

Abstract
Purpose
Tubular secretion of cisplatin is abolished in mice deficient for the organic cation transporters Oct1 and Oct2 [Oct1/2(−/−) mice], and these animals are protected from severe cisplatin-induced kidney damage. Since tubular necrosis is not completely absent in Oct1/2(−/−) mice, we hypothesized that alternate pathways are involved in the observed injury.
Experimental Design
Studies were done in wildtype, Oct1/2(−/−), or p53-deficient animals, all on an FVB background, receiving i.p. cisplatin at 15 mg/kg. The cisplatin metabolites were analyzed using mass spectrometry, and gene expression was assessed using Affymetrix microarrays and RT-PCR arrays.
Results
KEGG pathway analyses on kidneys from mice exposed to cisplatin revealed that most significantly altered genes were associated with the p53 signaling network, including Cdnk1a and Mdm2, in both wildtype (P=2.40×10–11) and Oct1/2(−/−) mice (P=1.92×10-8). This was confirmed by demonstrating that homozygosity for a p53-null allele partially reduced renal tubular damage, while loss of p53 in Oct1/2(−/−) mice [p53(−/−)/Oct1/2(−/−)] completely abolished nephrotoxicity. We found that pifithrin-α, an inhibitor of p53-dependent transcriptional activation, inhibits Oct2 and can mimic the lack of nephrotoxicity observed in p53(−/−)/Oct1/2(−/−) mice.
Conclusions
These findings indicate that (i) the p53 pathway plays a crucial role in the kidney in response to cisplatin treatment and (ii) clinical exploration of OCT2 inhibitors may not lead to complete nephroprotection unless the p53 pathway is simultaneously antagonized.
Keywords: Cisplatin, OCT2, p53, pifithrin-α, nephrotoxicity
Introduction
Cisplatin is among the most widely used chemotherapeutic agents for the treatment of various cancer types. Despite its extensive clinical use, therapy with cisplatin is limited by the onset of several detrimental side effects, of which the responsible mechanisms remain poorly understood. Kidney damage to the S3 segment of the renal proximal tubules is of particular concern, since it affects more than 40% of patients, and this is considered the major dose-limiting toxicity associated with cisplatin treatment (1). Current evidence suggest that the organic cation transporter 2 (OCT2), encoded by the solute carrier gene SLC22A2, facilitates the renal secretion of cisplatin and various other, structurally diverse compounds, at the basolateral membrane of the proximal tubules (2–6). This is borne out by data indicating that the urinary excretion of cisplatin is drastically reduced in mice lacking the murine ortholog transporters Oct1 and Oct2 [Oct1/2(−/−)] compared to wildtype animals, and that these animals are simultaneously resistant to severe cisplatin-induced renal tubular necrosis (7, 8). Similarly, patients carrying a genetic variant of OCT2 that is associated with reduced function appear to be protected from cisplatin-induced nephrotoxicity (7, 9, 10). On the basis of these findings, strategies involving the use of OCT2 inhibitors, such as cimetidine, in combination with cisplatin-based therapy have been explored, and have provided promising results both in vitro and in vivo (10–13).
Although the severity of cisplatin-induced nephrotoxicity is reduced following genetic or pharmacological knockout of Oct1 and Oct2, renal tubular damage is not completely abolished. This suggests the existence of a secondary pathway that contributes, independently of Oct1/Oct2-mediated renal tubular drug uptake, to cisplatin-induced renal damage. A proper understanding of additional mechanisms contributing to cisplatin-nephrotoxicity in Oct1/2(−/−) mice is of fundamental relevance to the discovery and development of translational strategies that are completely renoprotective and in turn improve treatment outcome. The aim of the current study was to identify common genetic factors contributing to cisplatin-induced kidney damage in both wildtype mice and Oct1/2(−/−) mice. We further sought to assess whether inhibition of the identified multiple molecular regulators of renal tubular damage could completely abolish cisplatin-induced nephrotoxicity.
Materials and Methods
Animal experiments
Male adult wildtype mice (8–12 weeks old), and sex- and age-matched mice with a deficiency of Oct1 and Oct2 [Oct1/2(−/−)], or p53 [p53(−/−)], all on an FVB background strain, were obtained from Taconic and bred in-house. The p53(−/−) mice were cross-bred with Oct1/2(−/−) mice to generate mice deficient of p53, Oct1 and Oct2 [p53(−/−)/Oct1/2(−/−)].Animals were housed in a temperature-controlled environment with a 12-hour light cycle and given a standard diet and water ad libitum. All animals were housed and handled in accordance with the Institutional Animal Care and Use Committee of St. Jude Children's Research Hospital or approved by a governmental committee overseeing animal welfare at the University of Münster and performed in accordance with national animal protection laws.
For experiments involving collection of urine, animals were single-housed in metabolic cages in a temperature-controlled environment with a 12-hour reverse light cycle. Following acclimation, a 24-hour baseline urine sample was collected, after which mice received administration of cisplatin (15 mg/kg, i.p.) and urine was collected for up to 72 hours. Platinum concentrations were determined by a validated assay based on flameless atomic absorption spectrometry (7).
Quantitative determination of GSH1 and GSH2
Frozen urine samples were thawed at room temperature, centrifuged at 16,000×g for 10 min and 20 µl of supernatant was injected for analysis. Concentrations of the glutathione-monoplatinum conjugate (GSH2) and glutathione-diplatinum conjugate (GSH1) were assessed with a Waters 2692 separation system and Micromass Quattro LC triple-quadrupole system. Separation was achieved on a Synergi 4 µm Polar-RP 80A column (150×4.6 mm using a column heater operating at 40°C with a Waters X-Terra RP18 guard column (3.5 µm, 10 × 2.1 mm). The gradient mobile phase was composed of 0.1% formic acid in acetonitrile and 10 mM ammonium acetate in water (pH 3.8). The flow rate was 0.9 ml/min and the separation was completed within 10 min. The retention times for GSH1 and GSH2 were 4.95 min and 5.05 min, respectively. The mass spectrometer was operated in the positive mode, and controlled by Masslynx 4.1 software. The analysis was performed in MRM mode and the following mass ions (m/z) were used for detection: m/z 835>778.1 for GSH1 and m/z 572>554 for GSH2. The MS/MS conditions were as follows: capillary voltage: 3.5 kV; cone voltage: 45 V; source temperature: 130 °C; desolvation temperature: 350 °C; cone gas flow: 10 L/h; desolvation gas flow: 900 L/h and collision energy: 15 V.
Gene expression analysis
Kidneys from wildtype or Oct1/2(−/−) mice were harvested either before or 72 hours post treatment with cisplatin, after which RNA was isolated for gene expression using the RNeasy mini kit (Qiagen). Gene expression was analyzed using the Mouse 430v2 .0 GeneChip array (Affymetrix) and significance analysis of microarrays (SAM), using the criteria of a false-discovery rate of 5%, to identify altered expression with an average fold change of ≥ 2.0. Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to interrogate potentially affected pathways. Confirmation of alterations in the p53 pathway was assessed using the Mouse p53 Signaling Pathway RT2 Profiles PCR array system (SABiosciences), and evaluation of transporter gene expression was done using the Mouse Transporter RT2 Profiles PCR array system (SABiosciences). Relative gene expression was determined using the ΔΔCt method, and normalized to a housekeeping gene (Gapdh or PPC).
Evaluation of nephrotoxicity
Mice received a single dose of saline, cisplatin (15 mg/kg, i.p.), or a combination of cisplatin and pifithrin-α (2.2 mg/kg, i.p.; Enzo Life Sciences). Pifithrin-α was solubilized in 1% ethanol to circumvent the protective effects observed when cisplatin is administered in the presence of its commonly used solvent, DMSO (14). After 72 hours, kidneys were collected, fixed overnight in 10% neutral-buffered formalin, embedded in paraffin, sectioned (4 µm), and stained with hematoxylin and eosin (Lab Vision). Microscopic evaluation of nephrotoxicity was done independently by an experienced veterinary pathologist who was blinded to the treatment of the animals, as described (7). Nephrotoxicity was assessed from the percentage of observed damaged tubules and graded on a five-point scale as 0 (<10%; absent), 1 (11–25%; minimal), 2 (26–50%; mild), 3 (51–75%; moderate), and 4 (>75%; severe). BUN and serum creatinine were measured as described (11).
Activation of p53, caspase-3, and p21 was assessed using formalin-fixed, paraffin-embedded sections of kidneys after deparaffinization in xylene, and staining as previously described (15) using the primary rabbit anti-p53 antibody (1:200 for 90 min; Vector Laboratories), the primary rabbit anti-mouse cleaved caspase-3 antibody (1:100 for 60 min; BioCare Medical), or the primary rabbit anti-p21 antibody (1:400 for 30 min; Abcam). Ten random high-power fields (10×) were selected per slide for immunohistochemistry scoring.
Assessment of transporter function
HEK293 cells obtained from Invitrogen (Aug 2006; no authentication was conducted by the authors upon receipt) and transfected with human OCT2, mouse Oct1, mouse Oct2 or a control vector were seeded in 6-well plates with complete DMEM containing 10% fetal bovine serum and hygromycin B (100 µg/mL) at 37°C. Wells were washed once with warm PBS then pre-incubated for 15 min with DMEM containing pifithrin-α (0–100 µM), followed by a 30 minute incubation with the inhibitor and [14C]TEA (0.5, 1.0 and 2 µM), or cisplatin (500 µM). After incubation, cells were washed twice with cold PBS, collected, and solubilized in 1N sodium hydroxide. Radioactivity was assessed by liquid scintillation counting and platinum concentration was measured using atomic absorption spectroscopy. Uptake data were normalized to protein concentration, determined using a bicinchoninic acid protein-assay kit (Pierce Biotechnology).
Normal human kidney samples were obtained from a patient undergoing resection of a kidney tumor, as described (16). The procedure was approved by the ethics commission of the Universitätsklinikum Münster, and written consent was obtained from the patient. Proximal tubules from the human kidney sample or from wildtype mice were isolated for functional analyses (16). Organic cation transporter function and its sensitivity to inhibitory properties of pifithrin-α were evaluated according to an established protocol (16).
The contribution of OCT2 to the intracellular uptake of pifithrin-α or pifithrin-β (1 µM and 10 µM) was assessed using HEK293 cells transfected with OCT2 or an empty vector at pH 7.4 or 6.0 in the presence or absence of 125 mM NaCl. After incubation, cells were washed twice with cold PBS, collected, and lysed in 1 N sodium hydroxide. Levels of pifithrin- α and pifithrin-β were measured using liquid chromatography-tandem mass spectrometry (17).
Statistical analysis
Data are presented as mean values ± standard error, with n referring to the number of animals, cell monolayers or isolated tubules used in the experiments. An unpaired two sided Student’s t-test, and analysis of variance (with Tukey post-test) were used to demonstrate statistical significance of the effects, where P < 0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software Inc.).
Results
Phenotypic characterization of Oct1/2(−/−) mice
Since tubular necrosis is not completely absent in Oct1/2(−/−) mice treated with cisplatin (Fig. 1A), we hypothesized that alternate pathways are involved in the observed injury. Analysis of livers and kidneys from Oct1/2(−/−) mice revealed that the expression of 84 ATP-binding cassette transporter and solute carrier genes was not substantially changed compared with tissues obtained from wildtype animals, with the exception of a downregulation of Slc22a1 and Slc22a2 “transcripts” (Fig. 1B and C). In addition, γ-glutamyltranspeptidase (GGT; Ggt1 in mice) activity (Fig. 1D) and expression levels of the GGT-pathway genes Ggt1, Anpep (diaminopeptidase N) and Ccbl1 (cysteine-S-conjugate-β-lyase), previously implicated in cisplatin nephrotoxicity (Supplementary Fig. S1) (18), were unaffected by Oct1/Oct2-deficiency (Fig. 1E). Expression levels of other putative cisplatin transporters, such as Abcc2 (Mrp2) and Slc31a1 (Ctr1), Slc47a1 (Mate1) or the glutathione transporter Slc22a8 (Oat3) were also not altered in kidneys of Oct1/2(−/−) mice (Fig. 1E). In line with this finding, urinary ratios of the cisplatin-glutathione metabolite GSH2 to total platinum were unchanged in Oct1/2(−/−) mice compared to wildtype mice (Fig. 1F). These observations suggest that the GGT pathway is not differentially contributing to cisplatin nephrotoxicity in wildtype mice and Oct1/2(−/−) mice.
Figure 1.

Phenotypic characterization of Oct1/2(−/−) mice. (A) Degree of nephrotoxicity based on histology scores observed in kidneys isolated from wildtype (n = 30), Oct1/2(−/−) (n = 18) 72 hours following administration of cisplatin. Toxicity scores are based on percentage of damaged tubules: 0 (<10%; absent), 1 (11–25%; minimal), 2 (26–50%; mild), 3 (51–75%; moderate), and 4 (>75%; severe). Comparative expression of 84 transporter genes in kidneys (B) and livers (C) of wildtype and Oct1/2(−/−) mice (n = 3 each). (D) Activity of γ-glutamyltranspeptidase (GGT) in kidneys of wildtype and Oct1/2(−/−) mice (n = 4 each). (E) Relative expression of putative cisplatin transporter and GGT-pathway genes in kidneys of wildtype and Oct1/2(−/−) mice (n = 3 each). (F) Concentration of GSH2 (glutathione-monoplatinum conjugate) in urine of wildtype and Oct1/2(−/−) mice (n = 11 and 12, respectively). Levels of the glutathione-diplatinum conjugate, GSH1, were undetectable (<0.1 µg/mL). P-values above the bars denote statistical comparison between wildtype and Oct1/2(−/−) mice as determined by an unpaired t-test. All data are represented by mean values and standard error (error bars).
Transcriptional profiling of Oct1/2(−/−) mouse kidney
As a next step toward understanding the molecular mechanisms contributing to cisplatin-induced nephrotoxicity in Oct1/2(−/−) mice, we performed microarray analyses on kidney biopsies following cisplatin administration in vivo. Transcriptional profiling identified complex gene expression changes and a drug-response signature comprising of 1063 up-regulated and 1072 down-regulated genes in wildtype mice that was largely qualitatively similar in Oct1/2(−/−) mice, albeit not quantitatively. Further characterization of the expression signature using a gene ontology analysis identified 10 out of 193 analyzed pathways that showed significant (P < 0.001) alteration in wildtype mice exposed to cisplatin (Fig. 2A). These changes were largely absent in Oct1/2(−/−) mice, although the most significantly altered pathway in both wildtype mice (P = 2.40×10−11) and Oct1/2(−/−) mice (P = 1.92×10−8) involved genes associated with the p53 signaling network. This pathway has been implicated previously as a central component in DNA damage in response to cisplatin in various tissues, including the kidney (19).
Figure 2.

Transcriptional profiling of mouse kidneys after cisplatin treatment. (A) Significance of expression changes in 10 of 193 signaling pathways represented by -log2 of the P-value, as measured from 1063 up-regulated and 1072 down-regulated genes (fold change ≥2.0) in kidneys isolated from wildtype or Oct1/2(−/−) mice exposed to cisplatin and compared to untreated animals (n = 3). (B) Confirmatory fold expression changes of the p53-dependent genes Ccng2 (cyclin G1), Cdnk1a (p21), Mdm2, Pmaip1 (NOXA), Sesn2, and Trp53 (p53), as measured by PCR, in wildtype and Oct1/2(−/−) mice 72 hours following administration of cisplatin compared to baseline. All data are represented by mean values and standard error (error bars).
Using a p53 signaling pathway array, we confirmed renal expression changes of Trp53 (p53) itself and found strong induction of multiple other well-characterized p53 dependent genes, including Ccng2 (cyclin G1), Cdnk1a (p21), Mdm2, Pmaip1 (NOXA), and Sesn2, in response to cisplatin (Fig. 2B; Supplementary Table S1). None of the genes with altered expression in response to cisplatin differed among mouse genotypes in the absence of treatment. This suggests that the ability to mount an effective p53 response following cisplatin accumulation in renal tubular cells influences treatment sensitivity both in wildtype mice and Oct1/2(−/−) mice.
Contribution of p53 to cisplatin nephrotoxicity in Oct1/2(−/−) mice
To confirm that p53 acted as a mediator of cisplatin-induced nephrotoxicity, we first assessed renal tubular damage by histology scores following 3 days of cisplatin administration in p53-null mice [p53(−/−)]. C57BL/6 mice, the most commonly used strain for p53 knockout, are relatively resistant to cisplatin-induced nephrotoxicity compared with FVB mice, the strain used for Oct1/2 knockout (Supplementary Fig. S2A and B), presumably due to reduced renal expression of Oct1/2 and impaired urinary excretion of cisplatin (Supplementary Fig. 2C and D). A similar strain-dependence has been reported for bromodichloromethane-induced nephrotoxicity, which is more severe in FVB mice than C57BL/6 mice (20). Therefore, p53(−/−) mice on a FVB background were used in further studies and to generate a line lacking p53 as well as Oct1 and Oct2 [p53(−/−)/Oct1/2(−/−)]. These animals were phenotypically normal, with the exception of slightly increased alkaline phosphatase, as well as slightly reduced calcium and phosphorous levels (Supplementary Fig. S3A). As anticipated (21), the life span of p53(−/−) and p53(−/−)/Oct1/2(−/−) mice was significantly shorter compared to Oct1/2(−/−) mice due to the short latency period for malignant tumors in these animals (Supplementary Fig. S3B).
In regards to nephrotoxicity, wildtype mice and mice lacking one copy of p53 [p53(+/−) mice] experienced severe acute renal tubular necrosis following administration of cisplatin, whereas Oct1/2(−/−) and p53(−/−) mice experience only mild damage (Fig. 3A). Renal tubular necrosis was found to be entirely absent in p53(−/−)/Oct1/2(−/−) mice (Fig. 3A), suggesting that Oct1/2-mediated transport and p53 signaling are independently contributing to cisplatin-induced nephrotoxicity. As expected, loss of p53 by itself was not associated with altered urinary excretion of cisplatin (Fig. 3B).
Figure 3.

Influence of p53 loss on cisplatin-induced nephrotoxicity. (A) Degree of nephrotoxicity based on histology scores observed in kidneys isolated from wildtype, Oct1/2(−/−), p53(+/−), p53(−/−) or p53(−/−)/Oct1/2(−/−) mice 72 hours following administration of cisplatin. Toxicity scores are based on percentage of damaged tubules: 0 (<10%; absent), 1 (11–25%; minimal), 2 (26–50%; mild), 3 (51–75%; moderate), and 4 (>75%; severe). (B) Cumulative urinary excretion of platinum from wildtype, p53(−/−) or p53(−/−)/Oct1/2(−/−) mice within 72 hours after receiving cisplatin. Error bars represent standard error (n = number of mice) and significance was assessed by ANOVA (with Tukey post-test).
Identification of pifithrin-α as an OCT2 inhibitor
During the course of a recently performed library screen aimed at identifying novel inhibitors of OCT2 (22), we made the serendipitous observation that the organic cation pifithrin-α, a small molecule inhibitor of p53-dependent transcriptional activation (23), also inhibits OCT2 function as determined by its ability to reduce uptake of the OCT2 substrate, 4-[4-(dimethylamino)styryl]-N-methylpyridinium-iodide (ASP+) in HEK293 cells engineered to overexpress OCT2. We confirmed this observation by showing that pifithrin-α and its planar tricyclic degradation product pifithrin-β (24) inhibit OCT2-mediated transport of tetraethylammonium (TEA) in the same model (Fig. 4A). In order to identify the mechanism of this interaction, kinetic analyses were performed with and without pifithrin-α using varying concentrations of TEA as the OCT2 substrate. The resulting Dixon plot revealed that data for each substrate concentration fall on straight lines that intersect on the abscissa, suggesting that pifithrin-α is a non-competitive inhibitor of OCT2 function with an inhibition constant (Ki) of 3.0 µM (Fig. 4B). In line with this inhibitory mechanism, we found that neither pifithrin-α nor pifithrin-β were transported substrates of OCT2, irrespective of concentration, pH, or the presence of Na+ (Fig. 4C).
Figure 4.

Identification of pifithrin-α as an OCT2 inhibitor. (A) Inhibition of 2 µM tetraethylammonium (TEA) uptake in OCT2 transfected HEK293 cells with various concentrations of pifithrin-α or pifithrin-β (n = 3). Data are normalized to TEA uptake in the absence of pifithrin-α/β. Concentration-dependent inhibition of pifithrin-β was affected by constraints in aqueous solubility. (B) Potential of various concentrations of pifithrin-α to inhibit uptake of 0.5 – 2.0 µM TEA in OCT2 transfected HEK293 cells (Ki = 3.04 µM) (n = 3). (C) Cellular accumulation of pifithrin-α (PFT-α) or pifithrin-β (PFT-β) at various concentrations in OCT2 transfected HEK293 cells or vector control (VC) at pH 7.4 or 6.0 and in the presence or absence of 125 mM sodium (Na+) (n = 3). (D) Uptake of cisplatin (500 µM) in OCT2 transfected HEK293 cells in the presence or absence of pifithrin-α (100 µM) (n = 12). Data are normalized to the percent uptake of cisplatin in HEK293 cells transfected with OCT2 in the absence of pifithrin-α. (E) Uptake of ASP+ (1 µM) in isolated human proximal tubules in the presence or absence of 500 µM pifithrin-α (n = 4). Data are normalized to the percent uptake of ASP+ in human proximal tubules in the absence of pifithrin-α. Error bars represent standard error. Significance as measured by star (*) is representative of P < 0.05 versus uptake in OCT2 transfected HEK293 cells or human proximal tubular cells in the absence of pifithrin-α as determined by an unpaired t-test.
Pifithrin-α was also capable of completely blocking the cellular accumulation of cisplatin mediated by OCT2 in HEK293 cells (Fig. 4D). Moreover, to confirm the translational potential of these findings, we found that pifithrin-α inhibits uptake of ASP+ in freshly isolated human proximal tubular cells (Fig. 4E). Similar results were observed in murine-based models, where pifithrin-α reduced uptake of TEA in Oct1 and Oct2 overexpressing HEK293 cells (Fig. 5A) and impeded uptake of ASP+ in proximal tubular cells isolated from wildtype mice (Fig. 5B).
Figure 5.
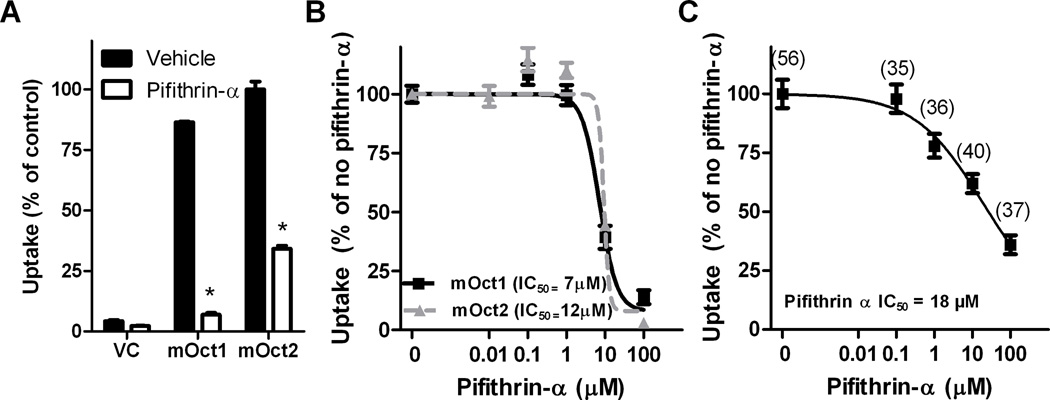
Inhibition of Oct1 and Oct2 function by pifithrin-α. (A) Uptake of 2 µM tetraethylammonium (TEA) in HEK293 cells transfected with vector control (VC), mOct1, or mOct2 for 30 minutes in the presence or absence of 10 µM pifithrin-α (n = 3). Data are normalized to the percent uptake of cisplatin in HEK293 cells transfected with mOct2 in the absence of pifithrin-α. (B) Uptake of ASP+ (1 µM) in HEK293 cells transfected with mOct1, or mOct2 for 30 minutes in the presence of various concentrations of pifithrin-α (n = 5 – 18) (C) Uptake of ASP+ (1 µM) in proximal tubules isolated from male FVB mice for 30 minutes in the presence of various concentrations of pifithrin-α. Numbers in parentheses represent the number of proximal tubules used per concentration. The estimated concentration associated with 50% inhibition of uptake is represented as IC50. Error bars represent standard error. Significance as measured by star (*) is representative of P < 0.05 versus vehicle as determined by an unpaired t-test.
Pifithrin-α ameliorates cisplatin-induced nephrotoxicity
To explore pifithrin-α further as a dual p53/OCT2-targeted agent for preventing cisplatin-induced nephrotoxicity, it was administered concomitantly with cisplatin in wildtype and Oct1/2(−/−) mice. Pifithrin-α was formulated in ethanol, since even relatively small amounts of its common solvent dimethylsufoxide (DMSO) can strongly reduce the cytotoxicity of cisplatin (14). Blood urea nitrogen (BUN) (Fig. 6A) and serum creatinine (Fig. 6B), two commonly used markers of cisplatin nephrotoxicity, were significantly reduced in wildtype and Oct1/2(−/−) mice when pifithrin-α was administered in combination with cisplatin compared to cisplatin given alone. In accordance with these findings, histological examination of kidney biopsies confirmed that cisplatin-induced nephrotoxicity could be completely abolished by pifithrin-α (Fig. 6C). This renoprotective phenomenon was also seen in Oct1/2(−/−) mice, which typically experience moderate renal tubular damage following administration of cisplatin (Fig. 6C). As expected, activation of p53 and p21, as well as cleavage of caspase-3 was observed in wildtype mice following exposure to cisplatin, but not in animals that also received pifithrin-α (Fig. 6D)
Figure 6.

Influence of pifithrin-α on cisplatin-induced nephrotoxicity. Levels of blood urea nitrogen (A) or serum creatinine (B) in wildtype or Oct1/2(−/−) mice 72 hours after receiving vehicle only, cisplatin alone, or a combination of cisplatin and pifithrin-α. (C) Degree of nephrotoxicity based on histology scores observed in kidneys isolated from wildtype or Oct1/2(−/−) mice following 72 hours of receiving vehicle only, cisplatin alone, or a combination of cisplatin and pifithrin-α. Numbers in parentheses represent the number of proximal tubules used per concentration. Toxicity scores are based on percentage of damaged tubules: 0 (<10%; absent), 1 (11–25%; minimal), 2 (26–50%; mild), 3 (51–75%; moderate), and 4 (>75%; severe). All data are represented by mean values and standard error (error bars). (D) Hematoxylin and eosin (H&E) staining or immunohistochemistry staining of phosphorylated p53, cleaved caspase-3, or p21 in kidneys isolated from wildtype or Oct1/2(−/−) mice 72 hours after receiving cisplatin in the presence or absence of pifithrin-α. Activation of p53, caspase-3, and p21 was based on antibody signal intensity: Rare, Low, Moderate and High.
Discussion
The current study identified the p53 signaling network as an important contributor to cisplatin-induced renal damage, which occurs despite loss of Oct1 and Oct2-dependent uptake of cisplatin into tubular cells. Our results show that mice with a deficiency of p53 are protected from severe cisplatin-induced nephrotoxicity, but renal damage is only completely abolished in mice with a concurrent deficiency of Oct1 and Oct2. In addition, we found that pifithrin-α, an investigational inhibitor of p53-dependent transcriptional activation and apoptosis, is a potent inhibitor of organic cation-mediated transport of cisplatin, and that concomitant use of pifithrin-α with cisplatin completely abrogates renal tubular damage by simultaneously inhibiting both drug transport and hindering the cisplatin-induced p53 activation.
The identification of the p53 pathway as a regulator of cisplatin-induced nephrotoxicity was not unanticipated, considering that p53 is the central mediator of DNA damage response which can be triggered by cisplatin (25). In fact, recent studies have reported that inflammation, oxidative stress and p53-related apoptosis in part explain the damage to proximal tubules caused by exposure to cisplatin (26–28). However, similar to Oct1/2(−/−) mice, loss of p53 alone provides only partial renoprotection, and damage to kidneys remains detectable. This suggests that strategies to inhibit the collective function of Oct1, Oct2 and p53 are necessary to completely alleviate the renal damage. Translational approaches aimed at inhibiting OCT2 function in humans are certainly attractive given the abundant evidence showing that the transporter is absent in the majority of human tumors, and that use of OCT2-inhibitors does not compromise anti-tumor activity or systemic levels of cisplatin in vitro or in vivo (7, 11–13).
In contrast, attempts to ameliorate renal tubular necrosis via inhibition of p53 signaling may be potentially worrisome based on the current understanding that the anti-tumor efficacy of cisplatin may be partially dependent on p53-mediated apoptosis in cancer cells. Nontheless, it should be noted that p53 is either lost or mutated in 50% of tumors, and previous reports have shown that loss of p53 function does not necessarily alter sensitivity to cisplatin chemotherapy (29). Additionally, p53 can actually function as a survival factor against various chemotherapeutics (30) and therefore, intentional inhibition of p53 as a strategy to ameliorate nephrotoxicity is unlikely to alter anti-tumor efficacy. Furthermore, while p53(−/−) mice are genetically predisposed to rapid tumor development (31), temporal blockade of p53 function with pifithrin-α is not known to be associated with an increased incidence in tumorgenesis (23).
Our findings concerning the ability of pifithrin-α to offer protection against cisplatin-induced nephrotoxicity is consistent with previous results reported by Wei et al. in C57BL/6 mice (32). A careful examination of these authors’ data reveals that the degree of renoprotection in wildtype mice receiving cisplatin with pifithrin-α is greater than that observed in p53(−/−) mice receiving cisplatin alone. This suggests that in this mouse model pifithrin-α can also affect cisplatin-induced nephrotoxicity by an additional, p53-independent mechanism, and it is possible that inhibition of cisplatin uptake into tubular cells by pifithrin-α contributed to the agent’s renoprotective properties. These collective data suggests that pifithrin-α could potentially be explored further as a unique dual OCT2/p53-inhibitor for prevention of cisplatin nephrotoxicity. In this context, it is worth pointing out that pifithrin-α has also shown potential to reduce cisplatin-dependent hair cell loss in cochlear and utricular cultures (33), suggesting that pifithrin-α use may ameliorate other side effects caused by cisplatin that are associated with OCT2, such as ototoxicity (8).
The notion that combinational inactivation of the p53 pathway and OCT function is necessary to offer complete protection from cisplatin-induced renal damage raises new questions. First of which concerns the p53-independent mechanism responsible for renal tubular damage that appears to occur downstream of Oct1/2-mediated intracellular accumulation of cisplatin. A candidate mechanism involved is the activation of PKCδ by cisplatin in renal tubular cells, which process leads to kidney cell apoptosis and tissue damage independently of p53 (34). The second involves the identity of the regulators that modulate cisplatin accumulation in the absence of Oct1 and Oct2 that are upstream of p53. Compensatory mechanisms involving putative cisplatin transporters are unlikely since their expression levels are unchanged in Oct1/2(−/−) mice. However, the possibility remains that other transporters regulate accumulation of cisplatin, which in turn generates a p53-mediated response. Indeed, Ctr1 has been implicated in cisplatin-induced renal toxicity and is known to mediate a p53-response (35), although its involvement as a carrier of cisplatin has recently been brought into question (36). Interestingly, probenecid has been shown to significantly reduce the tubular secretion of cisplatin without affecting glomerular filtration, and has been shown to reduce the severity of nephrotoxicity (37–40). Although the underlying mechanism is unclear, it is conceivable that platinum-sulfhydryl complexes formed in the GGT pathway accumulate into kidney cells through an organic anion transport mechanism that is probenecid-sensitive (41). Additional studies are currently ongoing to definitively address these possibilities.
In conclusion, the results of our study indicate that p53 plays a crucial role in the kidney in response to cisplatin independently of OCT2. This suggests that clinical exploration of OCT2 inhibitors as an adjunct to cisplatin-based chemotherapy is unlikely to lead to complete renoprotection unless the p53 pathway is simultaneously antagonized. Future investigation will focus on the exploration of pifithrin-α, and structurally-related compounds with improved pharmaceutical properties (42), as dual OCT2/p53-inhibitors for prevention of cisplatin nephrotoxicity.
Supplementary Material
Translational Relevance.
The human organic cation transporter OCT2 is known to be involved in cellular accumulation of cisplatin and contribute to cisplatin-induced nephrotoxicity. Pharmacological or genetic inhibition of OCT2 function has been associated with protection from cisplatin-induced nephrotoxicity, however previous in vivo studies have demonstrated that renal tubular damage is not completely abolished when OCT2 alone is targeted. Data from the current study indicates that the p53 pathway plays a crucial role in the kidney in response to cisplatin independently of OCT2, suggesting that clinical exploration of OCT2 inhibitors as an adjunct to cisplatin-based chemotherapy is unlikely to lead to complete renoprotection unless the p53 pathway is simultaneously antagonized with pharmacological inhibitors.
Acknowledgments
The authors thank Drs. David Finkelstein, Kelly Filipski, Ryan Franke, Zhaoyuan Chen, and Shuiying Hu (all St. Jude Children’s Research Hospital, Memphis, TN) for their various contributions to this work, and Dr. Paul Workman (Institute of Cancer Research, Sutton, Surrey, United Kingdom) for providing a protocol for measurement of pifithrin-α.
Grant Support
This work was supported in part by the American Lebanese Syrian Associated Charities (ALSAC), USPHS Cancer Center Support Grant 3P30CA021765, the National Institutes of Health grant NCI 5R01CA151633-01, and the IZKF of the Münster Medical Faculty and the Deutsche Forschungsgemeinschaft grant DFG CI 107/4-1. None of the funding bodies had a role in the preparation of the manuscript.
Footnotes
Disclosure of Potential Conflict of Interest
The authors declare no conflict of interest.
Authors’ Contributions
Concept and design: J.A. Sprowl, C.S. Lancaster, N. Pabla, and A. Sparreboom.
Development of methodology: J.A. Sprowl, C.S. Lancaster, G.Ciarimboli, and A. Sparreboom.
Acquisition of data: J.A. Sprowl, C.S. Lancaster, E. Hermann, A.M. Kosloske, A.A. Gibson, L. Li, D. Zeeh, E. Schlatter, L.J. Janke, G. Ciarimboli.
Analysis and interpretation of data: J.A. Sprowl, C.S. Lancaster, N. Pabla, G.Ciarimboli, and A. Sparreboom.
Writing, review, and/or revision of the manuscript: J.A. Sprowl, C.S. Lancaster, N. Pabla, G.Ciarimboli, and A. Sparreboom.
Study supervision: A. Sparreboom
References
- 1.de Jongh FE, van Veen RN, Veltman SJ, de Wit R, van der Burg ME, van den Bent MJ, et al. Weekly high-dose cisplatin is a feasible treatment option: analysis on prognostic factors for toxicity in 400 patients. Br J Cancer. 2003;88:1199–1206. doi: 10.1038/sj.bjc.6600884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yonezawa A, Masuda S, Nishihara K, Yano I, Katsura T, Inui K. Association between tubular toxicity of cisplatin and expression of organic cation transporter rOCT2 (Slc22a2) in the rat. Biochem Pharmacol. 2005;70:1823–1831. doi: 10.1016/j.bcp.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 3.Yonezawa A, Masuda S, Yokoo S, Katsura T, Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family) J Pharmacol Exp Ther. 2006;319:879–886. doi: 10.1124/jpet.106.110346. [DOI] [PubMed] [Google Scholar]
- 4.Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciarimboli G, Ludwig T, Lang D, Pavenstadt H, Koepsell H, Piechota HJ, et al. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol. 2005;167:1477–1484. doi: 10.1016/S0002-9440(10)61234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Filipski KK, Loos WJ, Verweij J, Sparreboom A. Interaction of cisplatin with the human organic cation transporter 2. Clin Cancer Res. 2008;14:3875–3880. doi: 10.1158/1078-0432.CCR-07-4793. [DOI] [PubMed] [Google Scholar]
- 7.Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86:396–402. doi: 10.1038/clpt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciarimboli G, Deuster D, Knief A, Sperling M, Holtkamp M, Edemir B, et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am J Pathol. 2010;176:1169–1180. doi: 10.2353/ajpath.2010.090610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwata K, Aizawa K, Kamitsu S, Jingami S, Fukunaga E, Yoshida M, et al. Effects of genetic variants in SLC22A2 organic cation transporter 2 and SLC47A1 multidrug and toxin extrusion 1 transporter on cisplatin-induced adverse events. Clin Exp Nephrol. 2012;16:843–851. doi: 10.1007/s10157-012-0638-y. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Zhou W. Ameliorative effects of SLC22A2 gene polymorphism 808 G/T and cimetidine on cisplatin-induced nephrotoxicity in Chinese cancer patients. Food Chem Toxicol. 2012;50:2289–2293. doi: 10.1016/j.fct.2012.03.077. [DOI] [PubMed] [Google Scholar]
- 11.Franke RM, Kosloske AM, Lancaster CS, Filipski KK, Hu C, Zolk O, et al. Influence of Oct1/Oct2-deficiency on cisplatin-induced changes in urinary N-acetyl-beta-D-glucosaminidase. Clin Cancer Res. 2010;16:4198–4206. doi: 10.1158/1078-0432.CCR-10-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katsuda H, Yamashita M, Katsura H, Yu J, Waki Y, Nagata N, et al. Protecting cisplatin-induced nephrotoxicity with cimetidine does not affect antitumor activity. Biol Pharm Bull. 2010;33:1867–1871. doi: 10.1248/bpb.33.1867. [DOI] [PubMed] [Google Scholar]
- 13.Sprowl JA, van Doorn L, Hu S, van Gerven L, de Bruijn P, Li L, et al. Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: influence on antitumor efficacy and systemic clearance. Clin Pharmacol Ther. 2013;94:585–592. doi: 10.1038/clpt.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer SJ, Benson LM, Fauq A, Naylor S, Windebank AJ. Cisplatin and dimethyl sulfoxide react to form an adducted compound with reduced cytotoxicity and neurotoxicity. Neurotoxicology. 2008;29:444–452. doi: 10.1016/j.neuro.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 15.Lancaster CS, Hu C, Franke RM, Filipski KK, Orwick SJ, Chen Z, et al. Cisplatin-induced downregulation of OCTN2 affects carnitine wasting. Clin Cancer Res. 2010;16:4789–4799. doi: 10.1158/1078-0432.CCR-10-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciarimboli G, Lancaster CS, Schlatter E, Franke RM, Sprowl JA, Pavenstadt H, et al. Proximal tubular secretion of creatinine by organic cation transporter OCT2 in cancer patients. Clin Cancer Res. 2012;18:1101–1108. doi: 10.1158/1078-0432.CCR-11-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walton MI, Wilson SC, Hardcastle IR, Mirza AR, Workman P. An evaluation of the ability of pifithrin-alpha and -beta to inhibit p53 function in two wild-type p53 human tumor cell lines. Mol Cancer Ther. 2005;4:1369–1377. doi: 10.1158/1535-7163.MCT-04-0341. [DOI] [PubMed] [Google Scholar]
- 18.Hanigan MH, Devarajan P. Cisplatin nephrotoxicity: molecular mechanisms. Cancer Ther. 2003;1:47–61. [PMC free article] [PubMed] [Google Scholar]
- 19.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 20.Torti VR, Cobb AJ, Everitt JI, Marshall MW, Boorman GA, Butterworth BE. Nephrotoxicity and hepatotoxicity induced by inhaled bromodichloromethane in wild-type and p53-heterozygous mice. Toxicol Sci. 2001;64:269–280. doi: 10.1093/toxsci/64.2.269. [DOI] [PubMed] [Google Scholar]
- 21.Anikin IV, Kozlov AP, Popov AV, Zabezhinskii MA, Tyndyk ML, Anisimov VN. [Susceptibility of wild and knockout p53 FVB/N line mice to benz(a)pyrene-induced subcutaneous sarcoma] Vopr Onkol. 2002;48:700–702. [PubMed] [Google Scholar]
- 22.Sprowl JALW, Du G, Ness RA, Baker SD, Chen T, Sparreboom A. Identification of tyrosine kinase inhibitors as modulators of OCT2 function. Eur J Cancer. 2012;48:45. (Abstr 146) (EORTC-NCI-AACR). [Google Scholar]
- 23.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 24.Gary RK, Jensen DA. The p53 inhibitor pifithrin-alpha forms a sparingly soluble derivative via intramolecular cyclization under physiological conditions. Mol Pharm. 2005;2:462–474. doi: 10.1021/mp050055d. [DOI] [PubMed] [Google Scholar]
- 25.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 26.Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110:835–842. doi: 10.1172/JCI15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santos NA, Catao CS, Martins NM, Curti C, Bianchi ML, Santos AC. Cisplatin-induced nephrotoxicity is associated with oxidative stress, redox state unbalance, impairment of energetic metabolism and apoptosis in rat kidney mitochondria. Arch Toxicol. 2007;81:495–504. doi: 10.1007/s00204-006-0173-2. [DOI] [PubMed] [Google Scholar]
- 28.Pabla N, Huang S, Mi QS, Daniel R, Dong Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J Biol Chem. 2008;283:6572–6583. doi: 10.1074/jbc.M707568200. [DOI] [PubMed] [Google Scholar]
- 29.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 30.Zuco V, Zunino F. Cyclic pifithrin-alpha sensitizes wild type p53 tumor cells to antimicrotubule agent-induced apoptosis. Neoplasia. 2008;10:587–596. doi: 10.1593/neo.08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 32.Wei Q, Dong G, Yang T, Megyesi J, Price PM, Dong Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol. 2007;293:F1282–F1291. doi: 10.1152/ajprenal.00230.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang M, Liu W, Ding D, Salvi R. Pifithrin-alpha suppresses p53 and protects cochlear and vestibular hair cells from cisplatin-induced apoptosis. Neuroscience. 2003;120:191–205. doi: 10.1016/s0306-4522(03)00286-0. [DOI] [PubMed] [Google Scholar]
- 34.Pabla N, Dong G, Jiang M, Huang S, Kumar MV, Messing RO, et al. Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest. 2011;121:2709–2722. doi: 10.1172/JCI45586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2009;296:F505–F511. doi: 10.1152/ajprenal.90545.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivy KD, Kaplan JH. A re-evaluation of the role of hCTR1, the human high affinity Cu transporter in Pt-drug entry into human cells. Mol Pharmacol. 2013;83:1237–1246. doi: 10.1124/mol.113.085068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein J, Bentur Y, Cheung D, Moselhy G, Koren G. Renal handling of cisplatin: interactions with organic anions and cations in the dog. Clin Invest Med. 1991;14:388–394. [PubMed] [Google Scholar]
- 38.Jacobs C, Coleman CN, Rich L, Hirst K, Weiner MW. Inhibition of cis-diamminedichloroplatinum secretion by the human kidney with probenecid. Cancer Res. 1984;44:3632–3635. [PubMed] [Google Scholar]
- 39.Ross DA, Gale GR. Reduction of the renal toxicity of cis-dichlorodiammineplatinum(II) by probenecid. Cancer Treat Rep. 1979;63:781–787. [PubMed] [Google Scholar]
- 40.Jacobs C, Kaubisch S, Halsey J, Lum BL, Gosland M, Coleman CN, et al. The use of probenecid as a chemoprotector against cisplatin nephrotoxicity. Cancer. 1991;67:1518–1524. doi: 10.1002/1097-0142(19910315)67:6<1518::aid-cncr2820670610>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Kolb RJ, Ghazi AM, Barfuss DW. Inhibition of basolateral transport and cellular accumulation of cDDP and N-acetyl- L-cysteine-cDDP by TEA and PAH in the renal proximal tubule. Cancer Chemother Pharmacol. 2003;51:132–138. doi: 10.1007/s00280-002-0537-0. [DOI] [PubMed] [Google Scholar]
- 42.Christodoulou MS, Colombo F, Passarella D, Ieronimo G, Zuco V, De Cesare M, et al. Synthesis and biological evaluation of imidazolo[2,1-b]benzothiazole derivatives, as potential p53 inhibitors. Bioorg Med Chem. 2011;19:1649–1657. doi: 10.1016/j.bmc.2011.01.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.