

Abstract
Engineered chimeric cytokines can generate gain-of-function activity in immune cells. Here we report potent antitumor activity for a novel fusion cytokine generated by N-terminal coupling of GM-CSF to IL-4, generating a fusokine termed GIFT4. B cells treated with GIFT4 clustered GM-CSF and IL-4 receptors on the cell surface and displayed a pan-STAT hyperphosphorylation associated with acquisition of a distinct phenotype and function described to date. In C57BL/6J mice, administration of GIFT4 expanded endogenous B cells and suppressed the growth of B16F0 melanoma cells. Further, B16F0 melanoma cells engineered to secrete GIFT4 were rejected immunologically in a B cell-dependent manner. This effect was abolished when GIFT4-expressing B16F0 cells were implanted in B cell-deficient mice, confirming a B cell-dependent antitumor effect. Human GIFT4-licensed B cells primed cytotoxic T cells and specifically killed melanoma cells in vitro and in vivo. Taken together, our results demonstrated that GIFT4 could mediate expansion of B cells with potent antigen-specific effector function. GIFT4 may offer a novel immunotherapeutic tool and define a previously unrecognized potential for B cells in melanoma immunotherapy.
Introduction
Cytokine monotherapy for treatment of pre-established cancer has been characterized by modest clinical benefit (1). This holds true for FDA approved GM-CSF (2–4) and IL-2 (5). Nevertheless, cytokine-based cancer immunotherapy remains an active area of clinical research. Leukines such as IL-7, IL-15 and IL-21 are now being studied in early phase clinical trials (6–10). An argument can be made that pharmacological dosing of single agent cytokine is unlikely to provide meaningful immune stimulus that will overcome the multiplicity of immune escape mechanisms deployed by prevalent tumor types (11).
To address this potential limitation, we have developed the notion that physical coupling of functionally unrelated cytokines may display biochemical properties for which naturally evolved immune checkpoints deployed by tumors are lacking. As proof-of-concept, we have previously demonstrated that coupling of GM-CSF and common γ chain interleukins (aka GIFT fusokines) leads to novel biochemical properties (12, 13). When compared to parental cytokines, GIFT fusokines initiate heretofore unheralded interleukin-receptor driven STAT hyperactivation which lead to enhanced immune anti-tumor activities via induction of tumor-killer NK cells by GIFT2 (derived from GM-CSF and IL-2) (14, 15) or tumoricidal dendritic cells by GIFT21 (from GM-CSF and IL-21) (16, 17).
The notion of coupling GM-CSF to IL-4 as a fusokine is appealing since it is known that their combined use can induce monocytes to differentiate into dendritic cells (18). Indeed, many clinical dendritic cell-based cancer immunotherapy protocols utilize GM-CSF and IL-4 as a means to generate autologous antigen-presenting cells (19). Herein, we derived a novel fusokine: GIFT4 (derived from GM-CSF and IL-4) and have discovered that GIFT4 promotes an entirely novel B-cell tumoricidal immune response characterized by both B-helper and effector functions. This observation reveals the potential of both fusokine GIFT4 and GIFT4-licensed B-cells (GIFT4-B cells) as meaningful tumoricidal agents.
Materials and Methods
GIFT4 gene and protein
GM-CSF and IL-4 genes (cDNA) (Invivogen, San-Diego, CA) were cloned in frame allowing the expression of the chimeric transgenes and GIFT4 protein. The crystal structures of human GM-CSF and IL-4 were used for homology modeling of GIFT4 three-dimensional structure on the software PROSPECT v2. GIFT4-encoding retroviral plasmid was introduced into authenticated GP2-293 packaging cells from Clontech. The retroparticles were used to genetically modify 293T and B16F0 cells from American Type Culture Collection (ATCC) authenticated by short tandem repeats (STR) analysis. GIFT4-expressing positive clones were selected by single cell culture in 96-well plates. Similarly, B16F0 cells were genetically modified to stably expressing murine GM-CSF or IL-4. Cytokine expression was confirmed by ELISA.
Cell culture
GIFT4-secreting 293T, B16F0 or non-transfected cells were cultured in DMEM medium. Culture supernatant was concentrated with sterile centrifugal filter units (Millipore Corporation, Billerica, MA). Splenic B-cells from C57BL/6J (B6) mice were purified with pan B-cell enrichment kit (StemCell, Montreal, Canada). Human B-cells or T cells were purified from peripheral blood mononuclear cells (PBMC) of melanoma patients with B-cell or T-cell enrichment kits (StemCell). B-cells were cultured in complete RPMI-1640 medium in 96-well plate (105 cells/well) in presence of GIFT4, GM-CSF and IL-4 (2ng/ml) (PeproTech, Rocky Hill, NJ) for 6 days. After wash with RPMI-1640 medium, B-cells were cultured for 48 hours. The supernatants were then collected for luminex assay performed in the Human Immunology Monitoring Center at Stanford University. GIFT4- or control cytokine-treated human B-cells were co-cultured with autologous T cells (1:1) in RPMI-1640 medium for 6 days. Human A375 melanoma cells (2×104 cells/well) from ATCC authenticated by STR analysis plus gender marker were co-cultured with human T cells for 48 hours in a 96-well plate. After removal of T cells, melanoma cells were subject to MTT (3-(4,5- dimethylhiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay as described (20).
ELISA and Western Blot
GIFT4, GM-CSF, IL-4 and IFN-γ were quantified with ELISA kits (eBiosciences, San Diego, CA). GIFT4 were profiled by Western blot (WB) with anti-GM-CSF or anti-IL-4 antibodies (R&D systems). Murine B-cells (106 cells/ml) treated with GIFT4, GM-CSF and IL-4 with or without JAK inhibitors were lysed with buffer as described (21). STAT phosphorylation in B-cells was determined by WB with anti-pSTAT1 (Tyr701, D4A7), anti-pSTAT3 (Tyr705, D3A7), anti-pSTAT5 (Tyr694, D47E7), anti-pSTAT6 (Tyr641, C11A12), or anti-STAT antibodies (Cell Signaling). For immunoprecipitation of GMCSFRβ by common γc receptor after GIFT4 or GM-CSF and IL-4 stimulation, the B-cells were lysed with buffer as described (21). One mg of protein per sample was incubated with 2 mg of anti-common γc or anti-GMCSFR antibodies, or IgG isotype control (Santa Cruz Biotechnology, CA) overnight at 4°C. The sample-antibody complexes were incubated with protein A agarose for 2 hour at 4°C. The bound proteins were eluted for WB with anti-GMCSFR or anti-IL4R antibodies.
Confocal microscopy
B-cells treated with GIFT4 or GM-CSF and IL-4 (20 minutes at 37°C) were fixed with 3.7% paraformaldehyde, and stained with goat anti-mouse GMCSFRβ (R&D systems) and rabbit anti-mouse IL4R antibodies (Santa Cruz Biotechnology) overnight at 4°C. The cells were reacted with Donkey anti-rabbit Alexa 488 and Donkey anti-goat Alexa 555 (Invitrogen, Grand Island, NY) at room temperature for 2 hours, and mounted with ProLong Gold Antifade Reagent with DAPI, and imaged under a Zeiss LSM 510 Confocal microscope.
Flow cytometry
GIFT4-treated splenocytes or B-cells were profiled by flow cytometry (FACS) with antibodies against murine B220, CD3, CD19, CD22, CD23, CD25, CD27, CD40, CD69, CD80, CD83, CD86, MHCII, IgM by FACS on a BD FACSCanto II flow cytometer. For GM-CSF staining, B cells were fixed and permeabilized with BD Cytofix/Cytoperm™ solution followed by anti-GM-CSF antibody staining. Human B-cells were treated with human GIFT4, or GM-CSF and IL-4 (20ng/ml) for 6 days, and analyzed by FACS with a panel of antibodies against human B-cells. Cytotoxic T cells were profiled with anti-human CD3, CD8, CD314, INF-γ, granzyme B, granulysin antibodies (BD). FACS data were analyzed with FlowJo 9.1 software.
Melanoma mouse model
B16F0, melan-a GNAQQ209L cells (from Dr. Boris Bastian (co-inventor) at University of California, San Francisco, authenticated by detecting a mutation in a Gnaq gene by PCR), B16-GIFT4 cells, or B16-GMCSF/IL4 cells (106/mouse) were subcutaneously implanted into syngeneic B6 mice or Rag1−/−, CD4−/−, CD8−/−, μMT, IFN-γ−/− (B6.129S7-Ifngtm1Ts/J) or IL-10−/− (B6.129P2-IL10tm1Cgn/J) mice. On day 5, B6 mice pre-established with B16F0 or melan-a GNAQQ209L melanoma tumors were intravenously administered with three doses of murine GIFT4 (100ng/day/mouse) with 2-day interval. Additionally, B16-GIFT4 cells were subcutaneously immunized into B6 mice for one month. Purified splenic B-cells (10×106 cells/mouse) from immunized mice were adoptively transferred into B16F0 tumor pre-established μMT mice (n=5 per group). B16-GIFT4 or B16F0 cells (106 cells/mouse) in MatriGel solution (BD) were subcutaneously injected into B6 mice for 7 days. Tumors were harvested and digested as described (22). Infiltrating lymphocytes were isolated with Lymphocyte Separation Medium (Mediatech, Manassas, VA). For examining the anti-melanoma function of human GIFT4, human A375 melanoma cells (2×105 cells/mouse) were subcutaneously implanted into NOD-scid IL2Rgammanull (NSG) mice. On day 5, human PBMC (10×106 cells/mouse) were intravenously injected into the mice, supplemented with three doses of human GIFT4, GM-CSF and IL-4 (100ng/day/mouse) or PBS with 2-day interval. Alternatively, human B-cell primed T cells were intravenously injected into NSG mice with pre-established A375 melanoma. Tumor growth was measured with a digital caliper. All mice (6–8 weeks old) were from Jackson Laboratory (Bar Harbor, ME). The use of mice and the recruitment of melanoma patients were approved by Emory University.
Statistical analysis
Data were shown as mean ± SEM. P values were calculated using the one-way analysis of variance (ANOVA) test, or two-tailed Student t-test in Fig. S3D. P value of less than 0.05 was considered significant (*: P<0.05; **: P<0.01; *** P<0.001).
Results
The effect of GIFT4 on murine B-cells
Murine GIFT4 has a single polypeptide chain of 282 amino acids (Fig. 1A), with a predicted three-dimension structure (Fig. 1B). GIFT4 (50kDa) produced by bioengineered 293T cells was detected by WB (Fig. 1C). To test the effect of GIFT4 on resting leukocytes, splenocytes from naïve B6 mice were stimulated with murine GIFT4 or control cytokines. We observed that GIFT4 triggers the expansion of splenocytes (Fig. S1A) predominantly in the B-cell compartment (Fig. S1B and C). Working with purified splenic B-cells, we confirmed that murine GIFT4 induced the robust proliferation of B-cells (Fig. 1D–F). Luminex analysis on the culture supernatant of B-cells showed that GIFT4-B cells produced significantly higher levels of cytokines IL-1α, IL-6, IL-12 (Fig. 1G) as well as IL-5 and VEGF (Fig. S2), compared with GM-CSF and IL-4 or PBS treatments. Intriguingly, we observed that murine GIFT4 B-cells secreted massive amounts of GM-CSF and CCL3 (Fig. 1H), with undetectable IL-10 and very low levels of IFN-γ (Fig. 1G), IL-2, IP-10, IL-17, IL-13, KC, RANTES and TNFα (Fig. S2). In contrast, there was no GM-CSF and CCL3 secretion from control B-cells (Fig. 1H). Intracellular cytokine staining further confirmed the secretion of GM-CSF by GIFT4-B cells (Fig. S3A and B), which is tenfold higher than controls. Phenotyping GIFT4-B cells by FACS with a panel of antibodies against murine B-cell surface markers demonstrated that akin to GM-CSF and IL-4-treated control B-cells, GIFT4-B cells express CD19, CD22 and IgM. Distinct to control B-cells, GIFT4-B cells down-regulated CD23 and substantially up-regulated CD19, CD25, CD27, CD40, CD69, MHCI/II, CD80, CD83 and CD86 (Fig. 1I). To test the effect of GIFT4 stimulation on B-cell STAT signaling, splenic B-cells were stimulated with GIFT4 and compared to controls. We observed that GIFT4 on its own leads to hyperphosphorylation of STAT1, STAT3, STAT5 and STAT6 contemporaneously in B-cells in a manner distinct to that observed with control cytokine treatment (Fig. 2A). To test whether JAKs are involved in GIFT4-induced STAT hyperphosphorylation, B-cells were incubated with JAK inhibitors before GIFT4 stimulation. We observed that inhibition of JAK2 or JAK3 completely abrogated STAT1 phosphorylation; suppression of JAK1 or JAK3 robustly reduced the phosphorylation of STAT3, STAT5 and STAT6. Collective Inhibition of JAKs completely interrupted STAT1, STAT3, STAT5 and STAT6 signaling (Fig. 2A). Consequently, blocking JAK1, JAK2 or JAK3 with its inhibitors dramatically suppressed GIFT4-induced B-cell proliferation (Fig. 2B). To test ability of GIFT4 to cluster GMCSFR and IL4R on B-cell surface, co-immunoprecipitation was performed with anti-GMCSFR or anti-IL4R antibodies on the lysates from treated B-cells. Indeed, GMCSFR was pulled down with IL4R after GIFT4 treatment (Fig. 2C), but not after GM-CSF and IL-4 stimulation. Confocal microscopy with anti-GMCSFR and anti-IL4R antibodies (Fig. 2D–E) further confirmed the co-localization of GMCSFR and IL4R on GIFT4-treated B-cells (Fig. 2E), but not on control cytokine-treated cells (Fig. 2D).
Fig. 1. Murine GIFT4 induced robust B-cell proliferation in vitro.

(A) Murine GIFT4 protein sequence. (B) Predicted 3-dimension structure of murine GIFT4. (C) Murine GM-CSF, IL-4 or GIFT4 was blotted onto a nitrocellulose membrane and detected by WB with anti-GM-CSF or IL-4 antibodies. (D) Splenic B-cells from B6 mice were stimulated with GIFT4, or GM-CSF and IL-4 or PBS for 6 days. Cells were photographed under a microscope (10×). (E) CFSE-labeled B-cells were treated with GIFT4 (black), GM-CSF and IL-4 (gray), or PBS (white) for 6 days, and subject to FACS analysis. (F) B-cell number increase was presented as fold change based on initial number. (G and H) The culture supernatants of murine B-cells stimulated with GIFT4 (white), GM-CSF and IL-4 (gray) or PBS (black) were collected for cytokine luminex assay. Data were from three independent experiments. (I) GIFT4- (black), GM-CSF and IL-4 treated (gray) B-cells were stained with antibodies against murine B-cell markers or with antibody isotype (white), and profiled by FACS. Data represented one of the three independent experiments.
Fig. 2. GIFT4 induced hyper STAT signaling on murine B-cells.

(A) Splenic B-cells were treated with murine GIFT4, GM-CSF and IL-4 with or without inhibitors for JAK2 (TG101348), JAK3 (CP690550), or JAK1/2 (INCB018424). Ten microgram of proteins in B-cell lysate was subject to WB with anti-pSTAT1, anti-pSTAT3, anti-pSTAT5, anti-pSTAT6 or anti-STAT antibodies. (B) B-cells (105 cells/well) cultured with murine GIFT4, GM-CSF and/or IL-4 with or without JAK inhibitors for 5 days, were harvested and calculated. (C) B-cells stimulated with GIFT4, or GM-CSF and IL-4 were lysed. The lysate was incubated with anti-common γc, anti-GMCSFR antibodies or IgG isotype control, then with protein-A agarose. The bound proteins were eluted and subject to WB with anti-GMCSFR or IL4R antibodies. (D and E) B-cells stimulated with GM-CSF and IL-4 (D), or GIFT4 (E) were fixed with paraformaldehyde and stained with goat anti-GMCSFRβ and rabbit anti-IL-4R antibodies, then with anti-rabbit Alexa 488 and anti-goat Alexa 555 secondary antibodies. After mounted, the cells were imaged under a confocal microscope (63×). All the data are representatives from two or three independent experiments.
GIFT4 robustly expands endogenous B-cells and promotes anti-tumor immunity
B-cells play important roles in host immunity against pathogens (23) and tumors (24, 25). We therefore investigated whether the hypermorph effects of GIFT4 on B-cells leads to an enhanced B-cell driven immune response. To test the immune effect of murine GIFT4 in vivo, GIFT4 or GM-CSF and IL-4 were intravenously administered into B6 mice. After one week, GIFT4-treated mice developed splenomegaly (Fig. 3A) in comparison with GM-CSF and IL-4- or PBS-treated mice, with marked expansion of splenocytes (Fig. 3B). Mice treated with GM-CSF and IL-4 showed normal sized spleens as well as the number of splenocytes (Fig. 3A and B). Profiling B220+ cells and CD3+ cells demonstrated that there was a threefold expansion of splenic B-cells in GIFT4-treated mice (Fig. 3C), and modest but significant expansion of T cells (Fig. 3C) in comparison with GM-CSF and IL-4- or PBS treated groups. Intracellular staining further confirmed the induction of GM-CSF-secreting B-cells by GIFT4 (Fig. S3C and D), akin to the recently-described innate response activator (IRA) B-cells (23). The percentage of GM-CSF+ B-cells in GIFT4-treated mice is more than 30 fold higher when compared to GM-CSF and IL-4-treated controls (Fig. S3C and D) and the latter is similar to untreated mice. To test whether GIFT4 protein promotes a therapeutic anti-tumor response in vivo, we implanted B16F0 melanoma cells into B6 mice and five days later treated mice with GIFT4, GM-CSF and IL-4, or PBS. We observed significant suppression of melanoma growth in GIFT4-treated mice (Fig. 3D). As an independent validation of anti-melanoma activity, we implanted B6 mice with syngeneic melan-a cells stably transduced with GNAQQ209L as described (26). In comparison with GM-CSG and IL-4 or PBS treatments, GIFT4 significantly inhibited melan-a GNAQQ209L tumor growth (Fig. 3E).
Fig. 3. Murine GIFT4 induced B-cell expansion and inhibited melanoma tumor growth in vivo.
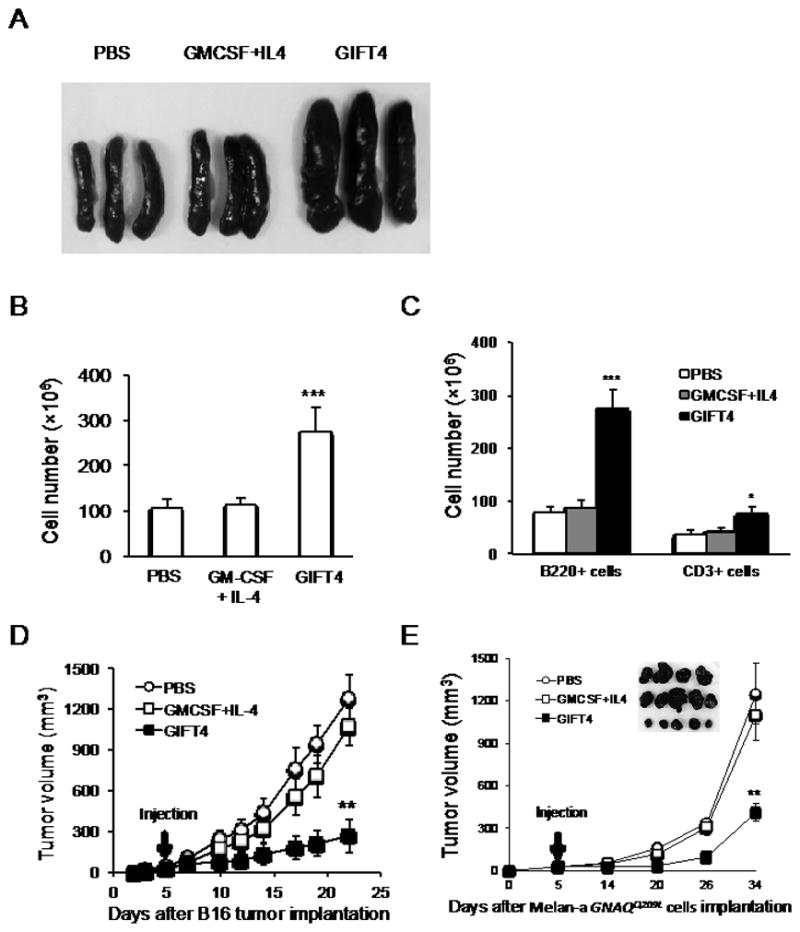
(A) B6 mice were injected with GIFT4, GM-CSF and IL-4, or PBS for 6 days. On day 7, spleens were harvested for photography. (B) Splenocytes were isolated and counted with an automated counter. (C) Splenic B-cells and T cells were profiled by FACS with anti-B220 and anti-CD3 antibodies. Total B-cells or T cells were calculated. B16F0 (D) or GNAQ (E) Melan-a GNAQQ209L cells (106 cells/mouse) were subcutaneously implanted into syngeneic B6 mice. On day 5, the mice were injected with three doses of murine GIFT4, GM-CSF and IL-4, or PBS with 2-day interval. Tumor size was measured. Data are represented from three independent experiments.
GIFT4-triggered anti-tumor immunity is B-cell dependent
To further test the anti-tumor function of GIFT4, we generated genetically modified B16F0 melanoma cell lines stably expressing murine GIFT4, GM-CSF or IL-4 cytokine. The tumor cells were implanted into B6 mice. Twenty days later, mice implanted with wild type B16F0 cells or with an admixture of B16F0-GMCSF and B16F0-IL4 cells developed melanoma tumors (Fig. 4A). However, tumor growth was substantially inhibited in mice implanted with B16-GIFT4 cells, indicating GIFT4 expression significantly suppressed melanoma tumor growth (Fig. 4A). To analyze whether GIFT4 expression affects the infiltration/expansion of B or T lymphocytes in tumor microenvironment, B16-GIFT4 cells, B16-GMCSF+IL4 cells, or B16F0 cells were embedded in Matrigel and implanted into B6 mice. One-week later, matrigel implants were surgically excised, enzymatically dissociated and cellular content analyzed by FACS. Consistent with GIFT4-induced splenic B-cell expansion, GIFT4 expression by B16F0 cells resulted in the robust increase of B-cell and T cell number in tumor microenvironment (Fig. 4B). To further determine the role of B-cells and T cells in GIFT4-triggered anti-melanoma immunity, we implanted B16-GIFT4 cells in Rag1−/− mice that lack functional B and T cells. We observed that B16-GIFT4 tumor grew quickly in Rag1−/− mice (Fig. 4C) confirming the pivotal role of lymphoid cells in anti-tumor effect of GIFT4. To test the hypothesis that B-cells play a pivotal role in GIFT4-triggered anti-tumor response, we implanted B16-GIFT4 cells into B-cell deficient μMT mice and observed a robust growth of melanoma tumors (Fig. 4C). To further test whether GIFT4-B cells can serve as T cell helpers and promote anti-tumor immunity, T cells were co-cultured with GIFT4-B cells or control B-cells. Quantification of IFN-γ secretion in the culture supernatant by ELISA showed that GIFT4-B cells markedly enhanced T cell IFN-γ production (Fig. S4), while GM-CSF and IL-4-treated B-cells has little effect (Fig. S4). Using knockout mice, we confirmed that mice deficient in IFN-γ, but not IL-10, could not suppress melanoma growth (Fig. 4D). To further define the relative contribution of T cell subsets in GIFT4-driven anti-melanoma response, we implanted B16-GIFT4 cells in CD4 or CD8 T cell deficient mice (Fig. 4E) and observed an absolute loss of melanoma tumoricidal activity. To investigate whether GIFT4-B cells from B16-GIFT4 cell-immunized mice could rescue anti-tumor immunity in B-cell deficient mice, we performed adoptive transfer experiments. We implanted B16-GIFT4 cells into μMT mice and when mice developed palpable melanoma tumors, splenic B-cells purified from immunized B6 mice were adoptively transferred. Measurement of tumor size showed that adoptive transfer of GIFT4-B cells from immunized mice significantly inhibited tumor growth in μMT mice (Fig. 4F). We were unable to detect acquisition of anti-GIFT4 antibodies in serum of B16-GIFT4 cell-immunized mice as well as GIFT4-treated B6 mice (Fig. S5).
Fig. 4. Murine GIFT4 elicited B-cell dependent tumoricidal activity.

(A) Murine B16F0 cells (B16), GIFT4-expressing B16F0 cells (B16-GIFT4 cells), or GMCSF-producing plus IL-4-secreting B16F0 cells (B16-GMCSF+IL4) were subcutaneously implanted into B6 mice. (B) B16-GIFT4, B16-GMCSF+IL4, or B16 cells (106 cells/mouse) in MatriGel solution were implanted into B6 mice. On day 7, the melanoma tumors were harvested. The infiltrated lymphocytes isolated from tumors were profiled by FACS with anti-murine B220 and CD3 antibodies. B220+ B-cells or CD3+ T cells were counted. (C–E) B16-GIFT4 cells were implanted into Rag1−/−, μMT mice, naive B6 mice (C); or IFN-γ−/−, IL-10−/− mice (D), or CD4, CD8 T cell-deficient mice (E). (F) μMT mice were implanted with B16-GIFT4 cells. On day 5, the mice were adoptively transferred with B-cells (107 cells/mouse) isolated from B16-GIFT4 immunized mice. Mice without B-cell transfer served as control. Tumor size was measured. Five mice were in each group. Data were represented from three independent experiments.
Human ortholog of GIFT4 induces B-effector immune response
To test whether human GIFT4 deploys similar B-cell stimulatory anti-melanoma function as murine GIFT4, we cloned human GIFT4 cDNA and genetically modified 293T cells. Human GIFT4 protein sequence is showed in Fig. 5A, and intact protein expression was confirmed by WB (Fig. 5B). To examine the effect of human GIFT4 on B-cells, we purified B-cells from PBMC isolated from melanoma patients (n=6). Treatment of B-cells with human GIFT4 revealed that GIFT4 induced robust proliferation of peripheral B-cells (Fig. 5C and D). Phenotyping by FACS showed that human GIFT4-B cells are CD20+, CD22+, CD80+, CD86+, MHCI+ and MHCII+, with enhanced expression of CD27, CD40 and CD83; but are absent of CD23 and CD138 (Fig. 5E). Profile of the secretome with luminex assay uncovered that human GIFT4-B cells from melanoma patients produced substantial amount of IL-2, IL-6, CCL3, CCL4, ICAM1 and GM-CSF (Fig. 5F).
Fig. 5. Human GIFT4 reprogramed human B-cells into antigen-presenting cells.

(A) Amino acids of human GIFT4, the underlined are signal peptides. (B) Human GIFT4 protein (55kDa) produced by bio-engineered 293T cells was detected by WB with anti-GM-CSF or IL-4 antibodies. There is no GIFT4 detected in 293T cell culture medium. (C) B-cells purified from PBMC of melanoma patients were stimulated with human GIFT4 or GM-CSF and IL-4 for 6 days. Cells were photographed under a microscope (10×). Alternatively, CFSE-labeled B-cells were treated with human GIFT4 (black), GM-CSF and IL-4 (gray), or PBS (White), and subject to FACS analysis. Division cycles were presented as individual peaks. (D) B-cell number change was calculated as fold change based on the initial number. (E) GIFT4- (black), GM-CSF and IL-4 treated (gray) B-cells (from melanoma patients) were stained with a panel of B-cell antibodies, or isotype controls (White), and profiled by FACS. (F) Human B-cells (106 cells/ml) from melanoma patients were treated with human GIFT4 (White), or GM-CSF and IL-4 (black) for 6 days. After wash, the B-cells were cultured in RPMI-1640 medium for additional 48 hours. Cytokines and chemokines in the culture supernatant were quantified with luminex assay. Data are represented from one of three repeated experiments.
Human GIFT4-B cells triggers melanoma-killing T cell response
To test the translational property of human GIFT4-B cells as T cell stimulators, we co-cultured GIFT4-B-cells, or GM-CSF and IL-4 treated B-cells with autologous CFSE-labeled T cells isolated from melanoma patient’s PBMC (n=8). Analysis of T cell proliferation by FACS demonstrated that GIFT4-B cells, but not control B-cells, promoted the proliferation of co-cultured T cells (Fig. 6A); the majority of those cells were CD8 T cells (Fig. 6B). To examine whether those T cells are cytotoxic cells, we performed surface and intracellular staining with anti-human NKG2D (CD314), granzyme B, granulysin and IFN-γ. FACS analysis showed that T cells primed by GIFT4-B cells mainly expressed tumor-killing NKG2D molecule (Fig. 6C), and produced granzyme B, granulysin and IFN-γ (Fig. 6D and E). To test the cytotoxicity of T cells, A375 melanoma cells or human umbilical vascular endothelial cells (HUVEC) were co-cultured with T cells (from melanoma patients) primed by GIFT4-B cells (GIFT4 T cells) or T cells primed with GM-CSF and IL-4 treated B-cells (GMCSF/IL-4 T cells). We have observed that GIFT4 T cells killed the majority of melanoma cells within 48-hour co-culture, without affecting the growth of HUVEC; in contrast, GMCSF/IL-4 T cells had little killing effect on melanoma cells (Fig. 6F). To test the hypothesis that human GIFT4 has similar anti-melanoma effect in vivo as murine GIFT4, we implanted A375 cells into NSG mice. On day 5, ten millions of PBMC from melanoma patients were delivered into the mice, supplemented with injection of human GIFT4, or GM-CSF and IL-4. In comparison with GM-CSF and IL-4 treatment or control group without PBMC, GIFT4 administration significantly reduced human melanoma growth in mice (Fig. 6G). To test if the GIFT4 T cells could kill human tumor cells in vivo, A375 melanoma cells were implanted into NSG mice, supplemented with an intravenous injection of GIFT4 T cells or GMCSF/IL4 T cells. Monitoring human melanoma growth demonstrated that adoptive transfer of GIFT4 T cells could directly kill human melanoma cells in the mice (Fig. 6H). However, the mice developed large human melanoma tumors on day 30 when delivered with GMCSF/IL4 T cells or without T cells (Fig 6H). Using B-cells and T cells from healthy donors, we found GIFT4-licensed B-cells and primed T cells have similar anti-melanoma functions to the ones from melanoma patients (Fig. S6A–F).
Fig. 6. Human GIFT4-B cells primed anti-tumor T cell response.

(A) CFSE-labeled T cells from melanoma patients were co-cultured with GIFT4-B cells (black), or GM-CSF and IL-4 treated (gray), or PBS-treated B-cells (white) for 6 days. The cells were subject to FACS analysis. (B–E) Profile of GIFT4 T cells by FACS with anti-CD3 and CD8 (B), CD314 (NKG2D) (C), granzyme B (D) and granulysin antibodies (E). (F) Human A375 melanoma cells or HUVEC (ATCC CRL-1730) were co-cultured with GIFT4 T cells or GMCSG/IL4 T cells at 1:1 or 3:1 ratios (T cells: A375 or HUVEC) for 48 hours. After removal of T cells, the melanoma cells were subject to MTT assay. Data were from three independent experiments. (G) Human A375 cells were implanted into NSG mice. On day 5, the mice were injected with 107 PBMC (from patients), supplemented with injection of human GIFT4 or GM-CSF and IL-4. Mice without PBMC served as control. (H) Alternatively, NSG mice implanted with A375 cells were transferred with GIFT4 T cells or GMCSF/IL4 T cells (2×106 cells/mouse), or without T cells. Tumor size was measured. Five mice were per group. Data were represented from three independent experiments.
DISCUSSION
B-cells are a population of heterogeneous lymphocytes where effector and suppressor subsets cohabitate (27, 28). B-cells can promote tumor growth (29) by inhibiting T cell tumoricidal activities (30), (31). Conversely, B-cells can secrete anti-tumor cytokines (25) and facilitate T cell anti-tumor function (32), (33). Virtually all studies here cited examining B-cell effector function in anti-tumor immunity examined the biology of primeval B-cells bereft of pharmacological activation. Herein lays the novelty of GIFT4 as a B-cell tropic immune stimulator. Distinct from IL-4 and GM-CSF, GIFT4 fusokine deploys a gain-of-function hyperphosphorylation of STATs through assembly of GMCSFR and IL4R on B-cell surface. The consequences of this pan STAT activation on B-cells are protean, including a robust mitogenic response and phenotypic conversion to a unique B-effector phenotype characterized by expression of B220, CD19, CD40, CD80, CD83, CD86, and IgM, but not CD23. Furthermore, GIFT4-B cells generate a unique secretome distinct from other known B-effector phenotypes described in mice, including: IL-1α, IL-6, IL-12, IL-5, VEGF, GM-CSF and CCL3. There is no report that STAT phosphorylation results in CCL3 secretion by normal B-cells; however, BCR stimulation can activate leukemic B-cells to produce CCL3 (34). GIFT4-B cells display functional feature reminiscent of IRA-B (23), producing GM-CSF. Unlike IRA-B cells, GIFT4-B cells are CD40+CD80+CD86+ and produce little IL-3. GIFT4-B cells are also distinct from B1 and B2 cells, of which B1 cells are B220low, and B2 cells are CD23+ (35). IL-12 has anti-tumor capacity to reconstitute the immune cellular microenvironment within tumors (36), and that produced by GIFT4-B cells may play a similar role. To our knowledge, it is the first report that activated normal B-cells can produce chemokine CCL3. Therefore, GIFT4-B cells have a distinct cytokine-secreting profile that can enhance T cell anti-tumor immunity.
Interestingly, GIFT4 expands B-cells including GM-CSF-secreting IRA-like B-cells. GM-CSF has been shown to increase the proliferation of antigen-activated cytotoxic T cells (37); and CCL3 is critical for recruiting CCR5+ T cells in anti-tumor immunity (38), suggesting that GM-CSF and CCL3-producing GIFT4-B cells may provide a direct helper effect towards T cell immunity against tumors. Since activated CD80+CD86+ B-cells are potent antigen-presenting cells that can generate autologous antigen-specific cytotoxic T cells against melanoma (39, 40), this feature may also be operative in GIFT4-B cells. The sum of these features may provide a mechanistic rationale for B-cell dependent anti-melanoma-driven effect of GIFT4. Indeed, we can map out a cascade of interaction where GIFT4-B cells license T cells to markedly increase IFN-γ production, cytotoxic factors granzyme B and granulysin secretion, and NKG2D/CD314 expression.
The human ortholog of GIFT4 behaves similarly to murine GIFT4 and functions as an immune stimulator to induce the proliferation of human peripheral B-cells. Like murine GIFT4-B cells, human GIFT4-B cells bear the typical surface co-stimulatory markers such as CD80, CD86, MHC I, MHC II; and enhance autologous T cell activation, proliferation and IFN-γ production. In addition, both murine and human GIFT4-B cells secrete common cytokines and chemokines such as IL-6, GM-CSF and CCL3. However, human GIFT4-B cells have a unique secretome profile, producing substantial amount of T cell-supportive cytokine IL-2 and driving a CD8 T cell response. B-cells can enhance granzyme-B+ cytotoxic CD8 T cell survival (41), and tolerize CD8 T cells (40, 42). We found that human GIFT4-B cells trigger an anti-tumor immune response by cytotoxic CD8 T cells derived from melanoma patients. Those GIFT4-B cell primed T cells express tumor-cytotoxic factors, and can directly kill human melanoma cells. The unique property of GIFT4-B cells and GIFT4-B cell primed autologous T cells from melanoma patients provides a potential novel strategy for personalized cell immunotherapeutic against melanoma.
In summary, our data demonstrate that GIFT4 fusokine deploys a unique STAT activation response in naive B-cells which converts them to helpers of T-cell activity as well tumoricidal effectors via their immune-stimulatory effects. These observations support the notion that chimeric cytokines such as the fusokine GIFT4 here described can lead to a hypermorph phenotype and function of B-cells and pharmacological utility in ailments amenable to an enhanced immune response.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by NIH (5R01AI093881) and Georgia Cancer Coalition (J. Galipeau); the Winship Robbins Scholar Award and the Winship Melanoma Research Fund (J. Deng).
We thank Julie Leff, RN for collecting blood samples from melanoma patients, Dr. Edmund Waller for providing IFN-γ−/− and IL-10−/− mice, Dr. Brian Pollack for preparing GNAQ cells, the Winship Integrated Cellular Imaging Microscopy Core for confocal imaging.
Footnotes
Conflicts of Interest: No
References
- 1.Atkins MB. Cytokine-based therapy and biochemotherapy for advanced melanoma. Clin Cancer Res. 2006;12:2353s–8s. doi: 10.1158/1078-0432.CCR-05-2503. [DOI] [PubMed] [Google Scholar]
- 2.Metcalf D. The colony-stimulating factors and cancer. Nat Rev Cancer. 2010;10:425–34. doi: 10.1038/nrc2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta R, Emens LA. GM-CSF-secreting vaccines for solid tumors: moving forward. Discov Med. 2010;10:52–60. [PMC free article] [PubMed] [Google Scholar]
- 4.Waller EK. The role of sargramostim (rhGM-CSF) as immunotherapy. Oncologist. 2007;12 (Suppl 2):22–6. doi: 10.1634/theoncologist.12-S2-22. [DOI] [PubMed] [Google Scholar]
- 5.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–27. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fewkes NM, Mackall CL. Novel gamma-chain cytokines as candidate immune modulators in immune therapies for cancer. Cancer J. 2010;16:392–8. doi: 10.1097/PPO.0b013e3181eacbc4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson JA, Curti BD, Redman BG, Bhatia S, Weber JS, Agarwala SS, et al. Phase I study of recombinant interleukin-21 in patients with metastatic melanoma and renal cell carcinoma. J Clin Oncol. 2008;26:2034–9. doi: 10.1200/JCO.2007.14.5193. [DOI] [PubMed] [Google Scholar]
- 8.Davis ID, Brady B, Kefford RF, Millward M, Cebon J, Skrumsager BK, et al. Clinical and biological efficacy of recombinant human interleukin-21 in patients with stage IV malignant melanoma without prior treatment: a phase IIa trial. Clin Cancer Res. 2009;15:2123–9. doi: 10.1158/1078-0432.CCR-08-2663. [DOI] [PubMed] [Google Scholar]
- 9.Sportes C, Babb RR, Krumlauf MC, Hakim FT, Steinberg SM, Chow CK, et al. Phase I study of recombinant human interleukin-7 administration in subjects with refractory malignancy. Clin Cancer Res. 2010;16:727–35. doi: 10.1158/1078-0432.CCR-09-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teague RM, Sather BD, Sacks JA, Huang MZ, Dossett ML, Morimoto J, et al. Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumors. Nat Med. 2006;12:335–41. doi: 10.1038/nm1359. [DOI] [PubMed] [Google Scholar]
- 11.Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol. 2011;23:286–92. doi: 10.1016/j.coi.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams P, Galipeau J. GM-CSF-based fusion cytokines as ligands for immune modulation. J Immunol. 2011 May 15;186(10):5527–32. doi: 10.4049/jimmunol.1003699. [DOI] [PubMed] [Google Scholar]
- 13.Rafei M, Hsieh J, Zehntner S, Li M, Forner K, Birman E, et al. A granulocyte-macrophage colony-stimulating factor and interleukin-15 fusokine induces a regulatory B cell population with immune suppressive properties. Nat Med. 2009;15:1038–45. doi: 10.1038/nm.2003. [DOI] [PubMed] [Google Scholar]
- 14.Stagg J, Wu JH, Bouganim N, Galipeau J. Granulocyte-macrophage colony-stimulating factor and interleukin-2 fusion cDNA for cancer gene immunotherapy. Cancer Res. 2004;64:8795–9. doi: 10.1158/0008-5472.CAN-04-1776. [DOI] [PubMed] [Google Scholar]
- 15.Penafuerte C, Bautista-Lopez N, Boulassel MR, Routy JP, Galipeau J. The human ortholog of granulocyte macrophage colony-stimulating factor and interleukin-2 fusion protein induces potent ex vivo natural killer cell activation and maturation. Cancer Res. 2009;69:9020–8. doi: 10.1158/0008-5472.CAN-09-2322. [DOI] [PubMed] [Google Scholar]
- 16.Williams P, Bouchentouf M, Rafei M, Romieu-Mourez R, Hsieh J, Boivin MN, et al. A dendritic cell population generated by a fusion of GM-CSF and IL-21 induces tumor-antigen-specific immunity. J Immunol. 2010;185:7358–66. doi: 10.4049/jimmunol.1002201. [DOI] [PubMed] [Google Scholar]
- 17.Williams P, Rafei M, Bouchentouf M, Raven J, Yuan S, Cuerquis J, et al. A fusion of GMCSF and IL-21 initiates hypersignaling through the IL-21Ralpha chain with immune activating and tumoricidal effects in vivo. Mol Ther. 2010;18:1293–301. doi: 10.1038/mt.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gluckman JC, Canque B, Chapuis F, Rosenzwajg M. In vitro generation of human dendritic cells and cell therapy. Cytokines Cell Mol Ther. 1997;3:187–96. [PubMed] [Google Scholar]
- 19.Vopenkova K, Mollova K, Buresova I, Michalek J. Complex evaluation of human monocyte-derived dendritic cells for cancer immunotherapy. J Cell Mol Med. 2012;16:2827–37. doi: 10.1111/j.1582-4934.2012.01614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rafei M, Deng J, Boivin MN, Williams P, Matulis SM, Yuan S, et al. A MCP1 fusokine with CCR2-specific tumoricidal activity. Mol Cancer. 2011;10:121. doi: 10.1186/1476-4598-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li P, Yuan S, Galipeau J. A fusion cytokine coupling GMCSF to IL9 induces heterologous receptor clustering and STAT1 hyperactivation through JAK2 promiscuity. PLoS One. 2013;8:e69405. doi: 10.1371/journal.pone.0069405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stagg J, Wu JH, Bouganim N, Galipeau J. Granulocyte-macrophage colony-stimulating factor and interleukin-2 fusion cDNA for cancer gene immunotherapy. Cancer Res. 2004;64:8795–9. doi: 10.1158/0008-5472.CAN-04-1776. [DOI] [PubMed] [Google Scholar]
- 23.Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335:597–601. doi: 10.1126/science.1215173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson BH. CD20+ B cells: the other tumor-infiltrating lymphocytes. J Immunol. 2010;185:4977–82. doi: 10.4049/jimmunol.1001323. [DOI] [PubMed] [Google Scholar]
- 25.Penafuerte C, Ng S, Bautista-Lopez N, Birman E, Forner K, Galipeau J. B effector cells activated by a chimeric protein consisting of IL-2 and the ectodomain of TGF-beta receptor II induce potent antitumor immunity. Cancer Res. 2012;72:1210–20. doi: 10.1158/0008-5472.CAN-11-1659. [DOI] [PubMed] [Google Scholar]
- 26.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lampropoulou V, Shen P, Hilgenberg E, Ries S, Opitz C, Fillatreau S. Functional interactions between B lymphocytes and the innate immune system. Infect Disord Drug Targets. 2012;12:191–9. doi: 10.2174/187152612800564374. [DOI] [PubMed] [Google Scholar]
- 28.Anolik JH, Looney RJ, Lund FE, Randall TD, Sanz I. Insights into the heterogeneity of human B cells: diverse functions, roles in autoimmunity, and use as therapeutic targets. Immunol Res. 2009;45:144–58. doi: 10.1007/s12026-009-8096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Fridlender ZG, Dunn R, Kehry MR, Kapoor V, Blouin A, et al. B-cell depletion using an anti-CD20 antibody augments antitumor immune responses and immunotherapy in nonhematopoetic murine tumor models. J Immunother. 2008;31:446–57. doi: 10.1097/CJI.0b013e31816d1d6a. [DOI] [PubMed] [Google Scholar]
- 30.Qin Z, Richter G, Schuler T, Ibe S, Cao X, Blankenstein T. B cells inhibit induction of T cell-dependent tumor immunity. Nat Med. 1998;4:627–30. doi: 10.1038/nm0598-627. [DOI] [PubMed] [Google Scholar]
- 31.Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006;66:7741–7. doi: 10.1158/0008-5472.CAN-05-3766. [DOI] [PubMed] [Google Scholar]
- 32.Chung Y, Kim BS, Kim YJ, Ko HJ, Ko SY, Kim DH, Kang CY. CD1d-restricted T cells license B cells to generate long-lasting cytotoxic antitumor immunity in vivo. Cancer Res. 2006;66:6843–50. doi: 10.1158/0008-5472.CAN-06-0889. [DOI] [PubMed] [Google Scholar]
- 33.Tomihara K, Shin T, Hurez VJ, Yagita H, Pardoll DM, Zhang B, et al. Aging-associated B7-DC+ B cells enhance anti-tumor immunity via Th1 and Th17 induction. Aging Cell. 2012;11:128–38. doi: 10.1111/j.1474-9726.2011.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyauchi K, Urano E, Yoshiyama H, Komano J. Cytokine signatures of transformed B cells with distinct Epstein-Barr virus latencies as a potential diagnostic tool for B cell lymphoma. Cancer Sci. 2011;102:1236–41. doi: 10.1111/j.1349-7006.2011.01924.x. [DOI] [PubMed] [Google Scholar]
- 35.Oliveira FL, Aguiar AM, Borojevic R, El-Cheikh MC. IgE expression on the surface of B1 and B2 lymphocytes in experimental murine schistosomiasis. Braz J Med Biol Res. 2005;38:1033–42. doi: 10.1590/s0100-879x2005000700006. [DOI] [PubMed] [Google Scholar]
- 36.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121:4746–57. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selleri S, Deola S, Pos Z, Jin P, Worschech A, Slezak SL, et al. GM-CSF/IL-3/IL-5 receptor common beta chain (CD131) expression as a biomarker of antigen-stimulated CD8+ T cells. J Transl Med. 2008;6:17. doi: 10.1186/1479-5876-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez-Martin A, Gomez L, Lustgarten J, Mira E, Manes S. Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4(+) and CD8(+) T cells. Cancer Res. 2011;71:5455–66. doi: 10.1158/0008-5472.CAN-11-1687. [DOI] [PubMed] [Google Scholar]
- 39.Schultze JL, Michalak S, Seamon MJ, Dranoff G, Jung K, Daley J, et al. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest. 1997;100:2757–65. doi: 10.1172/JCI119822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hollsberg P, Batra V, Dressel A, Hafler DA. Induction of anergy in CD8 T cells by B cell presentation of antigen. J Immunol. 1996;157:5269–76. [PubMed] [Google Scholar]
- 41.Brodie GM, Wallberg M, Santamaria P, Wong FS, Green EA. B-cells promote intra-islet CD8+ cytotoxic T-cell survival to enhance type 1 diabetes. Diabetes. 2008;57:909–17. doi: 10.2337/db07-1256. [DOI] [PubMed] [Google Scholar]
- 42.Bennett SR, Carbone FR, Toy T, Miller JF, Heath WR. B cells directly tolerize CD8(+) T cells. J Exp Med. 1998;188:1977–83. doi: 10.1084/jem.188.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.