12p40, serum NGAL and spleen NGAL are upregulated after pristane treatment
IL-12 is a pro-inflammatory cytokine instrumental in the pathogenesis of spontaneous as well as inducible models of lupus nephritis [39-41]; therefore, we measured serum levels of IL-12. Interestingly, IL-12 levels were significantly increased after pristane challenge in both groups of B6 and B6.NGAL−/− mice as compared to the respective control groups (Fig. 6D). Furthermore, pristane challenged B6 mice had significantly higher levels of IL-12 in serum as compared to the B6.NGAL−/− mice (Fig. 6D).
NGAL is induced in response to many forms of inflammation and tissue injury; therefore, we analyzed the levels of NGAL mRNA in spleen, and found a significant increase in pristane injected as compared to PBS treated B6 mice (Fig. 6E). Similar to the expression levels in spleen, we found that NGAL was significantly upregulated in the serum of pristane challenged as compared to the PBS treated B6 mice (Fig. 6F).
To determine the source of circulating NGAL in pristane challenged B6 mice, we examined splenocytes stimulated with pristane in vitro and found that pristane induces the release of NGAL in a dose dependent manner (Fig. 6G). To further evaluate whether peritoneal macrophages which are recruited locally at the sites of pristane induced inflammation can produce NGAL, we injected thioglycollate into the peritoneum of B6 mice. After 72 hours, macrophages were harvested by peritoneal lavage and stimulated for 48 hours to analyze the levels of NGAL in the supernatant. As seen in Fig. 6H, pristane induces the release of NGAL from macrophages in a dose dependent manner.
3.7. Inflammatory mediators are upregulated in the spleens of NGAL−/− mice
To explore if the inflammatory response in the spleen induced by pristane is regulated by NGAL, we analyzed several genes relevant to lupus pathogenesis. We found that the chemokine CXCL13 (B lymphocyte chemoattractant) and its receptor CXCR5, as well as CXCL10 (10 kDa interferon gamma-induced protein) and its receptor CXCR3, were upregulated in the spleen of pristane challenged B6.NGAL−/− mice as compared to B6 mice (Fig. 7A). Pristane challenged B6.NGAL−/− mice also had significantly higher levels of spleen mRNA expression for activation induced cytidine deaminase (AID) (Fig. 7B). Furthermore, significant upregulation in the levels of MCP-1 and IRF5 was observed in the spleen of pristane challenged B6.NGAL−/− mice as compared to B6 mice (Fig. 7A, B). Finally, pristane challenge in the B6.NGAL−/− strain, as compared to B6 mice, was associated with increased mRNA expression of transcription factors and cytokines associated with Th2 (GATA-3, IL-4) and Th17 (RORCγ, IL-21) type responses, respectively, rather than Th1 (data not shown).
Fig. 7.

NGAL deficiency leads to upregulation of inflammatory mediators in spleen. A, B. Spleen mRNA levels of selected genes were measured at 4 months post pristane treatment by real time PCR performed in duplicate (n=4-5 per PBS group, n=10-11 per pristane group). Relative expression of mRNA of genes of interest in relation to GAPDH is plotted (Y-axis scale is x 10−2 (in A) or x 10−3 (in B)). Data are expressed as mean ± SEM and analyzed by unpaired t-test, *p< 0.05, **p<0.01.
3.8. Diminished kidney pathology in NGAL−/− mice
To determine the effect of NGAL on renal disease in pristane induced lupus, we analyzed kidney sections stained by H&E and PAS. We found that mesangial proliferation, endocapillary proliferation, and PAS positive deposits were present in a significantly lower proportion of glomeruli in the pristane challenged B6.NGAL−/− group as compared to the B6 group (Fig. 8A, B). PAS positive deposits were predominantly observed in mesangial areas but occasional glomeruli with endocapillary proliferation also displayed subendothelial deposits that were visible by light microscopy (Fig. 8A). Significant tubulointerstitial lesions, including interstitial inflammation and fibrosis, were absent in all the groups of mice. PBS treated mice did not show any histopathological abnormalities (Fig. 8B). We did not observe any significant difference in kidney-infiltrating neutrophils between pristane challenged B6 and B6.NGAL−/− mice (data not shown).
Fig. 8.





NGAL deficiency restricts renal damage induced by pristane. A,B. Kidney sections from B6 and B6.NGAL−/− mice, 4 months after pristane or PBS challenge, were stained by PAS and H&E and analyzed for pathologic features including mesangial proliferation, endocapillary hypercellularity, and PAS positive deposits. Data was analyzed by Mann-Whitney test (n=4-5 per PBS group, n=10-11 per pristane group) and expressed as mean ± SEM, *p< 0.05. The figure (40x magnification) shows normal appearing glomeruli in PBS-injected B6 (left upper panel) and B6.NGAL−/− mice (left lower panel); pristane-injected B6 mice display global endocapillary proliferation (upper right panel), while glomeruli in pristane-injected B6.NGAL−/− mice are relatively preserved, showing only mild mesangial deposits and mild mesangial hypercellularity (lower right panel). C. Kidney mRNA levels (in duplicate) were measured at 4 months post pristane treatment by real time PCR (n=4-5 per PBS group, n=10-11 per pristane group). Relative expression of mRNA of genes of interest in relation to GAPDH is plotted (Y-axis scale is x 10−3). Data are expressed as mean ± SEM and analyzed by unpaired t-test, *p< 0.05, **p<0.01. D. Kidney mesangial and tubular cells were stimulated with either LPS (5 μg/ml) or TNFα +IFNγ (each at 5 ng/ml), and the mRNA expression of NGAL at 6, 12 and 24 hours was analyzed in 3-4 replicates by real time PCR. Relative expression of NGAL mRNA in relation to GAPDH was calculated. Fold change at each time point was calculated as the relative ratio to baseline just prior to stimulation. Data are representative of results from two independent experiments, performed in 3-4 replicates. Data are expressed as mean ± SEM and analyzed by unpaired t-test, *p< 0.05, **p<0.01, compared to medium treated group at each respective time point. E. Mesangial cells were stimulated with the NGAL at 10 μg/ml with or without anti-NGAL antibody or control IgG at 100 μg/ml in duplicate for 24 hours, and the degree of apoptosis analyzed by Annexin V and 7-AAD staining and flow cytometry. A total of 50,000 events were recorded per sample. Percentage of annexin-V and 7-AAD double positive cells were plotted against the treatment. Data are representative of results from two independent experiments, performed in 3-4 replicates. Data are expressed as mean ± SEM and analyzed by unpaired t-test, *p<0.05, **p<0.01, ***p<0.001. F. Mesangial cells were stimulated for 24 hours with NGAL at 1 μg/ml in presence and absence of anti-NGAL antibody or control IgG at 0.5 and 2 μg/ml (3-4 replicates), and the supernatant was analyzed for MCP-1 by ELISA. The percentage of NGAL induced MCP-1 was calculated by subtracting the value for media alone from each treatment group, and comparing this to the concentration of MCP-1 induced by NGAL alone which was set as 100%. Data are representative of results from two independent experiments, performed in 3-4 replicates. Data are expressed as mean ± SEM and analyzed by unpaired t-test, *p<0.05.
3.9. Inflammatory mediators in kidneys were regulated by NGAL
To explore a possible role of NGAL on renal inflammation we analyzed several relevant genes which have been studied in lupus nephritis, particularly in the pristane induced model [18-20]. There was no significant difference between the pristane challenged B6.NGAL−/− and B6 groups for the CXCL13, CXCL10, CXCR3, MCP-1, and IRF5 (Fig. 8C). The expression of TLR4 was significantly lower in kidneys of pristane challenged B6.NGAL−/− mice than B6 mice. Additionally, the proapoptotic gene APAF-1 and the adhesion molecule ICAM-1 were also found to be significantly lower in B6.NGAL−/− mice (Fig. 8C). Interestingly, we observed a trend towards increased levels of NGAL mRNA in kidneys of the pristane challenged B6 group as compared to PBS treated B6 group (Supplementary Fig. S1).
3.10. Exposure to inflammatory stimuli induces the expression of NGAL by renal cells
To determine which kidney cells express NGAL upon exposure to inflammatory mediators, we stimulated mesangial and tubular cells in vitro with either LPS or a combination of TNFα and IFNγ. We found that these inflammatory stimuli significantly increased the expression of NGAL in both mesangial and tubular cells (Fig. 8D). However, in contrast to what we described above for splenocytes and macrophages, pristane stimulation did not consistently increase NGAL mRNA in either mesangial cells or tubular cells (data not shown). To further investigate the effects of NGAL on mesangial cells, we stimulated mesangial cells with NGAL for 24 hours. As previously described [17], NGAL induced apoptotic cell death (Fig. 8E), and also increased the expression of the proinflammatory chemokine MCP-1 (Fig. 8F). These effects were significantly reversed in the presence of a monoclonal anti-NGAL antibody (Fig. 8E, 8F), indicating that the induction of apoptosis and MCP-1 in mesangial cells by NGAL is mediated by a specific receptor, rather than by an extraneous contaminant in the NGAL preparation.
3.11. NGAL receptor expression in spleen and kidney
To determine if the receptor for NGAL is expressed by spleen and kidney tissues in B6 mice, we analyzed the mRNA expression of 24p3R (also known as cation transporter BOCT or solute carrier family 22 member 17). 24p3R was expressed in spleen and kidneys in both PBS and pristane challenged B6 mice. We did not find an effect of pristane treatment on 24p3R levels in the kidneys. Interestingly, pristane injected mice had significantly lower levels of spleen 24p3R expression as compared to control treated mice (data not shown).
4. Discussion
In this study, we found that NGAL has a regulatory role in the onset of the autoantibodies in an inducible model of lupus. Post pristane challenge, mice deficient in NGAL as compared to wild type B6 mice had higher levels of specific autoantibodies as well as more pronounced hypergammaglobulinemia. Furthermore, pristane challenged B6.NGAL−/− mice had higher levels of expression of AID and inflammatory mediators in the spleen, as well as increased numbers of autoantibody secreting cells. Surprisingly, B6.NGAL−/− mice were partially protected from renal injury, and displayed less severe renal pathology than B6 pristane treated mice. Since our experimental design did not include littermate controls, it is important to acknowledge the possibility that some differences between the strains might have arisen from genetic heterogeneity. Nevertheless, the magnitude of differences in response to pristane observed here, together with the confirmatory in vitro studies, support the conclusion that these effects arose from differential expression of NGAL, rather than any minor background variations.
The immune response generated by B6 mice after pristane injection found in our study is consistent with previously reported observations in other strains of non-autoimmune mice [18-23]. Antibodies to nuclear antigens are a characteristic feature of SLE in both humans and mice. The pathological role of autoantibodies in progression of lupus related organ damage such as kidney, brain, and skin has been well established [4,27,42-44]. These autoantibodies are able to bind or cross react with nuclear antigens or antigens present on the cell surface [45-50], activating Fc receptors present on different cell types as well as complement pathways. This leads to expression of inflammatory mediators while promoting the infiltration of immune cells into the target organs, eventually leading to tissue injury. Here, we observed higher levels of autoantibodies to nuclear antigens in the serum of B6.NGAL−/− mice post pristane challenge, which suggests that the production of autoantibodies is, at least in part, regulated by NGAL. Interestingly, as has been observed in other pristane induced and spontaneous models (21,32,51), the increase in autoantibodies was not global but rather restricted to particular autoantigens and isotypes, suggesting a specific effect of NGAL. It should further be noted that the splenic cytokine profile did not perfectly correspond with the subclasses observed. While additional exploration of this point is outside the scope of the present study, this intriguing observation will require further elucidation. It will be interesting to determine if NGAL genetic polymorphisms may be associated with autoantibody production in human lupus.
Here, we provide evidence for a novel regulatory role of NGAL in limiting autoantibody titers by restricting the number of ASCs. These cells were more numerous in the spleen of pristane challenged B6.NGAL−/− mice, which would be consistent with the higher levels of autoantibodies. Our findings are in agreement with other studies in which NGAL deficiency led to the accumulation of myeloid and lymphoid cells in bone marrow and B cells in bone marrow and spleen, as well as hematopoietic cell types [52]. Therefore, lower antibody levels in NGAL sufficient mice following pristane is likely to be due to a regulatory role of NGAL in controlling the survival and number of autoantibody producing cells. Whether NGAL regulates the numbers of any other splenic cell subset remains to be determined.
NGAL is a sensitive biomarker of tissue inflammation and injury, including in the serum of lupus patients [16]. We found that NGAL was upregulated in B6 mice post pristane challenge, indicating that indeed NGAL is responsive to the triggering and development of systemic autoimmunity, including in the pristane model in which this had not been previously described. The source of serum NGAL is most likely activated macrophages and splenocytes, since we found that pristane stimulation directly upregulates NGAL expression in these cell types. The increase in NGAL expression in kidneys of pristane challenged mice may be due to a similar, direct effect on kidney cells, a cytokine driven effect, or a reflection of upregulated NGAL levels in infiltrating macrophages.
Consistent with the enhanced immune response and autoantibody levels in the pristane challenged B6.NGAL−/− mice, we observed higher levels of expression of AID in the spleen. AID is responsible for class switch recombination, is associated with human SLE, and is found in spleens and ectopic lymphoid tissues [25,31,53] in spontaneous and induced lupus. NGAL also influenced the levels of splenic CXCL13, a B lymphocyte chemokine that influences the migration of B cells into target organs and which is a known marker of disease activity in human SLE [54-56].
IRF5 is a major regulator of the type I IFN pathway, responsible for B cell activation and migration [22,25,57]. Previous studies [21,35,58,59] demonstrated that pristane induced lupus is type I interferon dependent; pristane injection leads to accumulation of peritoneal cells which express high levels of interferon stimulated genes (e.g. IRF5, IRF7, MCP-1, CXCL10), and peritoneal fluid rich in interferon-dependent cytokines (e.g. MCP-1, IL-6). Furthermore, IRF5 polymorphisms are associated with human SLE [60-62]. MCP-1 and IP-10, which are interferon responsive, play a major role in monocyte/macrophage recruitment [63-65] and are also associated with human SLE. While we found that IRF5, MCP-1, and CXCL10 levels were elevated in pristane treated NGAL−/− mice, levels of IRF3 and IRF7 were no different than in B6 NGAL sufficient mice (data not shown). Moreover, while IL-12 is interferon responsive and upregulated in spontaneous and pristane induced lupus [31,51,66], serum levels were actually lower in NGAL deficient mice. Thus, it would appear that the relationship between NGAL and the interferon signaling pathway is not straightforward. Moreover, our focus in these studies was the contribution of NGAL to the onset of autoantibodies and kidney disease. Therefore, more detailed analysis of the cellular composition of spleen and peritoneum and the interaction between NGAL and interferon signaling will need to be further explored in future studies.
Glomerular cellular proliferation and immune complex deposition were observed in both NGAL wild type and knockout pristane challenged groups, although the injury was less severe in the B6.NGAL−/− mice. Intracellular cell adhesion molecule-1 (ICAM-1) is a marker of kidney injury in human lupus nephritis [67], and responsible for kidney damage in lupus prone MRL/lpr mice [68,69], whereas apoptotic protease activating factor-1 (APAF-1) is a proapoptotic gene induced during glomerulonephritis [70,71]. TLR4 was found to be associated with the progression of murine lupus nephritis [19,72]. Pristane challenged B6.NGAL−/− mice had significantly lower kidney expression of ICAM-1, APAF-1, and TLR4, which is consistent with the decreased renal injury observed in this strain. It would be interesting to explore if NGAL levels can differentiate between lupus patients with high titers of autoantibodies but absent or mild kidney disease, as compared to patients with modest autoantibody titers but more severe kidney disease.
The current study supports previous findings from our lab in antibody mediated renal injury mediated by nephrotoxic serum, where NGAL deficient mice were protected while exogenous administration of NGAL further aggravated renal damage [17]. Here we have demonstrated that inflammatory stimuli/ligands such as those present in pristane induced lupus upregulate NGAL in kidney mesangial and tubular cells. NGAL could then directly contribute to the pathogenesis of renal disease via induction of caspase-3 mediated apoptosis in kidney resident cells, and a direct pro-inflammatory effect on kidney cells via activation of NF-kB [17, and above]. Therefore, it seems likely that the pristane mediated enhancement of systemic and local NGAL expression in NGAL wild type mice might explain their worse kidney disease as compared to NGAL deficient mice, despite higher autoantibody titers in the latter strain.
Recent reports similarly showing a proinflammatory effect of NGAL on structural cell types include the induction of INOS in chondrocytes, and IL-1β, iNOS, TNF, and CXCL10 in astrocytes [73,74]. An important role of NGAL in progression of renal disease was also reported in a partial nephrectomy model [75]. Additionally, a proinflammatory role of NGAL was recently demonstrated in the reverse passive arthus model of arthritis, wherein NGAL deficient mice had significantly reduced skin inflammation and immune cell infiltration [76]. Interestingly, in EAE however [12] disease severity was exacerbated in NGAL deficient mice. Furthermore, NGAL administration attenuated kidney injury in an ischemia-reperfusion model [77,78]. Therefore, NGAL expression apparently is not always detrimental, even in kidney disease, and may depend on the source of NGAL, localization of expression, and the particular disease context [8,17,75,77-79].
Conclusions
Pristane challenge upregulates the secretion of NGAL by macrophages and splenocytes, which then serves as a brake on the autoantibody response in this model by limiting the number of autoantibody producing cells in the spleen. In the context of early lupus, a basal level of NGAL would then have a regulatory, protective role. However, with progression of disease, a further increase in NGAL levels would be detrimental for the kidney, acting in concert with IgG and other local inflammatory mediators to exacerbate renal injury and damage. While NGAL inhibition may potentially be useful for the local kidney inflammatory process in lupus, it will be important to consider any possible effects on the adaptive immune response when considering modulating the NGAL pathway as a potential therapeutic approach for this disease.
Supplementary Material
Highlights.
NGAL regulates the anti-nuclear antibody response in pristane induced lupus.
NGAL deficient mice exhibit increased numbers of autoantibody secreting cells following pristane challenge.
Despite increased autoantibody titers, pristane-challenged NGAL deficient mice are partially protected from renal injury.
Pristane stimulates NGAL secretion by peritoneal macrophages and splenocytes.
Acknowledgements
This study was supported by grants from the NIH RO1-AR048692 and RO1-DK090319 (to C.P.) and an Arthritis Foundation fellowship (#5451) (to R.D.P.). Westley H. Reeves and Haoyang Zhuang were supported by grants RO1-AR44731 and T32AR007603 respectively. The authors thank Drs. Thorsten Berger and Tak W. Mak (Ontario Cancer Institute, Canada) for generously providing the NGAL−/− mice. The authors would like to acknowledge the laboratory of Dr. Richard Stanley (Albert Einstein College of Medicine) for their help in macrophage isolation.
Abbreviations
- NGAL
neutrophil gelatinase associated lipocalin
- dsDNA
double stranded DNA
- ssDNA
single stranded DNA
- SLE
systemic lupus erythematosus
- ASC
autoantibody secreting cells
- ANA
anti-nuclear antibodies
- RBC
Red blood cells
- AID
Activation induced cytidine deaminase
- IRF5
Interferon regulatory factor-5
- IP-10
IFNγ inducible protein-10
- MCP-1
Monocyte chemoattractant protein-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. Conflict of interest: None.
7. References
- [1].Kjeldsen L, Cowland JB, Borregaard N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim. Biophys. Acta. 2000;1482:272–83. doi: 10.1016/s0167-4838(00)00152-7. [DOI] [PubMed] [Google Scholar]
- [2].Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta. 2012;1826:129–69. doi: 10.1016/j.bbcan.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Saiga H, Nishimura J, Kuwata H, Okuyama M, Matsumoto S, Sato S, et al. Lipocalin 2-dependent inhibition of mycobacterial growth in alveolar epithelium. J. Immunol. 2008;181:8521–7. doi: 10.4049/jimmunol.181.12.8521. [DOI] [PubMed] [Google Scholar]
- [4].Qing X, Zavadil J, Crosby MB, Hogarth MP, Hahn BH, Mohan C, et al. Nephritogenic anti-DNA antibodies regulate gene expression in MRL/lpr mouse glomerular mesangial cells. Arthritis. Rheum. 2006;54:2198–210. doi: 10.1002/art.21934. [DOI] [PubMed] [Google Scholar]
- [5].Borkham-Kamphorst E, Drews F, Weiskirchen R. Induction of lipocalin-2 expression in acute and chronic experimental liver injury moderated by pro-inflammatory cytokines interleukin-1β through nuclear factor-κB activation. Liver. Int. 2011;31:656–65. doi: 10.1111/j.1478-3231.2011.02495.x. [DOI] [PubMed] [Google Scholar]
- [6].Rubinstein T, Pitashny M, Levine B, Schwartz N, Schwartzman J, Weinstein E, et al. Urinary neutrophil gelatinase-associated lipocalin as a novel biomarker for disease activity in lupus nephritis. Rheumatology. 2010;49:960–71. doi: 10.1093/rheumatology/kep468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hinze CH, Suzuki M, Klein-Gitelman M, Passo MH, Olson J, Singer NG, et al. Neutrophil gelatinase-associated lipocalin is a predictor of the course of global and renal childhood-onset systemic lupus erythematosus disease activity. Arthritis. Rheum. 2009;60:2772–81. doi: 10.1002/art.24751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia reperfusion injury. Proc. Natl. Acad. Sci. 2006;103:1834–9. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bachman MA, Miller VL, Weiser JN. Mucosal lipocalin 2 has pro-inflammatory and iron-sequestering effects in response to bacterial enterobactin. PLoS. Pathog. 2009;5:e1000622. doi: 10.1371/journal.ppat.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang L, Li H, Wang J, Gao W, Lin Y, Jin W, et al. C/EBP Δ targets to neutrophil gelatinase-associated lipocalin (NGAL) as a repressor for metastasis of MDA-MB-231 cells. Biochim. Biophys. Acta. 2011;1813:1803–13. doi: 10.1016/j.bbamcr.2011.06.010. [DOI] [PubMed] [Google Scholar]
- [11].Yang J, McNeish B, Butterfield C, Moses MA. Lipocalin 2 is a novel regulator of angiogenesis in human breast cancer. FASEB. J. 2013;27:45–50. doi: 10.1096/fj.12-211730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Berard JL, Zarruk JG, Arbour N, Prat A, Yong VW, Jacques FH, et al. Lipocalin 2 is a novel immune mediator of experimental autoimmune encephalomyelitis pathogenesis and is modulated in multiple sclerosis. Glia. 2012;60:1145–59. doi: 10.1002/glia.22342. [DOI] [PubMed] [Google Scholar]
- [13].Devireddy LR, Teodoro JG, Richard FA, Green MR. Induction of apoptosis by a secreted lipocalin that is transcriptionally regulated by IL-3 deprivation. Cell. 2005;123:1293–305. doi: 10.1126/science.1061075. [DOI] [PubMed] [Google Scholar]
- [14].Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Science. 2001;293:829–34. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- [15].Miharada K, Hiroyama T, Sudo K, Danjo I, Nagasawa T, Nakamura Y. Lipocalin 2-mediated growth suppression is evident in human erythroid and monocyte/macrophage lineage cells. J. Cell. Physiol. 2008;215:526–37. doi: 10.1002/jcp.21334. [DOI] [PubMed] [Google Scholar]
- [16].Pitashny M, Schwartz N, Qing X, Hojaili B, Aranow C, Mackay M, et al. Urinary lipocalin-2 is associated with renal disease activity in human lupus nephritis. Arthritis. Rheum. 2007;56:1894–903. doi: 10.1002/art.22594. [DOI] [PubMed] [Google Scholar]
- [17].Pawar RD, Pitashny M, Gindea S, Tieng AT, Levine B, Goilav B, et al. Neutrophil gelatinase-associated lipocalin is instrumental in the pathogenesis of antibody-mediated nephritis in mice. Arthritis. Rheum. 2012;64:1620–31. doi: 10.1002/art.33485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Savarese E, Steinberg C, Pawar RD, Reindl W, Akira S, Anders HJ, et al. Requirement of Toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis. Rheum. 2008;58:1107–15. doi: 10.1002/art.23407. [DOI] [PubMed] [Google Scholar]
- [19].Summers SA, Hoi A, Steinmetz OM, O’Sullivan KM, Ooi JD, Odobasic D, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J. Autoimmun. 2010;35:291–8. doi: 10.1016/j.jaut.2010.05.004. [DOI] [PubMed] [Google Scholar]
- [20].Urbonaviciute V, Starke C, Pirschel W, Pohle S, Frey S, Daniel C, et al. Toll-like receptor 2 is required for autoantibody production and development of renal disease in pristane-induced lupus. Arthritis. Rheum. 2013;65:1612–23. doi: 10.1002/art.37914. [DOI] [PubMed] [Google Scholar]
- [21].Xu Y, Lee PY, Li Y, Liu C, Zhuang H, Han S, et al. Pleiotropic IFN-dependent and - independent effects of IRF5 on the pathogenesis of experimental lupus. J. Immunol. 2012;188:4113–21. doi: 10.4049/jimmunol.1103113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends. Immunol. 2009;30:455–64. doi: 10.1016/j.it.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Satoh M, Richards HB, Shaheen VM, Yoshida H, Shaw M, Naim JO, et al. Widespread susceptibility among inbred mouse strains to the induction of lupus autoantibodies by pristane. Clin. Exp. Immunol. 2000;121:399–405. doi: 10.1046/j.1365-2249.2000.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Satoh M, Reeves WH. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J. Exp. Med. 1994;180:2341–6. doi: 10.1084/jem.180.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nacionales DC, Weinstein JS, Yan XJ, Albesiano E, Lee PY, Kelly-Scumpia KM, et al. B cell proliferation, somatic hypermutation, class switch recombination, and autoantibody production in ectopic lymphoid tissue in murine lupus. J. Immunol. 2009;182:4226–36. doi: 10.4049/jimmunol.0800771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–21. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- [27].Gao HX, Sanders E, Tieng AT, Putterman C. Sex and autoantibody titers determine the development of neuropsychiatric manifestations in lupus-prone mice. J. Neuroimmunol. 2010;229:112–22. doi: 10.1016/j.jneuroim.2010.07.020. [DOI] [PubMed] [Google Scholar]
- [28].Zhao Z, Burkly LC, Campbell S, Schwartz N, Molano A, Choudhury A, et al. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J. Immunol. 2007;179:7949–58. doi: 10.4049/jimmunol.179.11.7949. [DOI] [PubMed] [Google Scholar]
- [29].Deocharan B, Zhou Z, Antar K, Siconolfi-Baez L, Angeletti RH, Hardin J, et al. Alpha-actinin immunization elicits anti-chromatin autoimmunity in nonautoimmune mice. J. Immunol. 2007;179:1313–21. doi: 10.4049/jimmunol.179.2.1313. [DOI] [PubMed] [Google Scholar]
- [30].Xia Y, Pawar RD, Nakouzi AS, Herlitz L, Broder A, Liu K, et al. The constant region contributes to the antigenic specificity and renal pathogenicity of murine anti-DNA antibodies. J. Autoimmun. 2012;39:398–411. doi: 10.1016/j.jaut.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nacionales DC, Kelly KM, Lee PY, Zhuang H, Li Y, Weinstein JS, et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane) Am. J. Pathol. 2006;168:1227–40. doi: 10.2353/ajpath.2006.050125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pawar RD, Castrezana-Lopez L, Allam R, Kulkarni OP, Segerer S, Radomska E, et al. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating Toll-like receptor 2 at the glomerular filtration barrier. Immunology. 2009;128:e206–21. doi: 10.1111/j.1365-2567.2008.02948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Herlitz LC, Bomback AS, Markowitz GS, Stokes MB, Smith RN, Colvin RB, et al. Pathology after eculizumab in dense deposit disease and C3 GN. J. Am. Soc. Nephrol. 2012;23:1229–37. doi: 10.1681/ASN.2011121186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xia Y, Campbell SR, Broder A, Herlitz L, Abadi M, Wu P, et al. Inhibition of the TWEAK/Fn14 pathway attenuates renal disease in nephrotoxic serum nephritis. Clin. Immunol. 2012;145:108–21. doi: 10.1016/j.clim.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J. Exp. Med. 2008;205:2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Satoh M, Hamilton KJ, Ajmani AK, Dong X, Wang J, Kanwar YS, Reeves WH. Autoantibodies to ribosomal P antigens with immune complex glomerulonephritis in SJL mice treated with pristane. J Immunol. 1996;157:3200–6. [PubMed] [Google Scholar]
- [37].Satoh M, Mizutani A, Behney KM, Kuroda Y, Akaogi J, Yoshida H, Nacionales DC, Hirakata M, Ono N, Reeves WH. X-linked immunodeficient mice spontaneously produce lupus-related anti-RNA helicase A autoantibodies, but are resistant to pristane-induced lupus. Int. Immunol. 2003;15:1117–24. doi: 10.1093/intimm/dxg110. [DOI] [PubMed] [Google Scholar]
- [38].Frisoni L, McPhie L, Kang SA, Monestier M, Madaio M, Satoh M, Caricchio R. Lack of chromatin and nuclear fragmentation in vivo impairs the production of lupus anti-nuclear antibodies. J. Immunol. 2007;179:7959–66. doi: 10.4049/jimmunol.179.11.7959. [DOI] [PubMed] [Google Scholar]
- [39].Kitching AR, Turner AL, Semple T, Li M, Edgtton KL, Wilson GR, Timoshanko JR, et al. Experimental autoimmune anti-glomerular basement membrane glomerulonephritis: a protective role for IFN-gamma. J. Am. Soc. Nephrol. 2004;15:1764–74. doi: 10.1097/01.asn.0000128968.27705.5e. [DOI] [PubMed] [Google Scholar]
- [40].Kikawada E, Lenda DM, Kelley VR. IL-12 deficiency in MRL-Fas(lpr) mice delays nephritis and intrarenal IFN-gamma expression, and diminishes systemic pathology. J. Immunol. 2003;170:3915–25. doi: 10.4049/jimmunol.170.7.3915. [DOI] [PubMed] [Google Scholar]
- [41].Calvani N, Satoh M, Croker BP, Reeves WH, Richards HB. Nephritogenic autoantibodies but absence of nephritis in Il-12p35-deficient mice with pristane-induced lupus. Kidney. Int. 2003;64:897–905. doi: 10.1046/j.1523-1755.2003.00178.x. [DOI] [PubMed] [Google Scholar]
- [42].Zhuang H, Narain S, Sobel E, Lee PY, Nacionales DC, Kelly KM, et al. Association of anti-nucleoprotein autoantibodies with upregulation of Type I interferon inducible gene transcripts and dendritic cell maturation in systemic lupus erythematosus. Clin. Immunol. 2005;117:238–50. doi: 10.1016/j.clim.2005.07.009. [DOI] [PubMed] [Google Scholar]
- [43].Wen J, Xia Y, Stock A, Michaelson JS, Burkly LC, Gulinello M, et al. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J. Autoimmun. 2013;43:44–54. doi: 10.1016/j.jaut.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lucas JA, Menke J, Rabacal WA, Schoen FJ, Sharpe AH, Kelley VR. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J. Immunol. 2008;181:2513–21. doi: 10.4049/jimmunol.181.4.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Koren E, Koscec M, Wolfson-Reichlin M, Ebling FM, Tsao B, Hahn BH, et al. Murine and human antibodies to native DNA that cross-react with the A and D SnRNP polypeptides cause direct injury of cultured kidney cells. J. Immunol. 1995;154:4857–64. [PubMed] [Google Scholar]
- [46].Vlahakos D, Foster MH, Ucci AA, Barrett KJ, Datta SK, Madaio MP. Murine monoclonal anti-DNA antibodies penetrate cells, bind to nuclei, and induce glomerular proliferation and proteinuria in vivo. J. Am. Soc. Nephrol. 1992;2:1345–54. doi: 10.1681/ASN.V281345. [DOI] [PubMed] [Google Scholar]
- [47].Putterman C. New approaches to the renal pathogenicity of anti-DNA antibodies in systemic lupus erythematosus. Autoimmunity Rev. 2004;3:7–11. doi: 10.1016/S1568-9972(03)00082-X. [DOI] [PubMed] [Google Scholar]
- [48].Zhao Z, Weinstein E, Tuzova M, Davidson A, Mundel P, Marambio P, et al. Cross-reactivity of human lupus anti-DNA antibodies with alpha-actinin and nephritogenic potential. Arthritis. Rheum. 2005;52:522–30. doi: 10.1002/art.20862. [DOI] [PubMed] [Google Scholar]
- [49].Deocharan B, Qing X, Lichauco J, Putterman C. Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J. Immunol. 2002;168:3072–8. doi: 10.4049/jimmunol.168.6.3072. [DOI] [PubMed] [Google Scholar]
- [50].Putterman C, Diamond B. Immunization with a peptide surrogate for double-stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J. Exp. Med. 1998;188:29–38. doi: 10.1084/jem.188.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J. Am. Soc. Nephrol. 2007;18:1721–31. doi: 10.1681/ASN.2006101162. [DOI] [PubMed] [Google Scholar]
- [52].Liu Z, Yang A, Wang Z, Bunting KD, Davuluri G, Green MR, et al. Multiple apoptotic defects in hematopoietic cells from mice lacking lipocalin 24p3. J. Biol. Chem. 2011;286:20606–14. doi: 10.1074/jbc.M110.216549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, et al. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J. Immunol. 2007;178:7422–31. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, et al. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Invest. 2010;120:4251–72. doi: 10.1172/JCI42213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ishikawa S, Sato T, Abe M, Nagai S, Onai N, Yoneyama H, et al. Aberrant high expression of B lymphocyte chemokine (BLC/CXCL13) by C11b+CD11c+ dendritic cells in murine lupus and preferential chemotaxis of B1 cells towards BLC. J. Exp. Med. 2001;193:1393–402. doi: 10.1084/jem.193.12.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Schiffer L, Kumpers P, Davalos-Misslitz AM, Haubitz M, Haller H, Anders HJ, et al. B-cell-attrating chemokine CXCL13 as a marker of disease activity and renal involvement in systemic lupus erythematosus (SLE) Nephrol. Dial. Transplant. 2009;24:3708–3712. doi: 10.1093/ndt/gfp343. [DOI] [PubMed] [Google Scholar]
- [57].Yang L, Feng D, Bi X, Stone RC, Barnes BJ. Monocytes from Irf5−/− mice have an intrinsic defect in their response to pristane-induced lupus. J. Immunol. 2012;189:3741–50. doi: 10.4049/jimmunol.1201162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lee PY, Weinstein JS, Nacionales DC, Scumpia PO, Li Y, Butfiloski E, van Rooijen N, Moldawer L, Satoh M, Reeves WH. A novel type I IFN-producing cell subset in murine lupus. J. Immunol. 2008;180:5101–8. doi: 10.4049/jimmunol.180.7.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, Nacionales DC, Butfiloski EJ, van Rooijen N, Akira S, Sobel ES, Satoh M, Reeves WH. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am. J. Pathol. 2009;175:2023–33. doi: 10.2353/ajpath.2009.090328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Stone RC, Du P, Feng D, Dhawan K, Rönnblom L, Eloranta ML, et al. RNA-Seq for enrichment and analysis of IRF5 transcript expression in SLE. PLoS. One. 2013;8:e54487. doi: 10.1371/journal.pone.0054487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang C, Sandling JK, Hagberg N, Berggren O, Sigurdsson S, Karlberg O, et al. Genome-wide profiling of target genes for the systemic lupus erythematosus-associated transcription factors IRF5 and STAT4. Ann. Rheum. Dis. 2013;72:96–103. doi: 10.1136/annrheumdis-2012-201364. [DOI] [PubMed] [Google Scholar]
- [62].Cherian TS, Kariuki SN, Franek BS, Buyon JP, Clancy RM, Niewold TB. Brief Report: IRF5 systemic lupus erythematosus risk haplotype is associated with asymptomatic serologic autoimmunity and progression to clinical autoimmunity in mothers of children with neonatal lupus. Arthritis. Rheum. 2012;64:3383–7. doi: 10.1002/art.34571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Lyons R, Sobel E, et al. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis. Rheum. 2007;56:3770–83. doi: 10.1002/art.23023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lee PY, Weinstein JS, Nacionales DC, Scumpia PO, Li Y, Butfiloski E, et al. A novel type I IFN-producing cell subset in murine lupus. J. Immunol. 2008;180:5101–8. doi: 10.4049/jimmunol.180.7.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rose T, Grützkau A, Hirseland H, Huscher D, Dähnrich C, Dzionek A, et al. IFNα and its response proteins, IP-10 and SIGLEC-1, are biomarkers of disease activity in systemic lupus erythematosus. Ann. Rheum. Dis. 2013;72:1639–45. doi: 10.1136/annrheumdis-2012-201586. [DOI] [PubMed] [Google Scholar]
- [66].Lu LD, Stump KL, Wallace NH, Dobrzanski P, Serdikoff C, Gingrich DE, et al. Depletion of autoreactive plasma cells and treatment of lupus nephritis in mice using CEP-33779, a novel, orally active, selective inhibitor of JAK2. J. Immunol. 2011;187:3840–53. doi: 10.4049/jimmunol.1101228. [DOI] [PubMed] [Google Scholar]
- [67].Daniel L, Sichez H, Giorgi R, Dussol B, Figarella-Branger D, Pellissier JF, et al. Tubular lesions and tubular cell adhesion molecules for the prognosis of lupus nephritis. Kidney. Int. 2001;60:2215–21. doi: 10.1046/j.1523-1755.2001.00055.x. [DOI] [PubMed] [Google Scholar]
- [68].Bullard DC, King PD, Hicks MJ, Dupont B, Beaudet AL, Elkon KB. Intercellular adhesion molecule-1 deficiency protects MRL/MpJ-Fas(lpr) mice from early lethality. J. Immunol. 1997;59:2058–67. [PubMed] [Google Scholar]
- [69].McHale JF, Harari OA, Marshall D, Haskard DO. TNF-alpha and IL-1 sequentially induce endothelial ICAM-1 and VCAM-1 expression in MRL/lpr lupus-prone mice. J. Immunol. 1999;63:3993–4000. [PubMed] [Google Scholar]
- [70].Tzang BS, Hsu TC, Kuo CY, Chen TY, Chiang SY, Li SL, et al. Cystamine attenuates lupus-associated apoptosis of ventricular tissue by suppressing both intrinsic and extrinsic pathways. J. Cell. Mol. Med. 2012;16:2104–11. doi: 10.1111/j.1582-4934.2011.01511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pereira E, Tamia-Ferreira MC, Cardoso RS, Mello SS, Sakamoto-Hojo ET, Passos GA, et al. Immunosuppressive therapy modulates T lymphocyte gene expression in patients with systemic lupus erythematosus. Immunology. 2004;113:99–105. doi: 10.1111/j.1365-2567.2004.01929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lartigue A, Colliou N, Calbo S, François A, Jacquot S, Arnoult C, et al. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J. Immunol. 2009;183:6207–16. doi: 10.4049/jimmunol.0803219. [DOI] [PubMed] [Google Scholar]
- [73].Gómez R, Scotece M, Conde J, Lopez V, Pino J, Lago F, et al. Nitric oxide boosts TLR-4 mediated lipocalin 2 expression in chondrocytes. J. Orthop. Res. 2013;7:1046–52. doi: 10.1002/jor.22331. [DOI] [PubMed] [Google Scholar]
- [74].Jang E, Kim JH, Lee S, Kim JH, Seo JW, Jin M, et al. Phenotypic polarization of activated astrocytes: the critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J. Immunol. 2013;191:5204–19. doi: 10.4049/jimmunol.1301637. [DOI] [PubMed] [Google Scholar]
- [75].Viau A, El. Karoui K, Laouari D, Burtin M, Nguyen C, Mori K, et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J. Clin. Invest. 2010;120:4065–76. doi: 10.1172/JCI42004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Shashidharamurthy R, Machiah D, Aitken JD, Putty K, Srinivasan G, Chassaing B, et al. Differential role of lipocalin 2 during immune complex-mediated acute and chronic inflammation in mice. Arthritis. Rheum. 2013;65:1064–73. doi: 10.1002/art.37840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K, Mukoyama M, Kunis C, D’Agati V, Devarajan P, Barasch J. Endocytic delivery of lipocalin siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J. Clin. Invest. 2005;115:610–21. doi: 10.1172/JCI23056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mishra J, Mori K, Ma Q, Kelly C, Yang J, Mitsnefes M, Barasch J, Devarajan P. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. J. Am. Soc. Nephrol. 2004;15:3073–82. doi: 10.1097/01.ASN.0000145013.44578.45. [DOI] [PubMed] [Google Scholar]
- [79].Paragas N, Qiu A, Zhang Q, Samstein B, Deng SX, Schmidt-Ott KM, Viltard M, Yu W, Forster CS, Gong G, Liu Y, Kulkarni R, Mori K, Kalandadze A, Ratner AJ, Devarajan P, Landry DW, D’Agati V, Lin CS, Barasch J. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat. Med. 2011;17:216–22. doi: 10.1038/nm.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.