

Abstract
Improved outcomes for children with cancer hinge on the development of new targeted therapies with acceptable short-term and long-term toxicity. Progress in basic, preclinical, and clinical arenas spanning cellular immunology, gene therapy, and cell-processing technologies have paved the way for clinical applications of chimeric antigen receptor-based therapies. This is a new form of targeted immunotherapy that merges the exquisite targeting specificity of monoclonal antibodies with the potent cytotoxicity, potential for expansion, and long-term persistence provided by cytotoxic T cells. Although this field is still in its infancy, clinical trials have already shown clinically significant antitumor activity in neuroblastoma, chronic lymphocytic leukemia, and B-cell lymphoma, and trials targeting a variety of other adult and pediatric malignancies are under way. Ongoing work is focused on identifying optimal tumor targets and elucidating and manipulating both cell- and host-associated factors to support expansion and persistence of the genetically engineered cells in vivo. In pediatric oncology, CD 19 and GD2 are compelling antigens that have already been identified for targeting pre-B acute lymphoblastic leukemia and neuroblastoma, respectively, with this approach, but it is likely that other antigens expressed in a variety of childhood cancers will also soon be targeted using this therapy. The potential to target essentially any tumor-associated cell-surface antigen for which a monoclonal antibody can be made opens up an entirely new arena for targeted therapy of childhood cancer.
Introduction
Cell therapy for cancer is crossing the threshold of clinical activity, as trials in both adult and pediatric oncology have now shown unquestionable clinical responses (1–7). Historically, cell-based therapies have focused on cytolytic T cells targeting MHC-restricted antigens. Although this approach remains promising, its efficacy and applicability are limited by the need to enhance the affinity of naturally occurring T-cell receptors (TCR) that target most tumor antigens, and the tendency for tumors to downregulate MHC molecules. Furthermore, targeting MHC-restricted antigens is particularly challenging for pediatric cancer and other rare tumors because immunodominant epitopes have not been defined for most MHC alleles, and the difficulty of targeting a rare tumor in a limited population that expresses a particular MHC allele severely affects feasibility and applicability. For these reasons, chimeric antigen receptors (CAR; Fig. 1), which harness the potent cytolytic capacity of expanded effector populations with MHC-unrestricted targeting, are receiving increasing attention and represent a promising new therapeutic modality for childhood cancer. Below, we discuss the progress that has been made in this young field, as well as the challenges that remain to be overcome.
Figure 1.
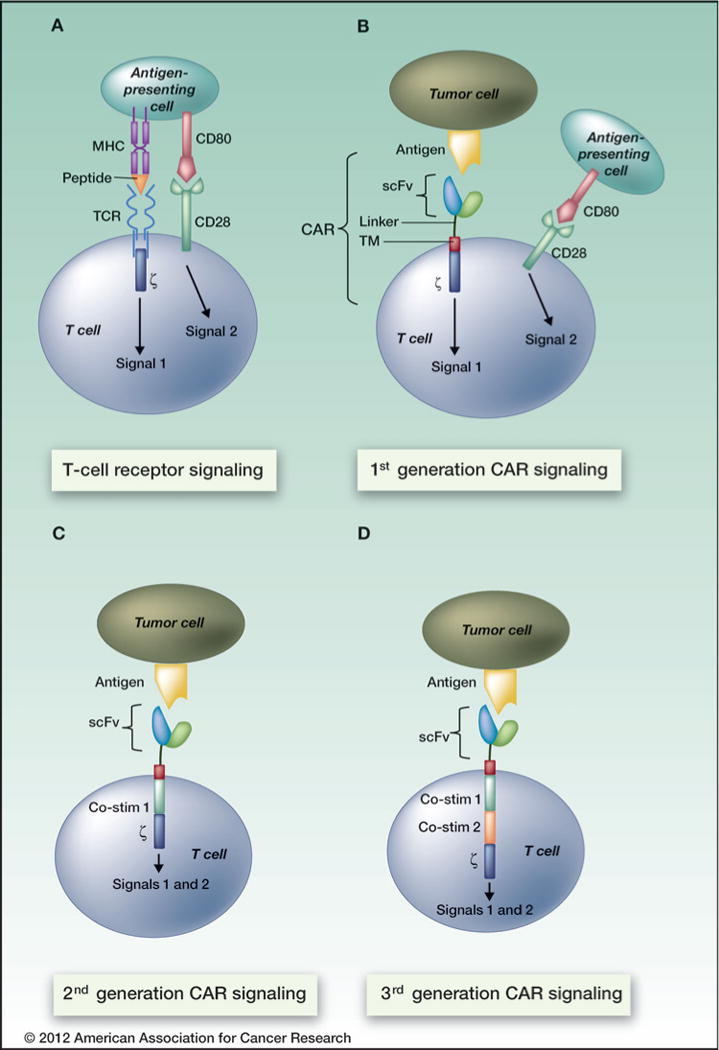
Current CAR design allows for MHC-independent antigen recognition and incorporates costimulatory signal (s) endowing the transduced T cell with potent cytotoxic activity. In contrast to the TCR, which recognizes peptide in the context of MHC and provides signal 1, CARs interact in an MHC-independent manner. All CARs must provide signal 1 in the form of the TCR z activating subunit (first-generation), but the addition of one (second-generation) or 2 (third-generation) costimulatory signals (CD28, 4-1BB, or OX40) provides the CAR-transduced T cell with both signals 1 and 2, leading to full activation, proliferation, and cytotoxicity.
CAR Design and Delivery
CARs (historically referred to as T-bodies) were first generated in 1989 by Eshhar and colleagues as fusions of antibody and TCR subunits that, when expressed on T cells, mediated MHC-independent T-cell activation (8–10). Although early results provided proof-of-principle, T-cell activation was not robust, and therefore CAR technologies were gradually optimized via modification and addition of subunits to increase signaling potency. The structure has evolved over the last 2 decades to most commonly incorporate a single-chain variable fragment (scFv) derived from a monoclonal antibody (mAb) plus the signaling motif from the TCR ζ chain [referred to as a first-generation CAR (Fig. 1; ref. 11)]. A major advance was made when 1 (second-generation CAR) or 2 (third-generation CAR) costimulatory activating motifs from CD28, 4-1BB (CD137), and/or CD134 (OX-40) were incorporated into CARs, leading to enhanced proliferation, cytotoxicity, and persistence in vivo (Fig. 1; refs. 3, 12–15). Beyond the general principle that CARs that incorporate costimulatory signals are more potent, it remains unclear whether any particular costimulatory molecule is superior to another, and to what extent the beneficial effects of any or all costimulators can be imparted by signaling during the expansion process without incorporating the molecule into the CAR itself (15–18). Of note, incorporation of the 4-1BB signaling motif into a CAR was recently reported to result in more robust proliferation in vitro compared with CD28 (15). These 2 costimulatory domains have not been compared in controlled clinical trials as yet; however, 2 of 3 patients with refractory chronic lymphocytic leukemia (CLL) treated with an anti-CD19 CAR that used 4-1BB as its costimulatory signal achieved durable (1 year) complete remissions, and the third achieved a partial remission (3, 19).
Multiple methods can be used to introduce CARs into effector cells. The most common approach uses g retro-viruses, which efficiently and stably integrate the receptor sequence into the target cell genome (6, 20, 21). Thus far, this approach has proved to be safe for transduction of mature T cells, although concerns remain that insertional mutagenesis or immune responses against retroviral antigens could occur (22, 23). Similarly, lentiviral transduction of the CAR sequence into expanding T cells has also been shown to be a safe and successful approach (3, 15). Non-viral methods, such as the use of transposon-based systems and direct RNA transfection (24–27), are also being explored. These methods are likely to be less costly to develop (25), and RNA transfection has the potential for self-limited expression in cases where permanent expression may be undesirable. In cases where permanent expression systems are used, some investigators have incorporated suicide genes, such as the herpes simplex thymidine kinase (TK) gene, or an inducible caspase 9 protein that can be activated by specific drugs and eradicate the genetically engineered cells if adverse effects occur following adoptive transfer (28–30).
Target Choice
CAR specificity is most commonly endowed by an scFv derived from phage display or from mAbs raised against cell-surface antigens, although in some cases receptor ligands have been used (31, 32). It is generally assumed that a high-affinity antigen-binding domain that targets a plentiful tumor cell-surface antigen with limited expression on normal tissue is desirable; however, the precise and relative importance of affinity and target expression levels have not been defined, and very few antigens show exclusive expression on tumors. The risk–benefit ratio of any particular CAR-expressing T lymphocyte is driven largely by properties of the target, because targets with vital organ expression will induce off-tumor, on-target effects. Depending on the tissues in which the target is expressed, this could result in minimal or unacceptable toxicity. B-cell malignancies provide several attractive CAR targets because mAbs are available to target several B-cell surface antigens, and the mature B-cell compartment is considered to be relatively expendable, at least temporarily, in patients with hematologic malignancies. Thus, CARs targeting CD20 (33, 34), CD19 (20), and CD22 (35) have been developed, and many trials for pediatric B-cell precursor acute lymphoblastic leukemia (pre-B ALL) are under way or anticipated (Table 1). CARs that target CD30 (18, 36) and ROR1 for lymphomas (37) are under study as well and may also be applicable to pediatric hematologic malignancies.
Table 1.
Active clinical trials of CAR therapy in children with cancer
| Location | Target | Malignancies | Comments | Trial number |
|---|---|---|---|---|
| Baylor/Texas Children’s Hospital/The Methodist Hospital | CD19 | pre-B ALL, B-cell lymphomas | 50% second generation (CD28)50% first generation | NCT00586391 |
| Baylor/Texas Children’s Hospital/The Methodist Hospital | CD19 | pre-B ALL, B-cell lymphomas | multivirus CTL transduced post-BMTsecond generation (CD28) | NCT00840853 |
| Children’s Hospital of Philadelphia/University of Pennsylvania | CD19 | pre-B ALL, B-cell lymphomas CLL (adults) | lentiviral transduction second generation (4-1BB) | NCT01029366 |
| Children’s Hospital of Philadelphia/University of Pennsylvania | CD19 | pre-B ALL, B-cell lymphomas CLL (adults) | donor T cells given for relapse after allo SCTsecond generation (4-1BB) | pending |
| University College of London | CD19 | pre-B ALL, B-cell lymphomas | donor EBV CTL post-BMT with EBV-LCL vaccinesecond generation (CD28) | NCT01195480 |
| Memorial Sloan-Kettering Cancer Center | CD19 | pre-B ALL, B-cell lymphomas | donor EBV CTL post-BMT second generation (CD28) | NCT01430390 |
| The University of Texas MD Anderson Cancer Center | CD19 | pre-B ALL, B-cell lymphomas | donor-derived modified T cells after umbilical cord transplant | NCT01362452 |
| National Cancer Institute Pediatric Oncology Branch | CD19 | pre-B ALL, B-cell lymphomas | second generation (CD28) | pending |
| Baylor/Texas Children’s Hospital/The Methodist Hospital | Her2 | Her2+ sarcomas | second generation (CD28) | NCT00902044 |
| glioblastoma | autologous CMV CTL (glioblastoma) | NCT01109095 | ||
| Baylor/Texas Children’s Hospital/The Methodist Hospital | CD30 | Hodgkin and CD30 + non-Hodgkin lymphoma | autologous EBV CTLs | NCT01192464 |
| first generation autologous T cells second generation (CD28) | NCT01316146 | |||
| Baylor/Texas Children’s Hospital | GD2 | neuroblastoma | autologous EBV CTLs first generation autologous polyclonal CTLs first generation | NCT00085930 |
Abbreviations: BMT, bone marrow transplant; CMV, cytomegalovirus; CTL, cytotoxic T lymphocyte; LCL, lymphoblastic cell line; SCT, stem cell transplantation.
Choosing CAR targets for solid tumors is potentially more challenging because solid-tumor–associated antigens often coexist on nonexpendable tissues. GD2 is a validated tumor antigen for which mAb-based targeting has proved to be safe (38). Efforts to target GD2 with mAbs are reviewed by Matthay and colleagues in this CCR Focus section (39). Not surprisingly, therefore, one of the first trials of CAR therapy used a GD2-CAR administered to children with advanced neuroblastoma. In that study, 3 of 11 patients with active disease experienced complete responses, no substantial toxicity was observed, and sustained clinical benefit was reported for several patients with long-term follow-up (1, 2). Of note, the CARs used in this trial were of the first-generation variety and incorporated only CD3 ζ chain signaling motifs. Current approaches that incorporate cost-imulatory signaling motifs may be even more active. In contrast to the favorable safety profile seen when targeting GD2, CAR therapy used to target Her2/Neu resulted in a patientdeath that was attributed to Her2/Neu expression on normal lung and/or cardiac tissue (40). It is important to note, however, that substantially higher numbers of CAR-transduced cells were administered in this case than were administered in many other trials, raising the prospect that lower doses of CARs targeting this antigen might also be safe. Indeed, a cautious dose-escalation study is under way in pediatric patients with osteosarcoma to determine whether Her2/Neu-CART cells have acceptable risk–benefit ratios (http://clinicaltrials.gov/ct2/show/NCT00902044). Nonetheless, this clinical experience illustrates that antigens that are safely targeted by antibody therapy cannot necessarily be assumed to be safe when targeted with CARs.
A full list of potential targets for CAR-based therapy of pediatric solid tumorsis beyond the scope of this report, but this is an area of very active investigation (for review, see ref. 41). Targets that are currently under study include epidermal growth factor receptor (EGFR) vIII (42) and interleukin (IL)-13 receptor a2 (32, 43), both of which are expressed on a substantial number of pediatric gliomas. B7H3 is expressed by many sarcomas and pediatric brain tumors (44), and fibroblast growth receptor 2 (FGFR2) is highly expressed by most rhabdomyosarcomas (45). Substantially more work is needed to prioritize targets for testing in early clinical trials of CAR therapy in pediatric oncology, and this will be based on preclinical studies of killing activity and the relative risk of off-target effects.
Manufacturing Cell Populations That Express CARs
Several groups have developed clinical-grade cell-manufacturing systems that are capable of producing large numbers of CAR-modified T cells. There is wide variation in the specific processes used, and to date there is no clear evidence that one is superior to another, although this remains an area of active investigation. One of the most commonly used methods employs anti-CD3 plus autologous or allogeneic feeder cells and high-dose IL-2 (Fig. 2A, I; refs. 46–49). Alternatively, artificial antigen-presenting cells (aAPC) that provide anti-CD3 plus costimulatory signals have been developed in an effort to replicate the physiologic process of T-cell activation by dendritic cells. Cell-based aAPCs, most often K562 cells modified to express costimulatory ligands (4-1BBL, CD80) and/or cytokines (IL-15 and IL-21), induce potent expansion of both natural killer cells and/or T cells (Fig. 2A, II; refs. 50–53). Bead-based aAPCs (Fig. 2A, III) employing anti-CD3 and anti-CD28 antibodies coupled to paramagnetic beads induce robust T-cell expansion without the need for exogenous cytokines (54, 55). All of these methods are sufficient to generate a several-hundred-fold expansion of cells over a 10- to 14-day period (Fig. 2B). In addition, investigators have developed approaches to expand donor or patient Epstein–Barr virus (EBV)- and cytomegalovirus (CMV)-specific T cells using viral antigens that engage endogenous TCRs and costimulatory pathways before and after CAR transduction (1, 18, 56). Such virus-specific T cells could prove useful for administering CAR T cells following allogeneic hematopoietic stem cell transplant because they are not predicted to induce graft-versus-host disease. In addition, the investigators hypothesize that exposure to intermittent viral reactivation in vivo will contribute to persistence following transfer. However, the production of CAR-modified, virus-specific cells generally requires a longer culture period compared with the polyclonal techniques described above (18).
Figure 2.
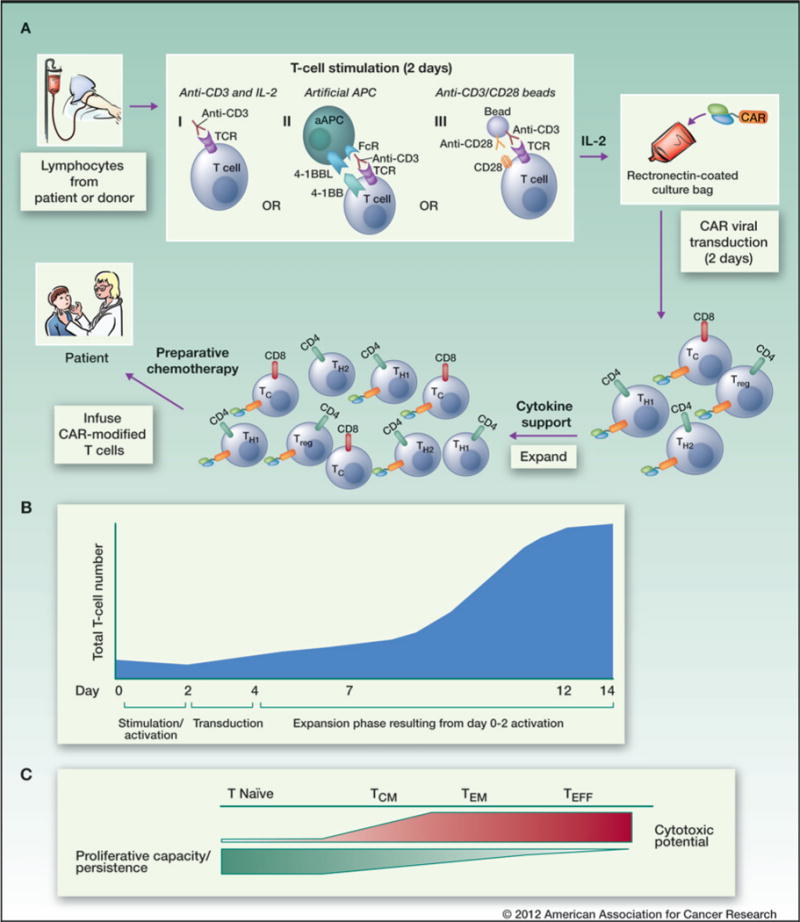
General schema for the preparation, transduction, and infusion of CAR-modified T cells. A, apheresed T cells from a patient or an allogeneic donor are activated. Three accepted methods are illustrated: (i) stimulation with the activating CD3 antibody, OKT3, in the presence of IL-2; (ii) stimulation with anti-CD3– and anti-CD28–coated paramagnetic beads in the presence of IL-2; and (iii) stimulation with aAPC (expressing 4-1BBL and an Fc receptor) with OKT3 and IL-2. Activated cells are then transduced with the CAR using a retro- or lentiviral platform. Because the CAR is integrated into the T-cell genome, all daughter cells that are generated (a mix of CD4+ [Th1/Th2/TH17/Treg] and CD8+ T cells) during this expansion also express the CAR. CAR T cells are then infused into the patient after preparative chemotherapy. B, generation of CAR-expressing T cells generally results in a several-hundred-fold expansion over 14 days. Such extensive proliferation may generate predominantly TEFF cells, which have cytotoxic capabilities but limited proliferative potential compared with T-effector and TCM cells. C, less intense stimulation and/or modulation of stimulation methods may produce more naïve T cells or T–central memory cells, which have an increased likelihood of persistence.
Cells that have been expanded in each of the above systems are amenable to transduction, secrete cytokines, and lyse antigen-expressing cells (57, 58); however, emerging studies suggest that the nature of the starting cell population that is genetically modified and/or the method of expansion may affect the likelihood that CAR T cells will persist in vivo and effectively mediate antitumor effects. In models that emphasized generation of larger doses of CAR T cells at the expense of driving them toward short-lived T-effector cells (Fig. 2C), recent studies of adoptive immunotherapy correlated clinical benefit with in vivo cell expansion and persistence (2, 59–61). In a recent clinical study, administration ofa very low number of CAR-specific T cells, calculated to yield an effector:target ratio of 1 CAR-transduced T cell to 93,000 CLL cells, yielded a dramatic clinical benefit (1, 3). This finding and the observation that administration of small numbers of T cells can mediate dramatic effects against large tumor burdens (62) emphasize the notion that cells that are capable of robust proliferation can overcome daunting tumor burdens.
Data gleaned from a variety of murine and human studies of adoptive T-cell immunotherapy show that cells bearing a terminally differentiated T-effector memory (TEM) pheno-type, which tendto dominate prolonged ex vivo cultures and systems using anti-CD3 plus IL-2, have a limited capacity to expand and persist in vivo following an encounter with the tumor (63). In contrast, T-central memory (TCM) cells are more potent in several models (64), and it is predicted that this subset may be more effective in the setting of CAR-transduced T cells as well. Some studies showed enhanced efficacy when TCM cells were usedasastartingpopulation for transduction (65), whereas in other studies investigators focused on expanding TCM cells using APC-based systems (66). Surprisingly, in some cases, TCM cells emerged in vivo following administration of a mixed population of predominantly TEM cells (64). Virus-specific T cells showed enhanced persistence compared with nonviral CAR T cells, perhaps due to an increased frequency of TCM in virus-specific populations (1). Recently, a new subset termed T stem cell memory (TSCM) performed better than TCM and TEM in a preclinical model of CARs directed against the tumor antigen mesothelin (67). Studies are under way to determine optimal approaches for generating or maintaining TSCM cells during expansion, and several agents, including rapamycin, IL-21 (26, 68), and Wnt signaling modulators (69), appear to be capable of limiting terminal differentiation and/or enhancing TSCM cells. Finally, recent data suggest that the efficacyof CAR T-cell products correlates with the number of CD4+ T cells infused (2), and historical data provide evidence that CD4+ T cells themselves support the persistence of CD8+ T cells (70). Of interest, bead-based aAPCs preferentially expand CD4+ T cells compared with most other approaches (26, 66). Although CD4+ regulatory T cells (Treg) theoretically could be transduced with a CAR or develop from a non-Treg that was CAR transduced (Fig. 2A), culture conditions do not seem to select for this subset, and there is no evidence thus far that CAR-transduced Tregs mediate clinically significant immunosuppression.
Given the multitude of cell subsets that can be targeted with CAR therapy, and the vast array of approaches that can be used to transduce and expand CAR T cells, it would be highly desirable to obtain accurate preclinical models to predict clinical efficacy. Unfortunately, for most CARs, such studies are limited to xenograft models that incorporate adoptive transfer of human tumors and human T cells into immunodeficient mice. Although such models provide some insights into clinical activity, they may not provide an accurate assessment of persistence in humans, and provide little insight into toxicity, including CAR T-cell–induced autoimmunity. Fully murine models may be preferable but are not available for most CARs currently under study in humans. Thus, optimizing preclinical models to prioritize targets and cell-manufacturing methods represents an important area for future efforts.
Preparing the Host
Early studies of adoptive immunotherapy focused on generating large quantities of antigen-specific effectors to overcome the perceived limitations posed by overwhelming tumor cell numbers compared with available effectors. In contrast, current efforts are more focused on administering cells with robust proliferative capacity, and modulating the host immune milieu to support in vivo expansion and persistence. Lymphopenia induces profound changes in T-cell physiology, due primarily to accumulation of IL-7 and IL-15, homeostatic cytokines that support T-cell expansion and survival (71–73). As a result, most adoptive cell therapy protocols incorporate lymphotoxic therapies prior to cell transfer to increase the availability of such homeostatic cytokines. The results of nonrandomized trials support this approach (7, 74), but no definitive data are available to confirm that it is necessary.
In pediatric oncology, most candidates for adoptive immunotherapy are already lymphopenic due to previous exposure to cytotoxic regimens. GD2-CAR therapy administered without a lymphodepleting regimen showed antitumor activity in patients with neuroblastoma (1, 2), although it remains unclear to what extent these patients were lymphopenic at the time cell therapy was initiated. Lymphodepleting regimens may also enhance the efficacy of adoptive cell therapies by transiently reducing Treg numbers (75), and by inducing mucosal damage that results in systemic exposure to lipopolysaccharide and other bacterial byproducts, which activate the innate immune system (Fig. 3; refs. 76, 77). Preclinical models show that administration of homeostatic cytokines combined with targeted therapies to induce Treg depletion is more effective than nonspecific cytotoxic regimens in supporting adoptive immunotherapy (78). Therefore, efforts are under way to induce or administer the essential host factors that will support adoptively transferred cells while limiting the toxicity of the regimen. This is particularly relevant for the field of pediatric oncology, where the desire to limit late effects is a major factor driving the development of targeted therapies such as CAR-based regimens.
Figure 3.
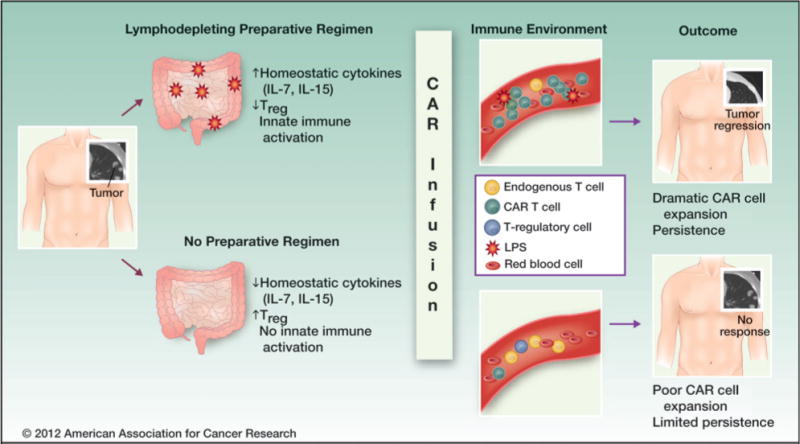
Lymphodepleting preparatory regimens can enhance the efficacy of adoptive cell therapy. Proposed mechanisms include (i) a reduction in endogenous lymphocytes, leading to accumulation of IL-7 and IL-15 (homeostatic cytokines that support cell expansion and persistence); (ii) a transient reduction in the number and frequency of Tregs, thereby diminishing suppression; and (iii) induction of gut damage, which can lead to the systemic release of bacterial byproducts [e.g., lipopolysaccharide (LPS)] that activate the innate immune system.
Brief Summary of Clinical Trials of CAR Therapy
At the present time, because of the complexity and cost of generating CAR-based therapies, only a few specialized centers are capable of delivering such products. Further, the approach is currently supported entirely by academia, government, and private sources; to date, no biotechnology or pharmaceutical company has sponsored a CAR trial. Despite these limitations, however, several clinical trials have already been completed and many others are currently under way or planned, including somein pediatrics. Several key elements of successful CAR therapy have been elucidated in these trials. First, a proof-of-principle that CAR therapy can mediate a clinical benefit against childhood cancers was provided in the first trial undertaken in pediatrics. Most trials that used CARs lacking costimulatory motifs were unsuccessful due to poor persistence and/or expansion of the transduced T cells; however, a clinical trial of anti-GD2 CAR T cells in 19 patients with neuroblastoma who received a mixture of autologous, CAR-modified EBV-cytotoxic T lymphocytes (CTL) and anti-CD3–activated polyclonal CAR-modified T cells proved to be an exception (1, 2). These patients did not receive a preparative regimen, although they may have already been lymphopenic due to extensive prior therapy. Three of 11 evaluable patients had a complete response, and improved clinical outcomes were observed in patients in whom CAR T cells could be detected beyond 6 weeks. In this trial, improved clinical outcomes also correlated with higher levels of CD4+ cells and higher levels of central memory cells in the infused adoptive cell product (2). Of interest, although pain associated with anti-GD2 mAb therapy is commonly seen, no significant GD2-CAR–associated pain was observed in this trial, raising the prospect that in some cases, CAR-based therapy could be less toxic than mAb therapy.
Clinical trials in adults have provided evidence that CD19 is a safe and effective target for CAR therapy of B-cell malignancies (3, 4, 19). In these studies, patients who experienced clinically meaningful responses with lysis of large tumor burdens and maintenance of complete responses showed massive expansion of the infused cells, and persistence of cells for at least 6 to 12 months. Thus, clinical activity has been observed in several CAR clinical trials, including those targeting CD19 and GD2, both of which are antigens expressed on pediatric tumors. Several groups have shown that incorporation of a costimulatory domain(s) into a CAR leads to increased persistence (17, 79, 80), and results from nonrandomized studies suggest that a lymphodepleting regimen improves outcomes (60, 77, 80). Therefore, most studies that are currently planned or under way in pediatrics will incorporate costimulatory elements in the CAR design and use a lymphodepleting regimen prior to adoptive transfer.
These early successes with CAR therapy, however, were not achieved without substantial toxicities. Flu-like symptoms such as fever, malaise, and myalgias are common after CAR T-cell infusion and likely relate to cytokine release by the infused cells, because they coincide with increases in IFN-g (19). As discussed earlier, one adult patient treated with 1010 highly activated Her2/Neu-CAR T cells developed fatal immune activation syndrome resulting from low-level target expression in the lungs shortly after CAR T-cell infusion (40, 81). A second patient who received cyclophosphamide conditioning followed by 3 × 107/kg anti-CD 19 CART cells developed exacerbation of preexisting immune activation, presumably from an underlying infection, 6 hours after cell infusion and ultimately died (81, 82). As a result of these toxicities, recent trials have decreased the number of infused CAR cells in the initial cohorts (generally starting at 106/kg), and incorporated close monitoring of clinical symptoms and serum cytokine levels. Other observed toxicities include autoimmunity mediated by on-target, off-tissue effects (e.g., hepatic toxicity) resulting from therapy with carbonic anhydrase IX-CAR, which has since been shown to be expressed on cells in the biliary tract (23). Furthermore, anti-CAR immune responses (23) may adversely affect the efficacy of the therapy itself.
Conclusions
Dramatic progress in our understanding of genomics (83, 84), cancer biology (85), and immunology (39) is fueling exciting new progress in the search for more effective and less toxic targeted therapies for childhood cancer (86). Among the most novel and promising of these approaches are CAR-based cell therapies that combine advances in genetic engineering and adoptive immunotherapy. Current research focused on optimizing this approach emphasizes (i) effective tumor targeting with limited off-tumor toxicity, (ii) optimized cell manufacturing to improve efficacy and render the therapies more exportable, and (iii) modulation of the host and/or cell product to increase in vivo expansion and/or persistence. In pediatric cancers, several candidate antigens have already been identified that provide acceptable tumor targeting, but much more work is needed in this arena, especially for pediatric solid tumors. Cell-surface antigens with robust tumor expression and limited normal tissue expression that could be effectively and safely targeted with this therapy must be systematically identified and prioritized. With regard to optimizing cell manufacturing, it is clear that many different approaches can reliably generate large numbers of CAR-expressingT cells. However, better preclinical models and surrogates for bioactivity are needed to compare manufacturing approaches, and carefully controlled clinical trials are needed to directly test the various CAR constructs and expansion techniques that are currently available. Finally, optimal modulation of host factors is also likely a key determinant of in vivo expansion and persistence, and recent insights suggest that lymphopenic hosts that receive cells bearing TCM or TSCM pheno-types experience optimal antitumor effects. Given the limitation in patient numbers that challenges early-phase pediatric trials, we anticipate that seminal insights in each of these arenas will be gleaned from clinical trials that are under way in adult oncology, or in trials that enroll both adult and pediatric patients. Ultimately, however, careful clinical trials in children will be needed to assess efficacy and oxicity. If favorable antitumor effects are observed, we anticipate that progress in manufacturing techniques will lead to reductions in the cost and complexity of generating these therapies, allowing them to be exported and/or made available to institutions where such approaches are currently not available.
We envision that CAR-based therapies will ultimately comprise multimodal regimens, likely incorporating host preparative regimens, novel elements in the cell expansion cocktail, and potentially cytokines to maximize in vivo expansion. Although most immunotherapies seem to be most active in the setting of minimal residual disease, impressive antitumor effects observed thus far in a small number of patients with bulky disease raise the prospect that CAR-based therapies may have activity in the setting of large tumor burdens. For studies undertaken in the setting of minimal residual disease, novel clinical trial endpoints are needed to determine whether results from early-phase trials warrant larger randomized trials with survival endpoints. Careful clinical trials will be needed to assess the optimal timing for incorporating CAR-based adoptive immunotherapies, and to compare CAR-based targeting of cell-surface antigens with mAb-based immunotherapies. In summary, CARs represent a novel and promising approach for targeted therapy of childhood cancer. Progress in this arena will be largely driven by academia and will require support for expensive early-phase clinical trials that promise to pave the way for a new form of targeted, exportable immunotherapy for children with cancer.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: D.W. Lee, D.M. Barrett, C.L. Mackall, R.J. Orentas, S.A. Grupp
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): D.W. Lee, R.J. Orentas, S.A. Grupp Writing, review, and/or revision of the manuscript: D.W. Lee, D.M. Barrett, C.L. Mackall, R.J. Orentas, S.A. Grupp,
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): D.W. Lee, R.J. Orentas
References
- 1.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblas-toma. Nat Med. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–9. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta sub-units of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–4. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedmann-Morvinski D, Bendavid A, Waks T, Schindler D, Eshhar Z. Redirected primary T cells harboring a chimeric receptor require costimulation for their antigen-specific activation. Blood. 2005;105:3087–93. doi: 10.1182/blood-2004-09-3737. [DOI] [PubMed] [Google Scholar]
- 10.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86:10024–8. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okur FV, Brenner MK. Cellular immunotherapy of cancer. Methods Mol Biol. 2010;651:319–45. doi: 10.1007/978-1-60761-786-0_19. [DOI] [PubMed] [Google Scholar]
- 12.Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172:104–13. doi: 10.4049/jimmunol.172.1.104. [DOI] [PubMed] [Google Scholar]
- 13.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–84. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 14.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 15.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1999;1:123–7. doi: 10.1038/sj.neo.7900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 18.Savoldo B, Rooney CM, Di Stasi A, Abken H, Hombach A, Foster AE, et al. Epstein Barr virus specific cytotoxic T lymphocytes expressing theanti-CD30zeta artificial chimericT-cell receptor for immunotherapy of Hodgkin disease. Blood. 2007;110:2620–30. doi: 10.1182/blood-2006-11-059139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kochenderfer JN, Feldman SA, Zhao YB, Xu H, Black MA, Morgan RA, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam JS, Reeves ME, Cowherd R, Rosenberg SA, Hwu P. Improved gene transfer into human lymphocytes using retroviruses with the gibbon ape leukemia virus envelope. Hum Gene Ther. 1996;7:1415–22. doi: 10.1089/hum.1996.7.12-1415. [DOI] [PubMed] [Google Scholar]
- 22.Burns WR, Zheng Z, Rosenberg SA, Morgan RA. Lack of specific gamma-retroviral vector long terminal repeat promoter silencing in patients receiving genetically engineered lymphocytes and activation upon lymphocyte restimulation. Blood. 2009;114:2888–99. doi: 10.1182/blood-2009-01-199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. 2011;117:72–82. doi: 10.1182/blood-2010-07-294520. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–9. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68:2961–71. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011;71:3516–27. doi: 10.1158/0008-5472.CAN-10-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22:1575–86. doi: 10.1089/hum.2011.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–70. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper LJ, Ausubel L, Gutierrez M, Stephan S, Shakeley R, Olivares S, et al. Manufacturing of gene-modified cytotoxic T lymphocytes for autologous cellular therapy for lymphoma. Cytotherapy. 2006;8:105–17. doi: 10.1080/14653240600620176. [DOI] [PubMed] [Google Scholar]
- 30.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stastny MJ, Brown CE, Ruel C, Jensen MC. Medulloblastomas expressing IL13Ralpha2 are targets for IL13-zetakine+ cytolytic T cells. J Pediatr Hematol Oncol. 2007;29:669–77. doi: 10.1097/MPH.0b013e3181468c68. [DOI] [PubMed] [Google Scholar]
- 32.Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–6. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–25. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, Press OW, Lindgren CG, Greenberg P, Riddell S, Qian X, et al. Cellular immunotherapy for follicular lymphoma using genetically modified CD20-specific CD8+ cytotoxic T lymphocytes. Mol Ther. 2004;9:577–86. doi: 10.1016/j.ymthe.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 35.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180:7028–38. doi: 10.4049/jimmunol.180.10.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116:4532–41. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Children’s Oncology Group Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–34. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clin Cancer Res. 2012;18:2740–53. doi: 10.1158/1078-0432.CCR-11-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orentas RJLD, Mackall C. Immunotherapy targets in pediatric cancer. Front Pediatr Oncol. 2012;2:1–16. doi: 10.3389/fonc.2012.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bax DA, Gaspar N, Little SE, Marshall L, Perryman L, Regairaz M, et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin Cancer Res. 2009;15:5753–61. doi: 10.1158/1078-0432.CCR-08-3210. [DOI] [PubMed] [Google Scholar]
- 43.Kioi M, Seetharam S, Puri RK. Targeting IL-13Ralpha2-positivecancer with a novel recombinant immunotoxin composed of a single-chain antibody and mutated Pseudomonas exotoxin. Mol Cancer Ther. 2008;7:1579–87. doi: 10.1158/1535-7163.MCT-07-2131. [DOI] [PubMed] [Google Scholar]
- 44.Luther N, Cheung NK, Souliopoulos EP, Karampelas I, Bassiri D, Edgar MA, et al. Interstitial infusion of glioma-targeted recombinant immunotoxin 8H9scFv-PE38. Mol Cancer Ther. 2010;9:1039–46. doi: 10.1158/1535-7163.MCT-09-0996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor JG, 6th, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119:3395–407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Padula SJ, Pollard MK, Lingenheld EG, Clark RB. Maintenance of antigen specificity by human interleukin-2-dependent T cell lines. Use of antigen-presenting cells and OKT3 antibody in the absence of antigen. J Clin Invest. 1985;75:788–97. doi: 10.1172/JCI111774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ochoa AC, Hasz DE, Rezonzew R, Anderson PM, Bach FH. Lymphokine-activated killer activity in long-term cultures with anti-CD3 plus interleukin 2: identification and isolation of effector subsets. Cancer Res. 1989;49:963–8. [PubMed] [Google Scholar]
- 48.Hoyle C, Bangs CD, Chang P, Kamel O, Mehta B, Negrin RS. Expansion of Philadelphia chromosome-negative CD3(+)CD56(+) cytotoxic cells from chronic myeloid leukemia patients: in vitro and in vivo efficacy in severe combined immunodeficiency disease mice. Blood. 1998;92:3318–27. [PubMed] [Google Scholar]
- 49.Goedegebuure PS, Douville LM, Li H, Richmond GC, Schoof DD, Scavone M, et al. Adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 in patients with metastatic malignant melanoma and renal cell carcinoma: a pilot study. J Clin Oncol. 1995;13:1939–49. doi: 10.1200/JCO.1995.13.8.1939. [DOI] [PubMed] [Google Scholar]
- 50.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–83. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee DA, Verneris MR, Campana D. Acquisition, preparation, and functional assessment of human NK cells for adoptive immunotherapy. Methods Mol Biol. 2010;651:61–77. doi: 10.1007/978-1-60761-786-0_4. [DOI] [PubMed] [Google Scholar]
- 52.Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010;17:147–54. doi: 10.1038/cgt.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15:981–8. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–30. [PubMed] [Google Scholar]
- 55.Garlie NK, LeFever AV, Siebenlist RE, Levine BL, June CH, Lum LG. T cells coactivated with immobilized anti-CD3 and anti-CD28 as potential immunotherapy for cancer. J Immunother. 1999;22:336–45. doi: 10.1097/00002371-199907000-00007. [DOI] [PubMed] [Google Scholar]
- 56.Biagi E, Marin V, Attianese GM, Pizzitola I, Tettamanti S, Cribioli E, et al. New advances in leukaemia immunotherapy by the use of chimeric artificial antigen receptors (CARs): state of the art and perspectives for the near future. Ital J Pediatr. 2011;37:46. doi: 10.1186/1824-7288-37-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carlens S, Gilljam M, Remberger M, Aschan J, Christensson B, Dilber MS. Ex vivo T lymphocyte expansion for retroviral transduction: influence of serum-free media on variations in cell expansion rates and lymphocyte subset distribution. Exp Hematol. 2000;28:1137–46. doi: 10.1016/s0301-472x(00)00526-9. [DOI] [PubMed] [Google Scholar]
- 58.Stockschläder M, Haiss M, Exner S, Schmah O, Veelken H, Follo M, et al. Expansion and fibronectin-enhanced retroviral transduction of primary human T lymphocytes for adoptive immunotherapy. J Hematother Stem Cell Res. 1999;8:401–10. doi: 10.1089/152581699320162. [DOI] [PubMed] [Google Scholar]
- 59.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–30. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99:16168–73. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Powell DJ, Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105:241–50. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paulos CM, Suhoski MM, Plesa G, Jiang T, Basu S, Golovina TN, et al. Adoptive immunotherapy: good habits instilled at youth have long-term benefits. Immunol Res. 2008;42:182–96. doi: 10.1007/s12026-008-8070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–7. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–33. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–13. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–44. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 71.Fry TJ, Connick E, Falloon J, Lederman MM, Liewehr DJ, Spritzler J, et al. A potential role for interleukin-7 in T-cell homeostasis. Blood. 2001;97:2983–90. doi: 10.1182/blood.v97.10.2983. [DOI] [PubMed] [Google Scholar]
- 72.Thiant S, Yakoub-Agha I, Magro L, Trauet J, Coiteux V, Jouet JP, et al. Plasma levels of IL-7 and IL-15 in the first month after myeloablative BMT are predictive biomarkers of both acute GVHD and relapse. Bone Marrow Transplant. 2010;45:1546–52. doi: 10.1038/bmt.2010.13. [DOI] [PubMed] [Google Scholar]
- 73.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–7. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11:1238–43. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 76.Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117:2197–204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. 2010;33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cui Y, Zhang H, Meadors J, Poon R, Guimond M, Mackall CL. Harnessing the physiology of lymphopenia to support adoptive immu-notherapy in lymphoreplete hosts. Blood. 2009;114:3831–40. doi: 10.1182/blood-2009-03-212134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183:5563–74. doi: 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lympho-blastic leukemia xenografts. Clin Cancer Res. 2007;13:5426–35. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 81.Ertl HC, Zaia J, Rosenberg SA, June CH, Dotti G, Kahn J, et al. Considerations for the clinical application of chimeric antigen receptor T cells: observations from a recombinant DNA Advisory Committee Symposium held June 15, 2010. Cancer Res. 2011;71:3175–81. doi: 10.1158/0008-5472.CAN-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18:666–8. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Loh ML, Mullighan CG. Advances in the genetics of high-risk childhood B-progenitor acute lymphoblastic leukemia and juvenile myelomonocytic leukemia: implications for therapy. Clin Cancer Res. 2012;18:2754–67. doi: 10.1158/1078-0432.CCR-11-1936. [DOI] [PubMed] [Google Scholar]
- 84.Pinto N, Cohn SL, Dolan ME. Using germline genomics to individualize pediatric cancer treatments. Clin Cancer Res. 2012;18:2791–800. doi: 10.1158/1078-0432.CCR-11-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lawlor ER, Thiele CJ. Epigenetic changes in pediatric solid tumors: promising new targets. Clin Cancer Res. 2012;18:2768–79. doi: 10.1158/1078-0432.CCR-11-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thiele CJ, Cohn SL. Genetically InFormed Therapies—a “GIFT” for children with cancer. Clin Cancer Res. 2012;18:2735–9. doi: 10.1158/1078-0432.CCR-11-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]