

Abstract
Serum amyloid A (SAA) is an apolipoprotein involved in poorly understood roles in inflammation. Upon trauma, hepatic expression of SAA rises 1000 times the basal levels. In the case of inflammatory diseases like rheumatoid arthritis, there is a risk for deposition of SAA fibrils in various organs leading to Amyloid A (AA) amyloidosis. Although the amyloid deposits in AA amyloidosis accumulate with the glycosaminoglycan (GAG) heparan sulfate, the role GAGs play in the function and pathology of SAA is an enigma. It has been shown that GAG sulfation is a contributing factor in protein fibrillation and for co-aggregating with a plethora of amyloidogenic proteins. Herein, the effects of heparin, heparan sulfate, hyaluronic acid, chondroitin sulfate A, and heparosan on the oligomerization and aggregation properties of pathogenic mouse SAA1.1 were investigated. Delipidated SAA was used to better understand the interactions between SAA and GAGs without the complicating involvement of lipids. The results revealed—to varying degrees—that all GAGs accelerated SAA1.1 aggregation, but had variable effects on its fibrillation. Heparan sulfate, hyaluronic acid, and heparosan did not affect much the fibrillation of SAA1.1. In contrast, chondroitin sulfate A blocked SAA fibril formation and facilitated the formation of spherical aggregates of various sizes. Interestingly, heparin caused formation of spherical SAA1.1 aggregates of various sizes, vast amounts of thin protofibrils, and few long fibrils of various heights. These results suggest that GAGs may have an intrinsic and divergent influence on the aggregation and fibrillation of HDL-free SAA1.1 in vivo, with functional and pathological implications.
Keywords: GAGs, reactive amyloidosis, inflammation, fibrillation kinetics
1. Introduction
Serum amyloid A (SAA) is an apolipoprotein that also interacts with carbohydrates known as glycosaminoglycans (GAGs) [1]. During acute inflammation SAA binds via its N-terminal domain to high density lipoprotein (HDL) and is involved in cholesterol transport, one of SAA's major functions [2]. In addition, it appears that SAA may be involved in various immunological functions [3]. Upon trauma or infection, pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) [4, 5] trigger hepatic overexpression of SAA to concentrations of up to 1 mg/mL, or 1000 times its basal level [6]. However, following persistent acute inflammation, as occur in rheumatoid arthritis, Crohn's disease, or tuberculosis, individuals may develop AA amyloidosis, a systemic amyloid disease characterized by deposition of SAA amyloid fibrils in the kidney, spleen, and liver [1]. The SAA amyloid deposits (also known as AA amyloid) largely comprise the N-terminal 76 residues of the SAA1.1 isoform.
For a long time it was known that amyloid deposits in vivo contain glycosaminoglycans (GAGs), in particular heparan sulfate (HS) [7]. GAGs are linear anionic polysaccharides, comprised of disaccharide repeating motifs of hexosamine and hexuronic acid, and their major functions range from anticoagulation, cell signaling and growth, to joint lubrication (Table 1) [8-10]. Although not appreciated at the time, the presence of carbohydrates in amyloid deposits dates to 1854, when Virchow characterized the “lardaceous” liver deposits first identified by Rokitansky (in 1842) as composed of starch material [11]. Virchow borrowed the botanical word when he designated these deposits “amyloid” (i.e. starch in Latin) [1, 12]. Five years later, Kekulé and Friedreich chemically characterized amyloid deposits as mostly proteinaceous [13]. Although amyloid research has focused on the protein/peptide component recent work has demonstrated increased interest in evaluating the intrinsic roles of GAGs in amyloid deposition for numerous proteins [14]. Various in vitro studies have shown that GAGs promote fibril formation by diverse proteins, presumably due to a scaffolding effect that is facilitated by anionic moieties [15, 16].
Table 1.
Chemical and biological characteristics of glycosaminoglycans (GAGs) employed in investigating SAA oligomerization and aggregation.
| GAG | Disaccharide structure1 | Chemical & biological properties |
|---|---|---|
| Heparin (Hep) |
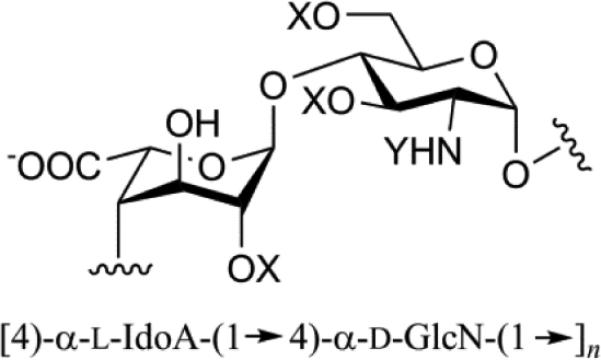
|
Mol. Wt. 5 – 40 kDa 90 % L-Iduronic acid High sulfation; 3 sulfates/disaccharide Anticoagulant produced in mast cells |
| Heparan Sulfate (HS) |

|
Mol. Wt. 5 – 40 kDa Varied sulfation; 1 sulfate/disaccharide Cell adhesion & growth factor regulator found in extracellular matrix (ECM) |
| Chondroitin Sulfate A (CSA) |
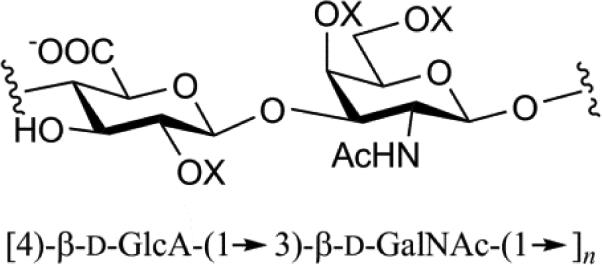
|
Mol. Wt. 5 – 50 kDa Mono sulfation on 4-O-GalNAc moiety Most abundant GAG, found in cartilage and biomechanically compressible via imbibing water |
| Hyaluronic Acid (HA) |
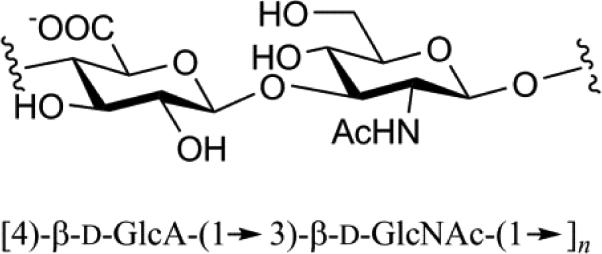
|
Mol. Wt. 4 – 8,000 kDa Non-sulfation Found in connective tissue and has size- dependent functionality (e.g. shock absorber) |
| Heparosan (Hepa) |

|
Mol. Wt. range 70,000 – 82,000 kDa Non-sulfation Biosynthetic precursor of Hep and HS |
X denotes H or SO3-and Y denotes SO3- or COCH3 (Ac).
The role of GAGs in AA amyloidosis is particularly intriguing. Interestingly, the original studies of Rotikansky included liver amyloid deposits from a tuberculosis patient, suggesting that the these deposits may have originated from SAA [11]. Subsequent studies have shown temporal co-deposition of spleen and liver amyloid, and the GAGs HS and HS proteoglycan (HSPG) in primary and secondary (i.e. AA) amyloidosis [17-20]. Heparin (Hep) and hyaluronic acid (HA) have also been found in trace amounts in liver and spleen amyloid [17, 21-23]. Additionally, it has been shown that chondroitin sulfate A (CSA) may colocalize in AA amyloid-laden liver and spleen [20, 24]. This is particularly interesting since individuals with rheumatoid arthritis have the highest incidence of developing AA amyloidosis. However, it should be noted that due to potential contamination of fibrils with CSA during their isolation from amyloid-rich tissue, it remains unclear whether CSA is an important component of AA deposits in vivo [25].
Most SAA-GAG studies carried out have focused on HS. In mouse models of AA amyloidosis and cell cultures, the fragmentation of HS through the overexpression of heparanase resulted in resistance to both amyloid accumulation and disease [26, 27]. It has been shown that HS induces the aggregation of HDL-bound human SAA1.1 by directly binding to SAA1.1 under mildly acidic conditions through a pH-sensitive motif involving a histidine residue [28, 29]. The evidence that HS plays a role in AA amyloidosis have led to strategies to arrest this disease using synthetic sulfated analogs of glucosamine to disrupt the interaction between GAGs and SAA [30-32]. In particular, a promising therapeutic candidate, eprodisate (sodium 1,3-propanedisulfonate), is currently being explored as a therapy for AA amyloidosis [33].
Motivated by the apparent pathological role of GAGs in AA amyloidosis, and the high in vitro propensity of SAA to self-assemble into various oligomers, aggregates, and fibril morphologies, we probed here the effect of various GAGs (Table 1) with different disaccharide moieties and sulfation patterns, on the self-assembly of mouse HDL-free SAA1.1. Because in vivo amyloid deposits do not contain HDL particles, suggesting that SAA undergoes misfolding and fibril formation after dissociation from HDL, here we investigate the inherent interactions of GAGs with lipid-free SAA, the form of SAA that is likely most susceptible to amyloid formation in vivo.
2. Materials and Methods
2.1. Materials
ThT dye was purchased from Sigma Aldrich. ThT fluorescence experiments were performed using a Hellma 10 mm quartz cuvette. For immuno-dot blots assay, the primary antibody, OC Anti-Amyloid fibril was purchased from EMD Millipore (Billerica, MD), and the secondary antibody, Goat anti-Rabbit horseradish peroxidase (IgG–HRP) was purchased from Invitrogen (Carlsbad, CA). CL-Xposure film and Erase-It background eliminator kit (Pierce), non-fat dry milk, Whatman Protran nitrocellulose blotting membranes, Thermo Scientific Super Signal West Pico chemiluminescent substrate, Tween-20, and autoradiography cassettes were all purchased from Fisher Scientific. Super Sharp Silicon-NCLR Atomic Force Microscopy cantilevers (nominal 2 nm ± 1nm) were purchased from Nano and More (Soquel, CA). Highest-grade (V1) mica sheets were purchased from Ted Pella through Fisher Scientific. Sensor SA chip was purchased from GE Healthcare (Uppsala, Sweden). Hep, HS, HA, and CSA (all sodium salts) were from Celsus Laboratories (Cincinnati, OH). Hepa was prepared through fermentation of Escherichia coli K5 and isolated as previously described [34].
2.2. SAA1.1 expression and purification
The mouse SAA1.1 gene, modified to include a N-terminal His-tag followed by a linker containing the tobacco etch virus (TEV) recognition site, was ordered from GenScript (Piscataway, NJ). The resulting SAA1.1 sequence is as follow: MAHHHHHHSAGENLYFQGFFSFIGEAFQGAGDMWRAYTDMKEAGWKDGDKYFHAR GNYDAAQRGPGGVWAAEKISDARESFQEFFGRGHEDTMADQEANRHGRSGKDPNYYR PPGLPAKY, where MAHHHHHHSAGENLYFQ represents the His-tag with the TEV recognition site that gets fully removed by the TEV protease during purification. Recombinant His-TEV-SAA1.1 was expressed at 37 °C in E. coli BL21(DE3)pLysS (Invitrogen) and purified with tandem immobilized metal affinity chromatography (IMAC) and preparative size exclusion chromatography (SEC). Briefly, after scaling up expression, SAA1.1 was induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3 h at 30 °C, and cells were harvested by centrifugation at 6000 RPM for 40 min and frozen overnight at −80 °C. Using a denaturing buffer (20 mM Tris, 6 M urea, 500 mM NaCl, pH 8.0) cells were stirred (lysed) and subsequently sonicated for 6 rounds of 30 s bursts. Next, centrifugation at 16,000 RPM for 45 min yielded a supernatant cell lysate that was passed through a 0.45 μm filter. His-SAA was separated by employing IMAC using a 5 mL HisTrap column (HP®, 5 GE Healthcare) coupled to a GE ÄKTA Prime® chromatography system (GE Life Sciences) at 22 °C. After concentration of His-SAA fractions, tandem purification was performed with SEC using a prep-grade column (HiLoad™ 16/60 Superdex 75, GE Healthcare) and the fractions were concentrated and dialyzed against refolding buffer (20 mM Tris pH 8.0 at 4 °C). The His-SAA was proteolyzed by TEV protease following overnight incubation at 22 °C, and quenched with 4 M Urea and 1 M guanidine hydrochloride. Subsequent purification with IMAC and SEC techniques produced pure SAA, which was dialyzed overnight against refolding buffer, dispensed, and used expediently or stored at −80 °C.
2.3. GAG solutions
The following lyophilized GAGs: Hep, HS, HA, CSA, and Hepa were each weighed in an analytical balance equipped with anti-static U-electrode (Mettler Toledo). Lyophilized GAGs were stored at 22 °C until needed. Refolding buffer was used to prepare stock solutions (10 – 30 mg/mL), and was then stored for short-term (4 °C) and long-term storage (−20 °C).
2.4. Preparation of SAA and SAA-GAG protofibrils and fibrils (aggregates)
SAA1.1 at 0.3 – 0.5 mg/mL (20 mM Tris pH 7.4) was thawed at 4 °C overnight. Next, SAA1.1 alone and SAA1.1 combined with each GAG to a final GAG concentration of 0.1 mg/mL was incubated at 4 °C (for oligomerization study) and 37 °C (for aggregation study) in an insulated incubator (to minimize condensation) in sealed Eppendorf tubes, without agitation. For binding studies, SAA1.1 alone (0.3 mg/mL) was dialyzed against 10 mM HEPES pH 7.4 at 4 °C and kept on ice (refolded oligomers) and/or subsequently incubated at 37 °C for 2 – 3 days to produce protofibrillar species (confirmed by AFM).
2.5. Analytical SEC of SAA-GAG oligomers
Oligomerization of SAA1.1 was monitored by employing a Superdex® 200 HR 10/300 column coupled to a GE ÄKTA Prime® chromatography system at 4 °C (GE Life Sciences). The analytical column was equilibrated with 20 mM Tris 150 mM NaCl, pH 8.0 prior to running SAA to monitor each oligomerization stage. SAA1.1 at 0.5 mg/mL concentration combined with GAG at a final GAG concentration of 0.1 mg/mL was incubated at 4 °C. Each aliquot (100 μL sample) was loaded, monitored at 280 nm, and eluted at a flow rate of 0.5 mL/min. This was repeated at 2, 5, 7, 10, 14 days for each sample. Three separate experiments were performed and herein only 14 days is represented for brevity and clarity. UNICORN 5.11 (GE Healthcare) software was used to analyze peaks.
2.6. Aggregation kinetics monitored by Thioflavin T (ThT) fluorescence
SAA1.1 and SAA1.1+GAG aggregation kinetics was monitored via the fluorescence of the ubiquitous fibril dye, ThT. To a working final volume of 300 μL (in 50 mM glycine-NaOH, pH 8.5) was added resuspended 20 μL aliquots of SAA1.1 (final concentration of 0.3 mg/mL), and ThT to a final concentration of 25 μM in a quartz cuvette (1 cm path length). ThT excitation was performed at 440 nm and emission maximum at 485 nm was recorded for 30 s on a Hitachi F-4500 fluorescence spectrophotometer (Danbury, CT). Bi-hourly readings, for up to the first 12-18 h were followed by 12-h readings for up to 8 days for SAA1.1. The acquired data was averaged from three separate experiments.
2.7. Immuno dot blot fibril kinetic experiments
At desired time intervals (e.g. 0, 2, 12, 24, 48 hr) SAA and SAA-GAG aggregates were resuspended, diluted (SAA1.1 ~ 0.1 mg/mL), and 2 μL were spotted on nitrocellulose membranes and allowed to air dry for 10 min at 22 °C, and stored at 4 °C. After spotting, the membrane was incubated overnight at 4 °C in a solution of 10 % nonfat dry milk (in 20 mM PBS pH 7.4) to block nonspecific binding. Washing was performed 3 consecutive times with solution of phosphate buffered saline (PBS) with 2 % Tween-20 detergent (Sigma) (PBST), followed by copious washing with PBS. OC primary antibody (Millipore), which detects amyloid fibrils and fibrillar oligomers, was diluted 1:1000 (in 5 % nonfat dry milk in 20 mM PBS pH 7.4) and the membrane was incubated with this solution at 22 °C for 2-4 h. After reaction, excess OC antibody was removed with PBST (3 washes) and PBS. Next, the membrane was incubated with goat-anti-rabbit horseradish peroxidase-conjugated secondary antibody (Invitrogen) at 22 °C for 1 h. After this secondary reaction, the membrane was again washed, then reacted with chemiluminescent substrate for 1 min (ECL Western blotting Substrate, Thermo Fisher) and developed using X-ray film (CL-XPosure Film, Thermo Scientific). Overexposed film and artifacts were removed with Background Eliminator (Thermo Scientific Pierce).
2.8. Probing particles with atomic force microscopy (AFM)
SAA and SAA-GAG aggregated samples after co-incubation for 7 days were diluted to 0.05 -0.1 mg/mL and 20 μL was deposited on freshly cleaved mica (Ted Pella) and allowed to air dry for 1 h. Mica samples were washed three times with 1 mL of 0.2 μm filtered and degassed Milli-Q water, and allowed to dry overnight in fume hood. Using super sharp cantilevers with a nominal radius of curvature of 2 nm (SSS-NCLR, Nanosensors) and AFM (MFP3D AFM, Asylum Research) triplicate scans were performed with the following parameters: 2, and 10 μm scan size, 512 pixel density, 4-channel AC (non-contact) mode. Using Asylum Research 6.22 AFM image quality control was performed by masking (determined by height boundaries of 100-200 pm ± 100 fm) and elimination of irregularities, and finally converting raw amplitude trace to 300 dpi TIFF. The same mask was used to perform particle size distribution analysis of the raw height trace scan, and the statistical raw data was used to obtain histograms.
2.9. Preparation of Heparin sensor chip and measuring interaction with SAA1.1 refolded oligomers
Hep (2 mg) was combined with Amine-PEG4-Biotin (2 mg, Pierce, Franklin, OH) in 200 μL H2O and NaCNBH3 (10 mg). The reaction was incubated at 70 °C for 24 h. Another aliquot of NaCNBH3 was added to the reaction and incubated an additional 24 h. The biotinylated Hep were purified using Amicon 3000 MWCO spin columns (Millipore, Billerica, MA) and lyophilized. BIAcore 3000 (GE Healthcare, Uppsala, Sweden) was used to measure the interaction of SAA 1.1 refolded oligomers with Hep. Briefly, 20 μL of biotinylated Hep in HBSEP (10mM 4-(2-hydroxyehtyl-1-piperazineethanesulfonic acid (HEPES), 150 mM sodium chloride, 3 nM ethylenediaminetatraacetic acid (EDTA), 0.005% polysorbate P20, pH 7.4) running buffer was injected over sensor chip flow cell 2, flow cell 3, and flow cell 4, respectively, at a flow rate of 10 μL/min. The successful immobilizations were confirmed by the observation of a ~100 resonance unit (RU) increase in the sensor flow cells. Flow cell 1 was reserved as a control cell using a 1 min injection of saturated biotin solution. SAA1.1 formation solutions prepared in 10 mM HEPES were diluted to different concentrations, and 90 μL was injected at a flow rate of 30 μL/min using HBS-EP as running buffer. A 3 min dissociation period was allowed after the injection by flowing running buffer through the sensor chip. The surface was then washed with 1 min injections of 2 M sodium chloride and HBS-EP buffer to aid in surface regeneration. The fitting of sensorgrams to a Langmuir binding model was used to obtain the SAA1.1-Hep dissociation constants.
2.10. Far-UV circular dichroism (CD) spectroscopy
SAA1.1 at 0.15 mg/L and SAA1.1 combined with the aforementioned GAGs to a final GAG concentration of 0.1 mg/mL were incubated at 37 °C for 1 – 3 weeks. Far-UV CD wavelength scan measurements from 195 to 260 nm were performed on an Olis DSM 1000 CD spectrophotometer (Bogart, GA) with a 1-mm path Hellma quartz cuvette. For each SAA1.1+GAG combination, triplicate wavelength scans were performed, averaged, subtracted from the GAG spectrum, and plotted against the molar ellipticity. AFM was performed to corroborate presence of aggregate formation.
3. Results
3.1 Monitoring the effects of GAGs on SAA1.1 folding and oligomerization
As an initial assessment of the inherent ability of SAA1.1 to interact with GAGs, we refolded SAA1.1 in the absence or presence of the various GAGs. We have previously shown that upon dialysis at 4 °C, urea-denatured SAA1.1 refolds into a mixture of quaternary structures, including dodecamer, tetramer, and monomer [35]. Therefore, we monitored the refolding of SAA1.1 at 4 °C in the presence of GAGs for 14 days and probed the oligomeric structure of SAA1.1 by size exclusion chromatography (SEC). As expected, in the absence of GAGs, SAA1.1 refolded into dodecamer (Ve - 12.5 mL), tetramer (Ve - 15.8 mL), and monomer (Ve - 17.7 mL) (Fig. 1, top trace). For SAA1.1+GAG, the dodecamer remained the highest peak overall (with the exception of SAA1.1+Hepa) in the SEC data (Fig. 1A), although the relative distribution of oligomers was not consistent from experiment to experiment. Interestingly, only Hepa showed some SAA1.1 aggregation in the SEC chromatogram, as noted by the void volume (~8 mL) peak. However, the overall oligomer peak area for most SAA+GAG decreased an average of ~ 20% (Fig. 1B), suggesting that insoluble aggregates may have formed and become trapped in the guard column frit. Overall, the SEC data show that for the most part, the presence of GAGs during the refolding of SAA1.1 at 4 °C does not interfere with the folding and oligomerization of SAA1.1.
Fig. 1.

Size exclusion chromatography of SAA1.1 refolded in the presence of GAGs. SEC data was collected after 14 days of incubation at 4 °C. The concentration of SAA1.1 and GAGs were 0.30 mg/mL and 0.10 mg/mL, respectively. The grey trace shows SAA1.1 without added GAGs for comparison.
3.2. GAGs accelerate the aggregation of SAA1.1
We previously studied the fibrillation kinetics of SAA1.1 by using the dye ThT, which fluoresces upon binding to specific motifs within proteins aggregates [36, 37]. We showed that when SAA1.1 was incubated at 37 °C it exhibited a ThT fluorescence (ThT fluo) lag phase of 3-4 days before forming amyloid-like fibrils [35]. To determine the effect of GAGs on the fibrillation kinetics of SAA1.1, we incubated SAA1.1 (0.3 mg/mL) with the various GAGs (0.1 mg/mL) at 37 °C, and monitored its aggregation using ThT fluo. In the absence of GAGs, SAA1.1 exhibited the expected lag phase of ~100 h (Fig. 2), whereas all GAGs significantly reduced the lag phase of SAA1.1 (Fig. 2). In particular, SAA1.1 in the presence of Hep eliminated the aggregation lag phase and almost doubled the ThT fluo intensity. In contrast, SAA1.1-CSA exhibited the lowest ThT fluo intensity, with a ~ 25% decrease. For samples containing HS, HA, and Hepa, the ThT fluo signal was similar to that of SAA1.1. Thus, whereas all GAGs shortened the ThT fluo lag phase of SAA1.1 aggregation, the differences in signal plateau suggest differences in the structure and/or amount of the SAA1.1 species formed [38].
Fig. 2.

Aggregation kinetics of SAA1.1 (0.3 mg/mL) monitored by Thioflavin-T fluorescence upon incubation at 37 °C in the presence of 0.1 mg/mL GAGs.
3.3 AFM shows that GAGs have major effects on the aggregation and fibrillation of SAA1.1
Atomic force microscopy (AFM) was used to assess the effect of GAGs on SAA1.1 aggregation, including whether the ThT fluo data was consistent with the formation of amyloid-like fibrils. SAA1.1 (0.3 mg/mL) was incubated at 37 °C in the absence or presence of 0.1 mg/mL GAG and then allowed to aggregate for 7 days. The AFM images showed that all GAGs tested had an effect on fibril formation ranging from minimal to complete inhibition. The AFM images of SAA1.1 showed predominant formation of long fibrils with an average particle height of ~ 5 nm (Fig. 3A). It should be noted from the plot of particle height distribution (Fig. 3A inset) that the count of SAA1.1 particles is low because the AFM software only takes into account separate particles, and considers the fibril itself as a single particle. SAA1.1 fibril formation in the presence of HS resulted in a similar particle height distribution profile as SAA1.1, although most of the fibrils were shorter (Fig. 3B). HA and Hepa, a precursor of Hep and HS, interfered significantly with SAA1.1 fibril formation (Fig. 3C and 3D). The AFM images showed a wide distribution of particles ranging from small oligomers to typical long SAA1.1 fibrils, although few of the latter. The diversity of aggregates induced by HA and Hepa is consistent with the particle height distribution observed for these samples (Fig. 3C inset) (Fig. 3D inset).
Fig. 3.

Atomic Force Microscopy (AFM) of SAA1.1 aggregates formed when co-incubated with GAGs (0.10 mg/mL) at 37°C for 7 days. Each AFM contains amplitude trace (large image), a particle height distribution plot for each trace (top figure), a 5X zoom of the black square in large image (middle, red scale bar 500 nm), and a height section of area underneath black arrow (bottom). AFM of SAA 1.1 alone (A) shows fibrils averaging to typical heights of 4 nm (black scale bar 1 μm). SAA1.1 incubated with HS, HA, and Hepa reveals that their morphologies are unique to each system, but produce “SAA1.1-like” fibrils. Conversely, fibrils grown with Hep show a unique bifurcated pathway of abundant protofibrillar populations with fibrillation. Incubation with CSA capped fibril formation and generated protofibrils and big oligomers ranging in sizes similar to full-length fibrils.
Hep is commonly used as a model of HS in amyloid studies [39]. The AFM for Hep+SAA1.1 samples showed an immense amount of small oligomers and curvilinear protofibrils while forming few long typical SAA1.1-like fibrils (Fig. 3E). The particle distribution profile showed that the protofibrils, a major component of the particle distribution profile, are thinner than the typical SAA1.1 fibrils. This suggests that they arise from the formation of a smaller oligomeric species. The persistence of the thin and small curvilinear fibrils even after 7 days of incubation, suggests that they are either unable to grow or they assemble very slowly. Zooming into these fibrils suggest that they may be capped by larger oligomers (Fig. 4).
Fig. 4.

Zoom-in and height analysis of SAA1.1+Hep AFM image to show putative capping of protofibrils. (A) AFM image (2 μm square) highlighting (black box) region of protofibrils surrounded by larger spherical particles. Blue arrows identify a few other cases where these larger particles are found at the end of protofibrils. (B) The region within the black box in panel A is magnified (~ 3X zoom) to better show the putative capping particles. (C) Height scans of the respective area under the red and green lines shown in panel A. (D) A 3-D rendered image of panel A showing the height distribution across the various SAA1.1 structures, including the greater height of spherical particles (4-6 nm) relative to the protofibrils (2-3 nm). Black scale bars represent 250 nm.
Remarkably, although the ThT fluo data suggested that CSA catalyzed the formation of SAA1.1 aggregates capable of binding ThT, the AFM data showed that these CSA-induced aggregates did not assemble into fibrils (Fig. 3F). The SAA1.1+CSA aggregates mainly consisted of spherical particles of various sizes and limited very short protofibrils. The particle distribution analysis of SAA1.1+CSA showed a broad distribution of particle height with an average of about 4 nm. At this time it is not clear why the short protofibrils seemed unable to extend fibrillation.
Overall, the AFM data showed that GAGs have modest to profound effects on SAA1.1 fibril formation under physiological-like temperature and pH. Interestingly, in contrast to what we observed with SAA1.1 (no GAG), the AFM data indicates that the ThT fluo is not directly monitoring the formation of fibrils in the SAA1.1+GAG samples, but rather the rapid formation of ThT-binding species, which in some cases may be precursor of fibrils (e.g. (HS, Hep, HA) or of protofibrils (e.g. Heparin). Together, the ThT fluo and AFM data suggest that the GAGs lower the kinetic barrier for SAA1.1 self-assembly into spherical aggregates of various size and different propensities for amyloid fibril formation. In the case of SAA-Hep, it is plausible some of these larger spherical aggregates may interact with amyloidogenic oligomers and protofibrils to cap fibril formation (Fig. 4), although further studies are required to confirm this possibility.
3.4. Surface plasmon resonance experiments reveal heparin binding to SAA1.1 refolded oligomers
The acceleration of SAA1.1 aggregation observed by ThT (Fig. 2) and the resulting effect on fibril formation suggest that Hep may be interacting with SAA1.1 early in the aggregation process. To gain further insight on the SAA1.1-Hep interaction we used surface plasmon resonance (SPR). Hep was covalently immobilized on a streptavidin chip, and refolded oligomers of SAA1.1 (as in Fig. 1) were injected into the SPR instrument. The SPR sensorgrams of SAA1.1 oligomer-heparin interaction were obtained (Fig. 5A). These sensorgrams fit well to a Langmuir 1:1 binding model with low concentration binding (Fig. 5B). The SPR data show that SAA 1.1 oligomer binds to heparin with dissociation constants KD of ~4.7 μM. The SPR competition assay was utilized to determine the binding preference of SAA1.1 oligomer to Hep, HS, and CSA. SAA 1.1 oligomer at 2.0 μM was pre-mixed with 1.0 μM of GAG and injected over the heparin chip. SPR competition sensorgrams and bar graphs of the GAG competition levels are displayed in Fig. 5C and D, respectively. Hep produced the strongest inhibition in SAA1.1 oligomer binding of immobilized Hep by competing >80% of the SAA1.1 oligomer binding to immobilized heparin on the chip surface. Modest inhibitory activities were observed for HS, and weak inhibitory activities were observed for CSA. Thus, the strong interaction of immobilized Hep with SAA1.1 oligomers is consistent with the elimination of the fibrillation lag phase observed by ThT fluo (Fig. 2), the dramatic impact on formation of protofibrils observed by AFM (Fig. 3), and previous studies showing that heparin binds to SAA peptides [28].
Fig. 5.

Interaction between SAA1.1 (0.15 mg/mL) refolded oligomers (4 °C) and heparin immobilized on a biochip measured with surface plasmon resonance (SPR). (A) SPR sensorgrams of SAA1.1-heparin interaction. (B) Sensorgrams fitting using models from BIAevaluate 4.0.1 to get dissociation constants KD of ~4.7 μM (the black curves are the fitting curves). (C) Sensorgrams of solution GAGs/surface heparin competition. SAA1.1 concentration was 2 μM, and concentrations of GAGs in solution were 1 μM). (D) Bar graphs (based on triplicate experiments with standard deviation) of normalized SAA1.1 binding preference to surface heparin by competing with different GAGs in solution.
3.5. All GAGs facilitate the formation of amyloidogenic oligomers and protofibrils
The accelerated increase in ThT fluo observed for all SAA1.1+GAG samples (Fig. 2) suggests that these GAGs facilitate the formation of SAA1.1 oligomers-aggregates that may be precursors to amyloid-like fibrils. However, the AFM data (Fig. 3) shows that the effect of GAGs produce different types of aggregates ranging from SAA1.1-like fibrils (e.g. HS) to short curvilinear protofibrils (e.g. Hep) to spherical aggregates (e.g. CSA). To probe whether SAA1.1 in the presence of GAGs still formed fibrillar species, we used the OC polyclonal antibody, which identifies generic epitopes present in many amyloid fibrils and fibrillar oligomers, but not in prefibrillar oligomers and natively folded proteins [40, 41]. Fig. 6 shows that all SAA1.1+GAG samples exhibit similar profile of OC-positive oligomers-aggregates. This observation is consistent with the ThT data suggesting the presence of amyloidogenic species. Furthermore, the AFM data show extensive presence of protofibrils and/or fibrils in all SAA1.1+GAG samples, including SAA1.1-CSA, which exhibit many short protofibrils (Fig. 3).
Fig. 6.
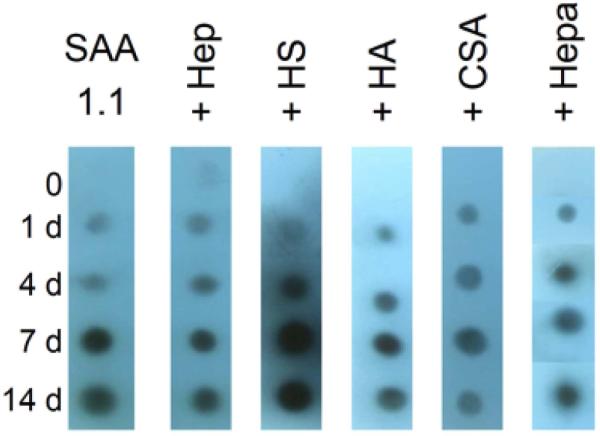
OC immuno dot blot aggregation kinetic experiments (37 °C) of SAA 1.1+GAGs. OC antibody binds to epitopes in fibrillar oligomers (light spots) and fibrils (dark spots). The concentration of SAA1.1 and GAGs were 0.20 mg/mL and 0.1 mg/mL, respectively.
To directly probe the presence of beta sheet structure on these SAA1.1+GAG samples, we carried out far-UV circular dichroism (CD) analysis of SAA samples after 7 days of incubation at 37 °C, pH 7.4. Refolded oligomers of SAA1.1 contain largely alpha helical structure with CD minima at 222 and 208 nm, and very little beta structure [35]. In contrast, SAA1.1 fibrils exhibit two CD minima at 222 (minor) and 205 nm (major), and together with deep UV Raman experiments, the data suggest SAA1.1 fibrils contain significant amounts of unordered secondary content with modest amounts of beta sheet structure [35]. The CD results shown in Fig. 7 revealed minimal changes in the 205 nm minima, but some changes in the 220-230 region minima. Overall the OC and CD experiments demonstrated that the GAGs do not seem to alter the inherent ability of SAA1.1 to form amyloidogenic oligomers-protofibrils that contain modest amounts of beta-sheet structure and the motif for OC binding.
Fig. 7.

Far UV circular dichroism (CD) spectroscopy of SAA1.1+GAGs. SAA1.1 (0.15 mg/mL) was incubated with GAGs (0.1 mg/mL) at 37 °C for 2 weeks and CD wavelength scans were performed at 37 °C. For SAA1.1 co-incubated with GAGs, the respective GAG spectra were subtracted. The CD spectra of SAA1.1 at 4 °C was included for reference.
4. Discussion
4.1. GAGs have diverse effects on the intrinsic aggregation and amyloid formation of lipid-free SAA1.1 in vitro
The presence of GAGs accelerated the aggregation of SAA1.1 into oligomeric, protofibrillar and/or fibrillar species that bound ThT (Fig. 2) and the OC antibody (Fig. 5), thereby suggesting that many of these species are on the amyloid-formation pathway. The AFM data demonstrate GAGs possess an intrinsic ability to affect the assembly of HDL-free SAA1.1 species into amyloid-like fibrils (Fig. 3), as illustrated in Fig. 8. We classified the effect of GAGs on SAA1.1 aggregation into three general categories representing the aggregation of SAA1.1 into mostly fibrils (HS, Hepa, and HA), short flexible protofibrils (Hep), and spherical particles (CSA) (Fig. 8). Although sulfation, carboxylation, and charge pattern of GAGs have been shown to be important for their effect on amyloid formation, this GAG structure-function correlation is not obvious in our results and warrants further investigation [42, 43]. However, our AFM data show that further studies are necessary to better understand the intrinsic effects of sulfation and carboxylation patterns, and saccharide conformation and content, on the aggregation propensity of SAA and other amyloidogenic proteins.
Fig. 8.

Illustration summarizing the effect of GAGs on the aggregation. The central pathways (green, magenta, and the presence of yellow curves) demonstrate prototypical SAA-like fibril formation; however, curvilinear protofibril production is heightened by interactions with HS/HA/Hepa. SAA-Hep (red curve) produced abundant protofibrils with caps on the ends, and parallel scaffolding of fibrils with caps on ends. Finally, SAA fibril formation was completely capped by interactions with CSA (blue curve), which produced large oligomers and protofibrils. Black scale bar 200 nm, red scale bar 500 nm.
Recent studies have suggested that GAGs may have scaffolding and capping effects on the fibrillation of some amyloidogenic proteins [44-46]. Heparin, the most sulfated and highly charged GAG known has been shown to accelerate fibril formation, presumably by serving as a scaffold where fibrillar oligomers easily orient to form protofibrils and subsequently fibrils [8, 46]. Interestingly, our results show that SAA1.1+Hep aggregation results in an abundance of protofibrillar structures that are much thinner (1-3 nm height) than those formed by SAA1.1 (Fig. 3 and Fig. 4). We have previously shown that mouse SAA2.2 and SAA1.1 form fibrils of different morphology [35], with the thinner SAA2.2 protofibrils and the early fibrils being similar in height as those seen for SAA1.1+Hep. Since SAA1.1+Hep is able to form oligomers of various sizes (Fig. 4), we speculate that perhaps Hep altered the early aggregation pathway of SAA1.1 to resemble that of SAA2.2 by favoring the formation of smaller SAA2.2-like oligomer precursor of thinner protofibrils. Furthermore, the presence of thinner SAA1.1-Hep protofibrils that do not progress towards full-length fibrils may be due to a capping effect caused by larger spherical particles (Fig. 4). Although it is not clear how the sulfation pattern and charge distribution (~3 per disaccharide) of Hep may be producing this effect, other studies have shown this capping phenomenon that inhibits fibril formation. Gupta et al. showed biophysical evidence that nucleobindin protein caps prefibrillar aggregates at the ends of IAPP [45]. In another report, Sievers et al. was able to use a structure-based approach to design a molecular cap made of synthetic amino acids and thereby inhibit tau protein fibrillation [47].
In contrast to Hep, CSA, the mono-sulfated and carboxylated GAG induced SAA1.1 to aggregate mostly into spherical particles with some very short protofibrils (Fig. 3 and Fig. 8). Interestingly, a similar morphological effect of CSA was reported by McLaughlin et al. where they found CSA induced spherical oligomers for the immunoglobulin light-chain protein, and suggested the effect may be driven by CSA's glucuronic acid moiety [48]. In another study, CSA caused the spontaneous fibrillation of β2-microglobulin, indicating the effect of CSA on fibril formation may be protein-dependent, and this may be the case for other GAGs [49]. CSA's single 4-O-sulfation of GalNAc (galactosamine N-acetyl) is the most notable difference in all the GAGs investigated in our study. As Castillo et al. suggest, conformational changes of GAG moieties, a reduction in sulfation, and their linear arrangement may influence the inhibition of fibrillation [42]. Thus, CSA's unique GalNAc moiety, its single sulfation pattern, or its glucuronic acid, either individually or collectively, may account for CSA's inhibition of SAA1.1 fibril formation in vitro, and warrant further studies to understand the structure-function relationship of this effect.
In Fig. 8, we grouped the remaining three GAGs for their modest effect on SAA1.1 aggregation formation. In each case there was significant amount of fibril formation that resembled those formed by GAG-free apo SAA1.1, mostly differing in the amount of spherical species present. HS exhibited the least spherical species, whereas HA had the most. HS, HA, and Hepa contain the same β-D-glucuronic acid, but HS also contains (in its variable sequence) a 2-O-sulfo-α-L-iduronic acid that is also present in Hep. Although not as highly sulfated as Hep, the sulfation pattern of HS (~1 per disaccharide) is more prominent in the glucosamine sugar moiety. These sulfation pattern differences may allow HS to elicit different effects than Hep on the aggregation of SAA1.1. HS, HA, and Hepa all contain the glucuronic acid present in CSA, but also the N-acetylated glucosamine moiety present in Hep. These carboxyl moieties (in HS, HA, and Hepa) have a decreased charge density in comparison with sulfation sites present in Hep, and may help explain why they don't behave like Hep in our study. It is important to note that experimental conditions may also influence the effect of GAGs on fibril formation. For example, in contrast to our results, prior SAA studies involving different solvent condition and HDL-SAA have shown similar effect of HS and Hep on SAA fibrillation [28, 29]. Nevertheless, our data highlight the intrinsic ability of GAGs to influence in different ways the aggregation and fibrillation of apo SAA1.1, an observation that is perhaps influenced by experimental conditions, the marginal stability of SAA, and SAA's innate propensity for forming different oligomers and morphologically distinct fibrils [35, 50-54].
4.2 Potential implications for the role of GAGs in reactive amyloidosis
A direct comparison of our results with many prior studies exploring the role of HS and Hep in AA amyloidosis is not trivial because of the different systems and experimental conditions involved. The aim of this study was to investigate the intrinsic effect of the interactions between HDL-free SAA1.1 and GAGs on SAA1.1 fibrillation. It is important to note that our studies provide unique information that complements other in vitro studies using lipidated SAA. Our study is the first to investigate the inherent effect of different GAGs on the aggregation of pathogenic mice SAA1.1 in the lipid-free form, and has yielded novel results on the potential role of GAGs on the aggregation, fibrillation, and fibril morphology of SAA1.1 that may be relevant to further understanding the role of GAGs in AA amyloidosis. Of particular relevance, our results show that HS and Hep, which is commonly used to mimic HS, do not have the same effect on SAA1.1 aggregation. The aforementioned reasons (e.g. sulfation pattern, experimental conditions) may be reasons for this in vitro behavior, but it may raise the question whether similar differences may occur in vivo. Furthermore, CSA has a remarkable inhibitory effect on the in vitro fibrillation of SAA1.1.
Although HS is the most abundant GAG present in AA deposits and has been the most widely investigated in SAA studies, various GAGs, in particular CS, have been identified in the amyloid deposits of afflicted individuals and in experimental models of the disease [55-57]. Interestingly, in a study of mice lacking serum amyloid P (SAP), a major component of the amyloid deposits found in AA amyloidosis, the amyloid deposits contained significant amounts of CS in the form of spherical particles [55]. Thus, it is possible that the interaction of CSA with SAA in vivo also influences the aggregation of SAA, although the pathological relevance of such an effect, if present, is not clear at this time. Nevertheless, it is worth noting that CSA is the most abundant GAG in connective tissue and is involved in joint lubrication. Furthermore, individuals with rheumatoid arthritis comprise 45% of all cases of AA amyloidosis [25]. Further studies are required to probe whether CSA may be related to the incidence of AA in patients with rheumatoid arthritis.
The effect of Hep on the structure of SAA1.1 fibrils is intriguing. Prior studies have shown that heparin accelerates fibril formation of various proteins/peptides, including alpha synuclein [58], gelsolin [46], β2-microglobulin [59] and A beta [60]. Here, we show that Hep significantly enhances the kinetics of SAA1.1 aggregation into thin curvilinear protofibrils, but delays the formation of fibrils. Thus, it appears that the catalytic effect of Hep on protein aggregation is a common effect but the effect on aggregate structure and fibril morphology may differ, especially for SAA [28, 29]. A recent study exploring the effect of Hep on peptides comprising the N-terminus (1-27), the central region (43-63), and the C-terminus (77-104) of human SAA1.1 showed the Hep enhanced amyloid formation of SAA (1-27) and SAA (43-63), but not SAA (77-104) [61]. Interestingly, it was shown that SAA(1-27) exhibited the strongest binding to Hep, resulting in the formation of short and straight fibrils, whereas Hep induced SAA(43-63) to form long and elastic fibrils. This morphological sensitivity of SAA is consistent with our recent findings that mice SAA2.2 and SAA1.1 form amyloid-like fibrils in vitro through distinct pathways leading to morphologically different fibrils [54].
Although, there have been many studies probing the role of GAGs on amyloid formation of various disease-related proteins and peptides most of these focus on HS and/or Hep and do not address effect of different GAGs on the ultrastructure of the various protein aggregates (i.e. oligomers, protofibrils, and fibrils) [14]. Also, unique to our GAG-dependent study, was the use of full-length lipid-free mouse SAA1.1 and pH 7.4. Other fibrillation studies of SAA have simulated the acidic endosome conditions (pH ~ 5), and in some cases involved peptides [61]. Our study shows that in addition to the known effects of GAGs on the fibrillation kinetics and proteolytic shielding of fibrils, GAGs could have a major effect in modulating the aggregation pathway, leading to different oligomers, aggregate heterogeneity, and fibrils of different morphology. In the case of SAA, further studies using relevant biological systems are needed to further understand the extent to which different GAGs contribute towards or protect from the development of AA amyloidosis.
All GAGs, mainly heparin, accelerated formation of Thioflavin T-binding SAA species
GAGs exhibited variable effects on the fibrillation of SAA
GAGs may have an intrinsic influence on the self-assembly of SAA in vivo
Acknowledgments
We acknowledge support from NIH Grant R01 AG028158 to W.C. and from NIH grant GM38060, HL096973 and HL62244 to R.J. L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations: AA, amyloid A; AFM, atomic force microscopy; apo, HDL-free; CD, circular dichroism; CSA, chondroitin sulfate A; GAG, glycosaminoglycan; HA, hyaluronic Acid; Hep, heparin; Hepa, heparosan; HS, heparan sulfate; HSPG, heparan sulfate proteoglycan; IPTG, isopropyl β-D-1-thiogalactopyranoside; PBS, phosphate buffered saline; SPR, surface plasmon resonance; SAA, Serum Amyloid A; SEC, size exclusion chromatography; ThT, thioflavin T.
References
- 1.Sipe JD. Amyloidosis. Annu. Rev. Biochem. 1992;61:947–975. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 2.van der Westhuyzen DR, de Beer FC, Webb NR. HDL cholesterol transport during inflammation. Curr. Opin. Lipidol. 2007;18:147–151. doi: 10.1097/MOL.0b013e328051b4fe. [DOI] [PubMed] [Google Scholar]
- 3.Eklund KK, Niemi K, Kovanen PT. Immune functions of serum amyloid A. Crit. Rev. Immunol. 2012;32:335–348. doi: 10.1615/critrevimmunol.v32.i4.40. [DOI] [PubMed] [Google Scholar]
- 4.Ganapathi MK, Rzewnicki D, Samols D, Jiang SL, Kushner I. Effect of combinations of cytokines and hormones on synthesis of serum amyloid A and C-reactive protein in Hep 3B cells. J. Immunol. 1991;147:1261–1265. [PubMed] [Google Scholar]
- 5.Tatsuta E, Sipe JD, Shirahama T, Skinner M, Cohen AS. Different regulatory mechanisms for serum amyloid A and serum amyloid P synthesis by cultured mouse hepatocytes. J. Biol. Chem. 1983;258:5414–5418. [PubMed] [Google Scholar]
- 6.McAdam KP, Sipe JD. Murine model for human secondary amyloidosis: genetic variability of the acute-phase serum protein SAA response to endotoxins and casein. J. Exp. Med. 1976;144:1121–1127. doi: 10.1084/jem.144.4.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hass G. Studies of Amyloid. II. The Isolation of a Polysaccharide from Amyloid-Bearing Tissues. Arch. Path. 1942;34:92–105. [Google Scholar]
- 8.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem. Int. Ed. Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 9.Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 10.Roth M, Papakonstantinou E, Karakiulakis G. Biological Function of Glycosaminoglycans. In: Garg MKCHG, Hales CA, editors. Carbohydrate Chemistry, Biology and Medical Applications. Elsevier; 2008. pp. 209–226. [Google Scholar]
- 11.Rokitansky C. Handbuch der Speciellen Pathologischen Anatomica, Braumuller und Seidel. Vienna. 1842:311–312. [Google Scholar]
- 12.Kyle RA. Amyloidosis: a convoluted story. Br. J. Haematol. 2001;114:529–538. doi: 10.1046/j.1365-2141.2001.02999.x. [DOI] [PubMed] [Google Scholar]
- 13.Friedreich N, Kekulé A. Zur Amyloidfrage. Archiv für Pathologische Anatomie und Physiologie und für Klinische Medicin. 1859;16:50–65. [Google Scholar]
- 14.Ariga T, Miyatake T, Yu RK. Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer's disease and related disorders: amyloidogenesis and therapeutic strategies--a review. J. Neurosci. Res. 2010;88:2303–2315. doi: 10.1002/jnr.22393. [DOI] [PubMed] [Google Scholar]
- 15.Motamedi-Shad N, Monsellier E, Chiti F. Amyloid formation by the model protein muscle acylphosphatase is accelerated by heparin and heparan sulphate through a scaffolding-based mechanism. J. Biochem. 2009;146:805–814. doi: 10.1093/jb/mvp128. [DOI] [PubMed] [Google Scholar]
- 16.Martin DJ, Ramirez-Alvarado M. Glycosaminoglycans promote fibril formation by amyloidogenic immunoglobulin light chains through a transient interaction. Biophys. Chem. 2011;158:81–89. doi: 10.1016/j.bpc.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bitter T, Muir H. Mucopolysaccharides of whole human spleens in generalized amyloidosis. J. Clin. Invest. 1966;45:963–975. doi: 10.1172/JCI105412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okuyama T, Turumi KI. Acid mucopolysaccharide from a spleen of primary amyloidosis. Clin. Chim. Acta. 1963;8:140–142. doi: 10.1016/0009-8981(63)90210-9. [DOI] [PubMed] [Google Scholar]
- 19.Lyon AW, Narindrasorasak S, Young ID, Anastassiades T, Couchman JR, McCarthy KJ, Kisilevsky R. Co-deposition of basement membrane components during the induction of murine splenic AA amyloid. Lab. Invest. 1991;64:785–790. [PubMed] [Google Scholar]
- 20.Snow AD, Willmer J, Kisilevsky R. Sulfated glycosaminoglycans: a common constituent of all amyloids? Lab. Invest. 1987;56:120–123. [PubMed] [Google Scholar]
- 21.Linker A, Hoffman P, Sampson P, Meyer K. Heparitin sulfate. Biochimica et Biophysica Acta. 1958;29:443–444. doi: 10.1016/0006-3002(58)90213-0. [DOI] [PubMed] [Google Scholar]
- 22.Ancsin JB, Kisilevsky R. The heparin/heparan sulfate-binding site on apo-serum amyloid A. Implications for the therapeutic intervention of amyloidosis. J. Biol. Chem. 1999;274:7172–7181. doi: 10.1074/jbc.274.11.7172. [DOI] [PubMed] [Google Scholar]
- 23.Nelson SR, Lyon M, Gallagher JT, Johnson EA, Pepys MB. Isolation and characterization of the integral glycosaminoglycan constituents of human amyloid A and monoclonal light-chain amyloid fibrils. Biochem. J. 1991;275( Pt 1):67–73. doi: 10.1042/bj2750067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linker A, Carney HC. Presence and role of glycosaminoglycans in amyloidosis. Lab. Invest. 1987;57:297–305. [PubMed] [Google Scholar]
- 25.Obici L, Raimondi S, Lavatelli F, Bellotti V, Merlini G. Susceptibility to AA amyloidosis in rheumatic diseases: a critical overview. Arthritis Rheum. 2009;61:1435–1440. doi: 10.1002/art.24735. [DOI] [PubMed] [Google Scholar]
- 26.Elimova E, Kisilevsky R, Szarek WA, Ancsin JB. Amyloidogenesis recapitulated in cell culture: a peptide inhibitor provides direct evidence for the role of heparan sulfate and suggests a new treatment strategy. FASEB J. 2004;18:1749–1751. doi: 10.1096/fj.03-1436fje. [DOI] [PubMed] [Google Scholar]
- 27.Li JP, Galvis ML, Gong F, Zhang X, Zcharia E, Metzger S, Vlodavsky I, Kisilevsky R, Lindahl U. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc. Natl. Acad. Sci. U S A. 2005;102:6473–6477. doi: 10.1073/pnas.0502287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elimova E, Kisilevsky R, Ancsin JB. Heparan sulfate promotes the aggregation of HDL-associated serum amyloid A: evidence for a proamyloidogenic histidine molecular switch. FASEB J. 2009;23:3436–3448. doi: 10.1096/fj.09-134981. [DOI] [PubMed] [Google Scholar]
- 29.Noborn F, Ancsin JB, Ubhayasekera W, Kisilevsky R, Li JP. Heparan sulfate dissociates serum amyloid A (SAA) from acute-phase high-density lipoprotein, promoting SAA aggregation. J. Biol. Chem. 2012;287:25669–25677. doi: 10.1074/jbc.M112.363895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kisilevsky R, Ancsin JB, Szarek WA, Petanceska S. Heparan sulfate as a therapeutic target in amyloidogenesis: prospects and possible complications. Amyloid. 2007;14:21–32. doi: 10.1080/13506120601116419. [DOI] [PubMed] [Google Scholar]
- 31.Kisilevsky R, Lemieux LJ, Fraser PE, Kong X, Hultin PG, Szarek WA. Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer's disease. Nature Medicine. 1995;1:143–148. doi: 10.1038/nm0295-143. [DOI] [PubMed] [Google Scholar]
- 32.Kisilevsky R, Szarek WA, Ancsin JB, Elimova E, Marone S, Bhat S, Berkin A. Inhibition of amyloid A amyloidogenesis in vivo and in tissue culture by 4-deoxy analogues of peracetylated 2-acetamido-2-deoxy-alpha- and beta-d-glucose: implications for the treatment of various amyloidoses. Am. J. Pathol. 2004;164:2127–2137. doi: 10.1016/s0002-9440(10)63771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, Livneh A, Lesnyak O, Puechal X, Lachmann HJ, Obici L, Balshaw R, Garceau D, Hauck W, Skinner M, A.A.A.T.G. Eprodisate for, Eprodisate for the Treatment of Renal Disease in AA Amyloidosis. N. Engl. J. Med. 2007;356:2349–2360. doi: 10.1056/NEJMoa065644. [DOI] [PubMed] [Google Scholar]
- 34.Lee SK, Lee KE, Hwang YH, Kida M, Tsutsumi T, Ariga T, Park JC, Kim JW. Identification of the DSPP mutation in a new kindred and phenotype-genotype correlation. Oral Dis. 2011;17:314–319. doi: 10.1111/j.1601-0825.2010.01760.x. [DOI] [PubMed] [Google Scholar]
- 35.Srinivasan S, Patke S, Wang Y, Ye Z, Litt J, Srivastava SK, Lopez MM, Kurouski D, Lednev IK, Kane RS, Colon W. Pathogenic Serum Amyloid A 1.1 Shows a Long Oligomer-rich Fibrillation Lag Phase Contrary to the Highly Amyloidogenic Nonpathogenic SAA2.2. J. Biol. Chem. 2013;288:2744–2755. doi: 10.1074/jbc.M112.394155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 37.Krebs MR, Bromley EH, Donald AM. The binding of thioflavin-T to amyloid fibrils: localisation and implications. J. Struct. Biol. 2005;149:30–37. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 38.Bazar E, Jelinek R. Divergent heparin-induced fibrillation pathways of a prion amyloidogenic determinant. Chembiochem. 2010;11:1997–2002. doi: 10.1002/cbic.201000207. [DOI] [PubMed] [Google Scholar]
- 39.Ramachandran G, Udgaonkar JB. Understanding the kinetic roles of the inducer heparin and of rod-like protofibrils during amyloid fibril formation by Tau protein. J. Biol. Chem. 2011;286:38948–38959. doi: 10.1074/jbc.M111.271874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glabe CG. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castillo GM, Lukito W, Wight TN, Snow AD. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J. Neurochem. 1999;72:1681–1687. doi: 10.1046/j.1471-4159.1999.721681.x. [DOI] [PubMed] [Google Scholar]
- 43.Ren R, Hong Z, Gong H, Laporte K, Skinner M, Seldin DC, Costello CE, Connors LH, Trinkaus-Randall V. Role of glycosaminoglycan sulfation in the formation of immunoglobulin light chain amyloid oligomers and fibrils. J. Biol. Chem. 2010;285:37672–37682. doi: 10.1074/jbc.M110.149575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bourgault S, Solomon JP, Reixach N, Kelly JW. Sulfated glycosaminoglycans accelerate transthyretin amyloidogenesis by quaternary structural conversion. Biochemistry. 2011;50:1001–1015. doi: 10.1021/bi101822y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta R, Kapoor N, Raleigh DP, Sakmar TP. Nucleobindin 1 caps human islet amyloid polypeptide protofibrils to prevent amyloid fibril formation. J. Mol. Biol. 2012;421:378–389. doi: 10.1016/j.jmb.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suk JY, Zhang F, Balch WE, Linhardt RJ, Kelly JW. Heparin accelerates gelsolin amyloidogenesis. Biochemistry. 2006;45:2234–2242. doi: 10.1021/bi0519295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Munch J, Baker D, Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McLaughlin RW, De Stigter JK, Sikkink LA, Baden EM, Ramirez-Alvarado M. The effects of sodium sulfate, glycosaminoglycans, and Congo red on the structure, stability, and amyloid formation of an immunoglobulin light-chain protein. Protein Sci. 2006;15:1710–1722. doi: 10.1110/ps.051997606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borysik AJ, Morten IJ, Radford SE, Hewitt EW. Specific glycosaminoglycans promote unseeded amyloid formation from beta2-microglobulin under physiological conditions. Kidney Int. 2007;72:174–181. doi: 10.1038/sj.ki.5002270. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, Lashuel HA, Walz T, Colon W. Murine apolipoprotein serum amyloid A in solution forms a hexamer containing a central channel. Proc. Natl. Acad. Sci. U S A. 2002;99:15947–15952. doi: 10.1073/pnas.252508399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang L, Colon W. Urea-induced denaturation of apolipoprotein serum amyloid A reveals marginal stability of hexamer. Protein Sci. 2005;14:1811–1817. doi: 10.1110/ps.051387005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang L, Lashuel HA, Colon W. From hexamer to amyloid: marginal stability of apolipoprotein SAA2.2 leads to in vitro fibril formation at physiological temperature. Amyloid. 2005;12:139–148. doi: 10.1080/13506120500223084. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Srinivasan S, Ye Z, Javier Aguilera J, Lopez MM, Colon W. Serum amyloid A 2.2 refolds into a octameric oligomer that slowly converts to a more stable hexamer. Biochem. Biophys. Res. Commun. 2011;407:725–729. doi: 10.1016/j.bbrc.2011.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patke S, Srinivasan S, Maheshwari R, Srivastava SK, Aguilera JJ, Colon W, Kane RS. Characterization of the oligomerization and aggregation of human serum amyloid a. PLoS One. 2013;8:e64974. doi: 10.1371/journal.pone.0064974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inoue S, Kawano H, Ishihara T, Maeda S, Ohno S. Formation of experimental murine AA amyloid fibrils in SAP-deficient mice: high resolution ultrastructural study. Amyloid. 2005;12:157–163. doi: 10.1080/13506120500232010. [DOI] [PubMed] [Google Scholar]
- 56.Inoue S, Kisilevsky R. In situ electron microscopy of amyloid deposits in tissues. Methods Enzymol. 1999;309:496–509. doi: 10.1016/s0076-6879(99)09034-5. [DOI] [PubMed] [Google Scholar]
- 57.Wien TN, Sorby R, Omtvedt LA, Landsverk T, Husby G. Kinetics of glycosaminoglycan deposition in splenic AA amyloidosis induced in mink. Scand. J. Immunol. 2004;60:600–608. doi: 10.1111/j.0300-9475.2004.01520.x. [DOI] [PubMed] [Google Scholar]
- 58.Cohlberg JA, Li J, Uversky VN, Fink AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry. 2002;41:1502–1511. doi: 10.1021/bi011711s. [DOI] [PubMed] [Google Scholar]
- 59.Yamamoto S, Yamaguchi I, Hasegawa K, Tsutsumi S, Goto Y, Gejyo F, Naiki H. Glycosaminoglycans enhance the trifluoroethanol-induced extension of beta 2-microglobulin-related amyloid fibrils at a neutral pH. J. Am. Soc. Nephrol. 2004;15:126–133. doi: 10.1097/01.asn.0000103228.81623.c7. [DOI] [PubMed] [Google Scholar]
- 60.McLaurin J, Franklin T, Zhang X, Deng J, Fraser PE. Interactions of Alzheimer amyloid-beta peptides with glycosaminoglycans effects on fibril nucleation and growth. Eur. J. Biochem. 1999;266:1101–1110. doi: 10.1046/j.1432-1327.1999.00957.x. [DOI] [PubMed] [Google Scholar]
- 61.Egashira M, Takase H, Yamamoto I, Tanaka M, Saito H. Identification of regions responsible for heparin-induced amyloidogenesis of human serum amyloid A using its fragment peptides. Arch. Biochem. Biophys. 2011;511:101–106. doi: 10.1016/j.abb.2011.04.019. [DOI] [PubMed] [Google Scholar]