

Abstract
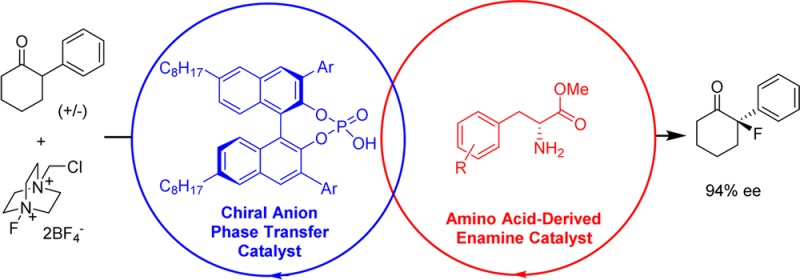
We report a study involving the successful merger of two separate chiral catalytic cycles: a chiral anion phase-transfer catalysis cycle to activate Selectfluor and an enamine activation cycle, using a protected amino acid as organocatalyst. We have demonstrated the viability of this approach with the direct asymmetric fluorination of α-substituted cyclohexanones to generate quaternary fluorine-containing stereocenters. With these two chiral catalytic cycles operating together in a matched sense, high enantioselectivites can be achieved, and we envisage that this dual catalysis method has the potential to be more broadly applicable, given the breadth of enamine catalysis. It also represents a rare example of chiral enamine catalysis operating successfully on α-branched ketones, substrates commonly inert to this activation mode.
The selective introduction of fluorine into small organic molecules has rapidly become a field of great interest, due to the beneficial pharmarcokinetic properties that judiciously placed fluorine atoms can confer.1 Great progress has been made employing chiral metal complexes for electrophilic fluorination of activated ketones1a,2 and using nucleophilic fluorine sources for enantioselective allylic fluorination.3 Chiral enamine catalysis has furnished a number of protocols for highly enantioselective α-fluorination of aldehydes4 and a single protocol for ketones.5 Cinchona alkaloids have been effective for fluorination of carbon nucleophiles6 and in a dual catalysis mechanism to enable the fluorination of acid chlorides.7
Within the burgeoning field of asymmetric catalysis controlled by chiral counterions,8 we have recently introduced chiral anion phase-transfer catalysis, employing chiral lipophilic BINOL-derived phosphates in combination with dicationic electrophilic reagents applied to enantioselective halogenation reactions.9 In our approach, the catalytic activation of the electrophilic reagent consists of its transport from the solid phase into the organic phase where its chiral anions enable enantioselective transformations. We envisage that this new approach to asymmetric fluorination could prove to be broadly applicable across a range of substrate classes given the proven versatility of chiral phosphoric acids and the powerful fluorinating ability of Selectfluor. However, in earlier reports, an amide group in the substrate was crucial for high enantioselectivity.9a,10
We hypothesized that this was due the need for a substrate amide to form a hydrogen bond with the BINOL-phosphoric acid catalyst in the transition state. We have since discovered that simple phenols can be employed as substrates in enantioselective fluorinative dearomatization11 and or as directing groups in enantioselective fluorination of alkenes.12 On the basis of these results and with the ultimate goal of expanding our methodology to encompass the most elementary of substrates, we targeted the enantioselective fluorination of simple ketones. We were aware that this is a challenging undertaking as evidenced by the fact that only one highly enantioselective ketone fluorination procedure has been reported to date.4b,5 This elegant example, using a cinchona-alkaloid-derived primary amine catalyst in combination with NFSI, was however limited to the fluorination of non-α-branched ketones.5,13
Specifically, we supposed that inclusion of a catalytic amount of a primary amine would form a transient enamine that should be reactive to electrophilic fluorination and act as a hydrogen-bond donor for binding to the chiral phosphate catalyst (Figure 1, cycle 1).14 This would also present the opportunity to introduce a chiral controlling element onto the enamine partner (e.g., an amino acid derivative, as depicted). Independently, the lipophilic chiral phosphate would undergo anion exchange with the achiral tetrafluoroborate counteranions of insoluble Selectfluor to catalytically generate a soluble chiral electrophilic fluorinating reagent (cycle 2). If compatible, the union of these two independent catalytic cycles should be capable of achieving direct asymmetric ketone α-fluorination (Figure 1, leading scheme), a general approach which has been referred to variously as ‘dual’, ‘synergistic’, or ‘cooperative’ catalysis.15 Such processes which involve two catalysts simultaneously activating two different reactants have the potential to be extremely powerful if the two catalytic cycles are able to operate in synchrony.15a They also offer the exciting opportunity to exert chiral influence from both catalysts to accomplish particularly challenging transformations, although few examples of such a scenario have been reported to date; most rely on one chiral and one achiral catalyst.16,17
Figure 1.
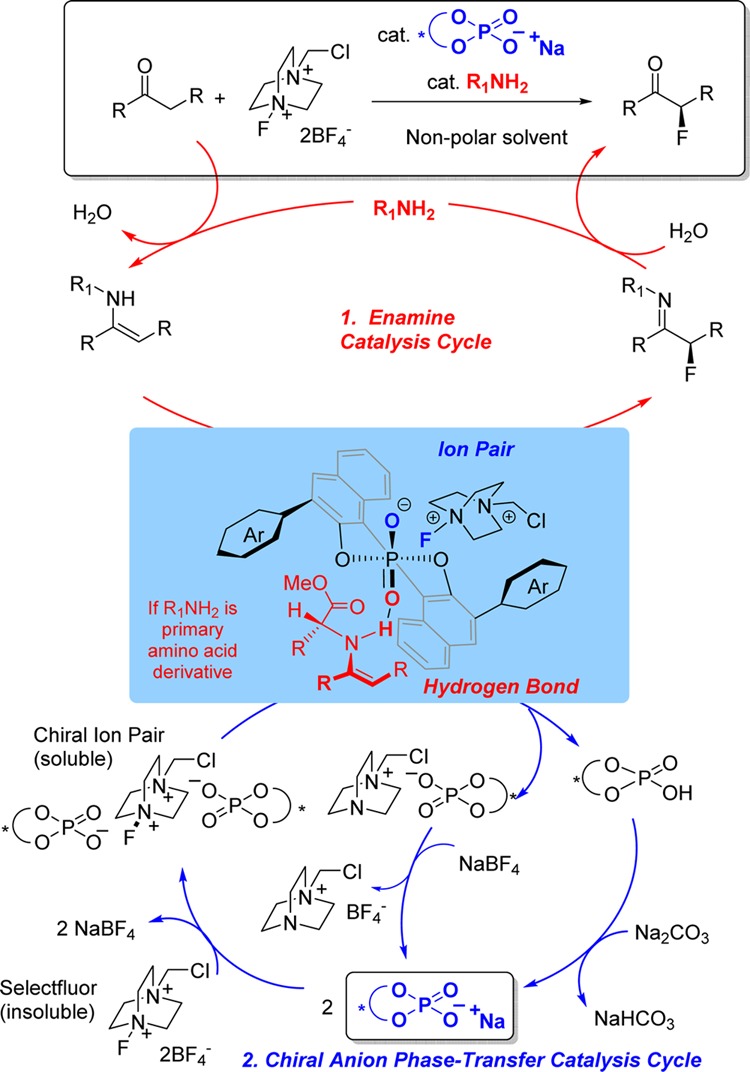
Proposed dual catalytic cycle for the enantioselective fluorination of ketones using (1) an enamine catalysis cycle and (2) a chiral anion phase-transfer catalysis cycle.
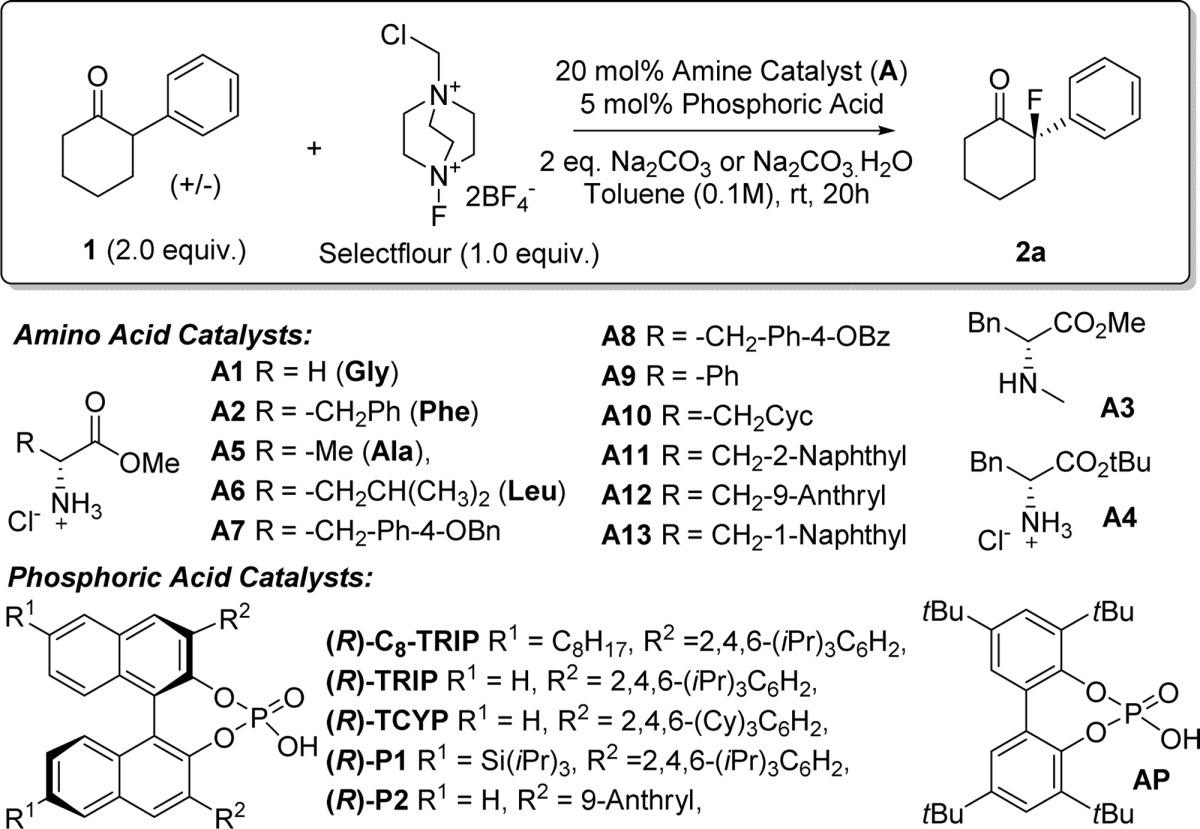
We selected 2-phenylcyclohexanone as an evaluation substrate, having previously observed that the benzoyl enamide derived from this ketone10b as well as geometrically similar 2-phenylphenol11 underwent highly enantioselective fluorination. Under our standard conditions (5 mol % chiral phosphoric acid, 2 equiv ketone, 1 equiv Selectfluor, 2 equiv Na2CO3, toluene, rt) in the absence of a primary amine cocatalyst, only trace amounts of the desired product were obtained (Table 1, entry 1). Encouragingly, inclusion of 20 mol % of benzylamine increased the yield of the fluorinated product to 29% and n-butylamine gave 24% yield based on Selectfluor, although both gave disappointingly low enantioselectivities (<5% ee, entries 2 and 3). Addition of catalytic glycine methyl ester gave the highest yield (entry 4), and although the enantioselectivity was low (10%), the effectiveness of this combination with regards to reactivity was encouraging.
Table 1. Investigation of the Fluorination of 2-Phenylcyclohexanone.
| entry | phosphoric acid | amine | yield based on: 1 (Selectfluor)a | eeb |
|---|---|---|---|---|
| 1c | (R)-C8-TRIP | none | 2 (5) | –2 |
| 2c | (R)-C8-TRIP | BnNH2 | 14 (29) | –3 |
| 3c | (R)-C8-TRIP | nBuNH2 | 12 (24) | –2 |
| 4c | (R)-C8-TRIP | A1 | 25 (50) | +10 |
| 5c | (R)-C8-TRIP | d-A2 | 31–36 (63–73) | +63–78 |
| 6d | (R)-C8-TRIP | d-A2 | 25 (50) | +32 |
| 7e | (R)-C8-TRIP | d-A2 | 37 (74) | +88 |
| 8e | (R)-C8-TRIP | l-A2 | 35 (70) | –40 |
| 9e | none | d-A2 | 5 (10) | +10 |
| 10e | AP | d-A2 | 28 (57) | +20 |
| 11e | (R)-C8-TRIP | d-A3 | 4 (8) | –24 |
| 12e | (R)-TRIP | d-A2 | 34 (69) | +82 |
| 13e | (R)-TCYP | d-A2 | 32 (65) | +67 |
| 14e | (R)-P1 | d-A2 | 26 (52) | +87 |
| 15e | (R)-P2 | d-A2 | 24 (48) | +21 |
| 16e | (R)-C8-TRIP | d-A4 | 39 (78) | +45 |
| 17e | (R)-C8-TRIP | d-A5 | 23 (47) | +35 |
| 18e | (R)-C8-TRIP | d-A6 | 26 (53) | +86 |
| 19e | (R)-C8-TRIP | d-A7 | 32 (64) | +87 |
| 20e | (R)-C8-TRIP | d-A8 | 35 (70) | +81 |
| 21e | (R)-C8-TRIP | d-A9 | 28 (56) | +61 |
| 22e | (R)-C8-TRIP | d-A10 | 13 (27) | +87 |
| 23e | (R)-C8-TRIP | d-A11 | 36 (72) | +83 |
| 24e | (R)-C8-TRIP | d-A12 | 14 (29) | +88 |
| 25e | (R)-C8-TRIP | d-A13 | 31 (62)f | +94 |
Yield determined by 19F-NMR spectroscopy by comparison with an internal standard.
Determined by chiral HPLC analysis of the crude reaction mixture after a short plug of silica gel.
Na2CO3 ground and dried under vacuum at 80 °C for 2 h.
Na2CO3 ground and dried at 200 °C for 4 h.
Commercially available Na2CO3.H2O used as received.
Isolated in 61% yield based on Selectfluor.
We next examined d-phenylalanine methyl ester as enamine catalyst and were excited to see that the enantioselectivity increased to 78% ee with good conversion and clean reaction (entry 5).18 The employment of Selectfluor as limiting reagent with excess ketone is due to the formation of trace amounts of difluorinated byproducts observed if Selectfluor was used in excess, which were very difficult to separate from the desired product. For clarity we have therefore displayed yields based on both components, with the yield based on the limiting reagent Selectfluor in parentheses. The high enantioselectivity initially obtained in this reaction was found to be unrepeatable, and after careful examination, we found that inconsistent levels of moisture in the Na2CO3 employed (dried under vacuum at 80 °C for 2h) led to this capriciousness. Using Na2CO3 dried at 200 °C for 4 h gave only 32% ee (entry 6), suggesting that a small amount of water is essential for the high enantioselectivity. Finally, we found using commercially available Na2CO3·H2O improved the ee to 88%, with excellent repeatability (entry 7). Recovery and analysis of the unreacted starting material showed the ee of the starting material to be <5% demonstrating that this is not a kinetic resolution. Evaluation of l-phenylalanine methyl ester under these optimized conditions showed that in this case the catalysts are mismatched, giving 40% ee of the opposite enantiomer in good yield based on Selectfluor (entry 8). Removing the chiral phosphoric acid resulted in low conversion and little enantioselectivity (entry 9), demonstrating that both catalysts are crucial; in the absence of either, both yield and enantiomeric excess are poor (<10% in each case).
In order to confirm that the chiral phosphate catalyst is playing an important role in enantioinduction, we used d-phenylalanine methyl ester in combination with the lipophilic achiral phosphoric acid AP. In this case the yield of fluorination was good, as expected, but the enantioselectivity was poor (20% ee, entry 10). This result, taken with those using achiral amines with a chiral phosphate (entries 2–4), demonstrates the requirement for chiral controlling elements on both catalysts in order to achieve high enantioselectivity. Use of the N-methylated d-phenylalanine methyl ester gave negligible conversion, as expected given that condensation of this secondary amine with the hindered ketone is likely disfavored (entry 11 vs entry 7), providing strong evidence for the proposed enamine mechanism, rather than an alternative mechanistic scenario where the amine may act as a base. A survey of a handful of chiral phosphoric acids showed C8-TRIP to be optimal (entries 12–15).19 Having established the indispensability of both the chiral phosphoric acid and chiral amino acid ester catalysts, we surveyed a range of amino acid esters.20 Use of the tert-butyl ester of phenylalanine (45% ee, entry 16) or the less bulky alanine methyl ester (35% ee, entry 17) was detrimental to the enantioselectivity. Leucine methyl ester performed almost as well as phenylalanine methyl ester (86% ee, entry 18), as did two O-substituted tyrosine derivatives (entries 19 and 20). Moving the side-chain aromatic ring closer to the amine in phenyl glycine proved to be detrimental (61% ee, entry 21), while replacing the phenylalanine aromatic ring with cyclohexyl resulted in no loss of enantioselectivity but low conversion (87% ee, entry 22). With the aim of increasing the steric bulk of the phenylalanine aromatic ring, we investigated naphthyl and anthryl variants (entries 23–25). This resulted in the identification of catalyst A13, delivering the fluorinated product with an excellent 94% ee in 61% isolated yield, based on the limiting Selectfluor (entry 25). Under our employed conditions using excess ketone, a substantial amount of unreacted ketone can be recovered. For example 81% of the excess ketone employed in synthesis of 2a (Table 1, entry 25) could be reisolated (see SI for details).
We next explored the scope and limitations of the transformation (Table 2). A two-fold excess of ketone is used, and the resulting yields calculated based on the limiting reagent Selectfluor are quoted in parentheses, after that based on the excess ketone. A range of substituted aryl groups are tolerated in the α-position of the cyclohexanone (2a–2j). This encompasses para and meta substituted arenes as well as a furan (2i) that is untouched by the Selectfluor and highlights the mild electrophilic fluorinating conditions. Similarly untouched in the α-fluorination is an aryl iodide (2j), despite the precedent for aryl iodides being readily oxidized to I(III) by Selectfluor under homogeneous conditions.21 Substitution (2k) and the presence of heteroatoms (2l and 2m) are well tolerated at the 4-position of the cyclohexanone. In three cases, phenylalanine catalyst A2 gave marginally (<5%) higher enantioselectivity than naphthyl variant A13. In the case of furyl substrate 2i, bulky 9-anthryl catalyst A12 gave an advantage of 10% ee over A13.
Table 2. Substrate Scope of the Asymmetric Fluorination of 2-Aryl Cyclohexanonesa,b,c.
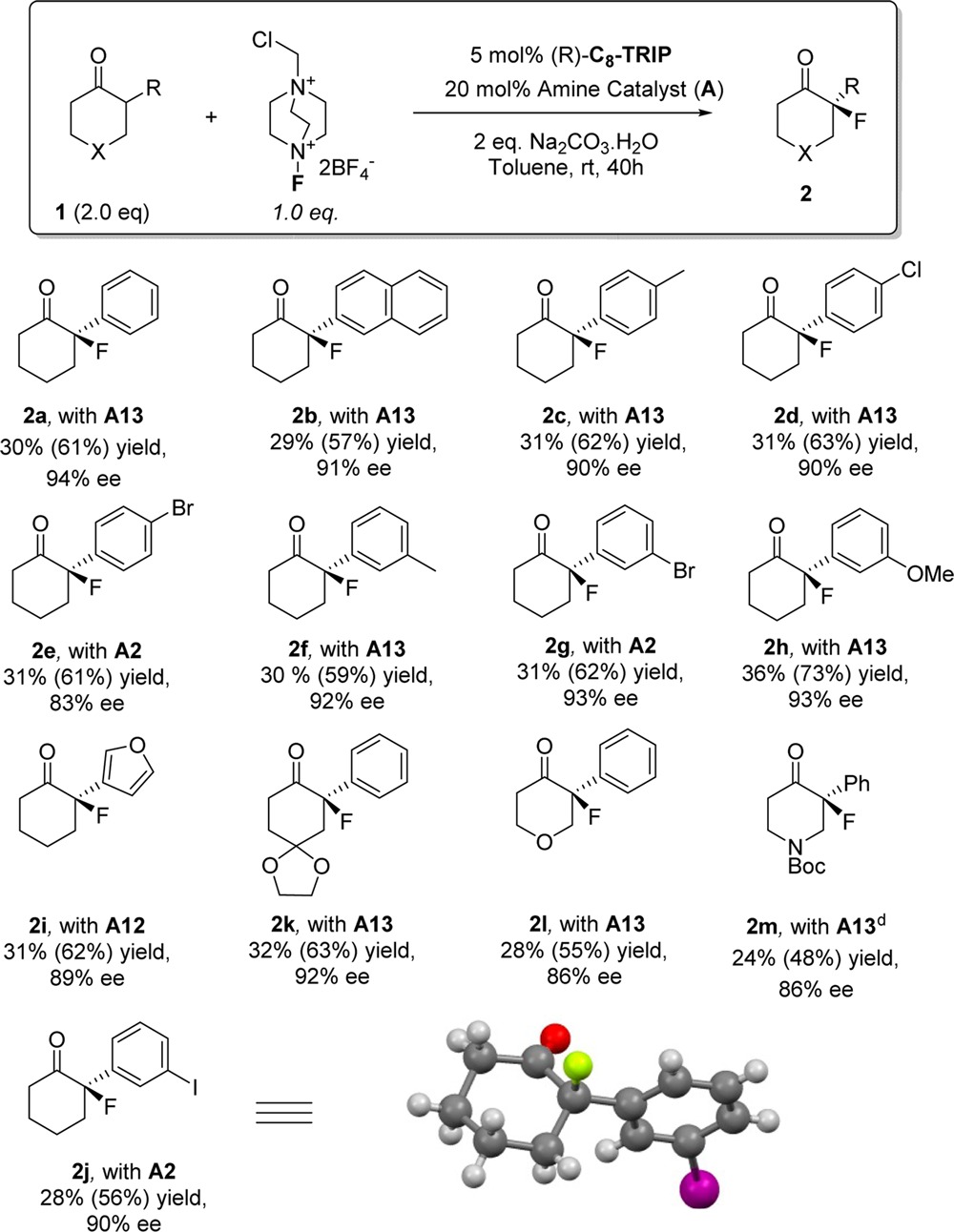
Reactions were carried out with ketone (0.2 mmol), Selectfluor (0.1 mmol) amine catalyst (0.02 mmol), phosphoric acid catalyst (0.005 mmol), and Na2CO3·H2O (0.2 mmol) in toluene (1.0 mL) for 40 h at rt.
Yields are isolated yields after chromatography. Primary yields calculated based on the excess ketone employed. Yields in parentheses calculated based on Selectfluor, the limiting reagent in all cases.
Absolute configurations assigned by analogy to 2j, determined by X-ray crystallography.
Characterized as the N-tosyl derivative.
We demonstrate that several 2-alkenyl and 2-alkynyl cyclohexanones are also viable substrates in our fluorination, delivering high enantioselectivies (Table 3). In these cases, complete regioselectivity is observed for fluorination at the α-position of the ketone, with no undesired fluorination of the alkene or alkyne. In the alkynyl substrates the enantioselectivities were somewhat reduced, and a catalyst survey revealed 9-anthryl catalyst A12 to be optimal (2o–2q). Disappointingly, no fluorination is observed for 2-alkylcyclohexanones under the present conditions, and we do not observe fluorination of closely related acyclic ketones under these conditions, demonstrating the present limitations of the methodology.22 Finally, selective transformation of the products into other chiral fluorinated derivatives was investigated (Scheme 1).
Table 3. Asymmetric Fluorination of 2-Alkenyl and 2-Alkynyl Cyclohexanonesa.
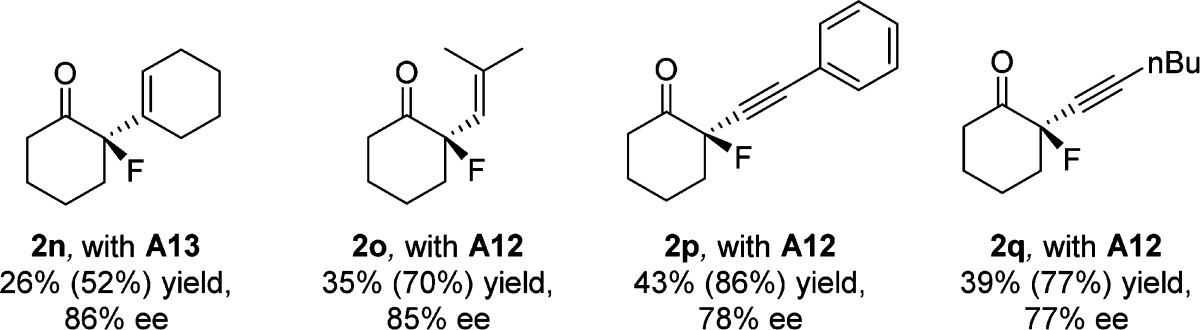
Conditions as in Table 2. Primary yields are isolated yields after chromatography, calculated based on excess ketone employed. Yield in parenthesis calculated based on the limiting reagent, Selectfluor.
Scheme 1. Further Transformations of Fluorinated Ketone Products.

In summary, we have developed an asymmetric fluorination of substituted cyclohexanones which generates quaternary fluorine-containing stereocenters. We propose that this is achieved through the combination of two separate catalytic cycles: enamine activation of the ketone and chiral anion phase-transfer activation of the Selectfluor. The two activated components are subsequently united most likely by direct interaction of the two chiral components via a hydrogen-bonded transition state which imparts high levels of stereoselectivity in the matched case. Successful combination of two catalytic cycles that both involve chiral catalysts is rare, and we believe that this study is among the first to combine two chiral organocatalytic cycles. While the current synthetic scope is limited to substituted cyclohexanones and requires careful matching of the two chiral catalysts, this also represents a rare example of chiral enamine catalysis operating successfully on α-branched ketones, substrates which are commonly inert to this activation mode.23
Acknowledgments
We gratefully acknowledge NIHGMS (RO1 GM104534), SIOC, Zhejiang Medicine and Pharmaron (Fellowship to X.Y.) and the European Commission (Marie Curie International Outgoing Fellowship to R.J.P.) for financial support. We gratefully acknowledge Ms. Z. Zhang for experimental assistance and Dr. A. DiPasquale for obtaining X-ray crystallographic data for 2j.
Supporting Information Available
Experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Lectard S.; Hamashima Y.; Sodeoka M. Adv. Synth. Catal. 2010, 352, 2708. [Google Scholar]; b Müller K.; Faeh C.; Diederich F. Science 2007, 317, 1881. [DOI] [PubMed] [Google Scholar]; c Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Chem. Soc. Rev. 2008, 37, 320. [DOI] [PubMed] [Google Scholar]
- a Hamashima Y.; Yagi K.; Takano H.; Tamás L.; Sodeoka M. J. Am. Chem. Soc. 2002, 124, 14530. [DOI] [PubMed] [Google Scholar]; b Hintermann L.; Togni A. Angew. Chem., Int. Ed. 2000, 39, 4359. [DOI] [PubMed] [Google Scholar]
- a Katcher M. H.; Sha A.; Doyle A. G. J. Am. Chem. Soc. 2011, 133, 15902. [DOI] [PubMed] [Google Scholar]; b Kalow J. A.; Doyle A. G. J. Am. Chem. Soc. 2010, 132, 3268. [DOI] [PubMed] [Google Scholar]
- a Beeson T. D.; MacMillan D. W. C. J. Am. Chem. Soc. 2005, 127, 8826. [DOI] [PubMed] [Google Scholar]; b Enders D.; Hüttl M. R. M. Synlett 2005, 991. [Google Scholar]; c Marigo M.; Fielenbach D.; Braunton A.; Kjærsgaard A.; Jørgensen K. A. Angew. Chem., Int. Ed. 2005, 44, 3703. [DOI] [PubMed] [Google Scholar]; d Steiner D. D.; Mase N.; Barbas C. F. Angew. Chem., Int. Ed. 2005, 44, 3706. [DOI] [PubMed] [Google Scholar]; e Pihko P. M. Angew. Chem., Int. Ed. 2006, 45, 544. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski P.; Beeson T. D.; Conrad J. C.; MacMillan D. W. C. J. Am. Chem. Soc. 2011, 133, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ishimaru T.; Shibata N.; Horikawa T.; Yasuda N.; Nakamura S.; Toru T.; Shiro M. Angew. Chem., Int. Ed. 2008, 47, 4157. [DOI] [PubMed] [Google Scholar]; b Lozano O.; Blessley G.; Martinez del Campo T.; Thompson A. L.; Giuffredi G. T.; Bettati M.; Walker M.; Borman R.; Gouverneur V. Angew. Chem., Int. Ed. 2011, 50, 8105. [DOI] [PubMed] [Google Scholar]; c Shibata N.; Suzuki E.; Takeuchi Y. J. Am. Chem. Soc. 2000, 122, 10728. [Google Scholar]
- a Erb J.; Paull D. H.; Dudding T.; Belding L.; Lectka T. J. Am. Chem. Soc. 2011, 133, 7536. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Paull D. H.; Scerba M. T.; Alden-Danforth E.; Widger L. R.; Lectka T. J. Am. Chem. Soc. 2008, 130, 17260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lacour J.; Moraleda D. Chem. Commun. 2009, 7073. [DOI] [PubMed] [Google Scholar]; b Phipps R. J.; Hamilton G. L.; Toste F. D. Nat. Chem. 2012, 4, 603. [DOI] [PubMed] [Google Scholar]; c Brak K.; Jacobsen E. N. Angew. Chem., Int. Ed. 2013, 52, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mahlau M.; List B. Angew. Chem., Int. Ed. 2013, 52, 518. [DOI] [PubMed] [Google Scholar]
- a Rauniyar V.; Lackner A. D.; Hamilton G. L.; Toste F. D. Science 2011, 334, 1681. [DOI] [PubMed] [Google Scholar]; b Hamilton G. L.; Kanai T.; Toste F. D. J. Am. Chem. Soc. 2008, 130, 14984. [DOI] [PubMed] [Google Scholar]
- a Honjo T.; Phipps R. J.; Rauniyar V.; Toste F. D. Angew. Chem., Int. Ed. 2012, 51, 9684. [DOI] [PubMed] [Google Scholar]; b Phipps R. J.; Hiramatsu K.; Toste F. D. J. Am. Chem. Soc. 2012, 134, 8376. [DOI] [PubMed] [Google Scholar]; c Shunatona H. P.; Früh N.; Wang Y.-M.; Rauniyar V.; Toste F. D. Angew. Chem., Int. Ed. 2013, 52, 7724. [DOI] [PubMed] [Google Scholar]; d Wang Y.-M.; Wu J.; Hoong C.; Rauniyar V.; Toste F. D. J. Am. Chem. Soc. 2012, 134, 12928. [DOI] [PubMed] [Google Scholar]
- Phipps R. J.; Toste F. D. J. Am. Chem. Soc. 2013, 135, 1268. [DOI] [PubMed] [Google Scholar]
- Wu J.; Wang Y.-M.; Drljevic A.; Rauniyar V.; Phipps R. J.; Toste F. D. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the synthesis of enantioenriched α-fluoroketones via decarboxylation, see:; Nakamura M.; Hajra A.; Endo K.; Nakamura E. Angew. Chem., Int. Ed. 2005, 44, 7248. [DOI] [PubMed] [Google Scholar]; and; Mohr J. T.; Nishimata T.; Behenna D. C.; Stoltz B. M. J. Am. Chem. Soc. 2006, 128, 11348. [DOI] [PubMed] [Google Scholar]
- a Xu L.-W.; Luo J.; Lu Y. Chem. Commun. 2009, 1807. [DOI] [PubMed] [Google Scholar]; b Melchiorre P. Angew. Chem., Int. Ed. 2012, 51, 9748. [DOI] [PubMed] [Google Scholar]
- a Allen A. E.; MacMillan D. W. C. Chem. Sci. 2012, 3, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Du Z.; Shao Z. Chem. Soc. Rev. 2013, 42, 1337. [DOI] [PubMed] [Google Scholar]; c Schindler C. S.; Jacobsen E. N. Science 2013, 340, 1052. [DOI] [PubMed] [Google Scholar]
- a Krautwald S.; Sarlah D.; Schafroth M. A.; Carreira E. M. Science 2013, 340, 1065. [DOI] [PubMed] [Google Scholar]; b Sammis G. M.; Danjo H.; Jacobsen E. N. J. Am. Chem. Soc. 2004, 126, 9928. [DOI] [PubMed] [Google Scholar]
- For elegant studies combining chiral phosphoric acids and chiral primary amines, see:; a Martin N. J. A.; List B. J. Am. Chem. Soc. 2006, 128, 13368. [DOI] [PubMed] [Google Scholar]; b Wang X.; Reisinger C. M.; List B. J. Am. Chem. Soc. 2008, 130, 6070. [DOI] [PubMed] [Google Scholar]
- For the addition of stoichiometric chiral cyclohexyl enamines to Michael acceptors, see:; a Pfau M.; Revial G.; Guingant A.; d’Angelo J. J. Am. Chem. Soc. 1985, 107, 273. [Google Scholar]; b d’Angelo J.; Desmaële D.; Dumas F.; Guingant A. Tetrahedron Asym. 1992, 3, 459. [Google Scholar]
- We propose that the C8H17 alkyl chains of C8-TRIP aid in the phase-transfer process, promoting the asymmetric fluorination over any background reaction.
- The quinidine-derived primary amine catalyst optimal in ref (5) was found to give low yield and poor enantioselectivity in our ketone fluorination. See SI for details of this catalyst and for a survey of solvents.
- Ye C.; Twamley B.; Shreeve J. M. Org. Lett. 2005, 7, 3961. [DOI] [PubMed] [Google Scholar]
- For example, neither 1,2-diphenylpropan-1-one nor propiophenone undergo fluorination under our presently employed conditions.
- Kang J. Y.; Carter R. G. Org. Lett. 2012, 14, 3178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.