

Abstract
Dysregulated TSC/mTOR signaling may play a pathogenetic role in forms of syndromic autism, such as autism associated with tuberous sclerosis, a genetic disorder caused by heterozygous TSC1 or TSC2 mutations. Environmental risk factors, such as gestational viral infections, may, in some cases, also contribute to the pathogenesis of autism and related neuropsychiatric disorders. We have recently found that a heterozygous Tsc2 mutation and the poly I:C model of maternal immune activation (MIA) interactively perturb fetal development and adult social behavior in mice, suggesting that these factors converge on shared pathways. TSC/mTOR signaling plays an important role in the modulation of immune responses, raising the possibility that the damage caused by MIA was greater in Tsc2+/− than in wildtype fetuses because of an exacerbated immune response in the mutants. Here, cytokine antibody arrays were employed to measure relative cytokine abundances in the fetal brain and the placenta during MIA. Cytokines were induced by gestational poly I:C but there was no obvious modulatory effect of Tsc2 haploinsufficiency. The data indicate that cytokine exposure during MIA is comparable in Tsc2 haploinsufficient and wildtype control fetuses, suggesting that downstream molecular and cellular processes may account for the interactive effects of Tsc2 haploinsufficiency and MIA.
1. Introduction
Dysregulated mTOR signaling and altered protein synthesis are emerging as common themes in the biology of autism [1–3]. In brief, heterozygous mutations in the TSC1 or TSC2 gene cause tuberous sclerosis, a multisystem disorder, which is associated with autism in 20–60% of cases [4–6]. Additionally, mutations in the genes encoding for the upstream regulators PTEN and NF1, as well as in the downstream effector eIF4E, have been linked to autism [7–9]. Moreover, fragile X syndrome, another single gene disorder, in which the regulation of mRNA translation is perturbed [1], is associated with autism [11]. These data collectively highlight the mTOR pathway and protein synthesis as one important theme in the pathogenesis of autism-related disorders.
In addition to genetic factors, environmental risk factors may also contribute to the pathogenesis of autism spectrum disorders. At least some prenatal viral infections elevate the risk for neuropsychiatric disorders (such as autism spectrum disorders and schizophrenia) in the offspring [10, 12–16]. Moreover, there is a growing literature suggesting the presence of inflammatory or immunological changes in the brains of at least a subset of individuals affected by autism [17–21], suggesting that immune activation/inflammatory processes may play a role (primary and/or secondary) in the pathogenesis of some cases of autism.
Prenatal viral infections have been modeled in mice by injection of poly I:C [19], a synthetic double-stranded RNA that elicits an immune response via activation of toll-like receptor 3. Gestational poly I:C injections in mice and rats elicit behavioral, neurochemical, and structural abnormalities in the developing offspring [19, 22, 23], which is consistent with the notion that maternal immune activation during gestational periods can alter proper neurodevelopment and, hence, results in behavioral consequences in the offspring generation.
Heterozygous Tsc2 mutations and the poly I:C paradigm of maternal immune activation (MIA) showed significant interactive effects in mice, indicating a cooperative influence on gestational survival and postnatal behavioral traits [24]: fewer Tsc2+/− pups than wildtype (WT) pups were born to poly I:C injected dams, while the number of Tsc2+/− pups and WT pups born to vehicle-injected mothers did not differ. Moreover, grown-up mice exposed to both gestational poly I:C and Tsc2 haploinsufficiency showed deficient social exploratory behavior, while this was not the case in the animals exposed to one of these factors alone.
Gestational immune activation may disrupt normal brain development at least in part because of crosstalk between cytokines and the developing CNS [19, 22, 25–27]. TSC/mTOR signaling regulates immunological and inflammatory processes [28, 29] and, accordingly, it is possible that an exacerbated immune response in the Tsc2 mutant fetus accounts for the more severe consequences of gestational poly I:C in the Tsc2+/− background. To address this possibility, Tsc2+/− fetuses and WT control fetuses were gestationally exposed to poly I:C or saline and cytokine responses were measured in the placenta, as well as the fetal Tsc2+/− and WT control brain.
2. Materials and Methods
2.1. Mice
Mice were generated as previously described [24]. In brief, in order to generate Tsc2+/− and WT offspring gestationally exposed to either poly I:C or saline, Tsc2+/− male breeders (on a C57BL6/Ncrl genetic background) were mated with C57BL6/J wildtype females. After overnight mating, female breeders with a vaginal plug were designated as E0, single-housed, and left undisturbed, except for weekly cage change. At E12.5, pregnant females received a single intraperitoneal injection of 20 mg/kg poly I:C (Sigma) or vehicle (see below). Poly I:C is supplied as potassium salt and poly I:C constitutes 10% of the total weight of the salt. The dosage was based on the actual weight of poly I:C itself. Poly I:C was dissolved in 0.9% sterile saline and a volume of 10 μL/g was injected. Local and federal regulations regarding animal welfare were followed.
2.2. Tissue Extraction
Two hours or 6.5 hours after poly I:C, pregnant females were anaesthetized with isoflurane and were decapitated. The uterus was extracted and briefly washed in ice-cold phosphate-buffered saline. Subsequently, fetal brains and placentas were isolated in fresh ice-cold phosphate-buffered saline under a dissection microscope. Tissue was flash frozen and stored at −80°C until processed for cytokine antibody arrays.
2.3. Cytokine Antibody Arrays
Cytokine antibody arrays (RayBio Mouse Cytokine Antibody Array 3 and RayBio Mouse Cytokine Antibody Array 2) were used according to the manufacturer's instructions. In brief, tissue was lysed in the cell lysis buffer provided after addition of proteinase inhibitors (Sigma, P8340), homogenized, and centrifuged at 5000 g for 10 min (4°C) and the supernatant was isolated. Protein concentrations were established with BCA assays. Next, the volume containing 125 μg protein was taken from each sample and pooled by group (WT/saline, WT/poly I:C, Tsc2+/−/saline, and Tsc2+/−/poly I:C; materials of 4 animals from 4 different litters were pooled per group for each experiment; at the 6.5 h time point, only fetal brain from the following groups was analyzed: WT/saline, WT/poly I:C, and Tsc2+/−/poly I:C). Pooled lysates were then diluted at least 10-fold in 1x blocking buffer to a total volume of 2.5 mL. After a blocking step, array membranes were incubated in pooled lysates at 4°C overnight. Next, membranes were washed 3 times in 2 mL wash buffer I for 5 min each followed by washing steps in wash buffer II (2 × 5 min; room temperature). Array membranes were then incubated in biotin-conjugated antibody solutions for 2 h at room temperature followed by washing steps in wash buffer I (3 × 5 min) and II (2 × 5 min). Next, membranes were incubated in HRP-conjugated streptavidin (1 : 1000) at room temperature for 2 h. After washing (wash buffer I, 3 × 5 min; wash buffer II, 2 × 5 min), array membranes were incubated in ECL-plus and imaged on a Typhoon 9400 scanner. Image analyses were performed in ImageQuant 5.2. Array membranes contained a total of 62 or 32 different cytokine antibody probes (RayBio Mouse Cytokine Antibody Array 3 and RayBio Mouse Cytokine Antibody Array 2, resp.), in addition to positive controls and negative controls. Fluorescent intensities were measured for cytokine probes and positive and negative controls. After subtraction of background, data for cytokine probes were normalized to the average fluorescence intensity of the positive controls. All data are expressed as percentage of the corresponding value of the WT/saline group and are based on duplicate measurements for each cytokine probe (shown as mean +/− SEM).
2.4. Statistics
In order to compare relative cytokine abundances across genotypes and treatment groups, multifactorial ANOVAs were performed (with genotype and/or treatment as between-subjects factor and/or cytokine as within-subjects factor) as described in the main text.
3. Results
To evaluate if fetal Tsc2 haploinsufficiency modifies gestational cytokine responses during a maternal immune activation paradigm [22], pregnant C57BL6/J female mice were subjected to a single poly I:C injection (20 mg/kg, i.p.) or saline control at E12.5. Fetal brains and placental tissue were extracted 2 h or 6.5 h after the injection and the relative cytokine abundance in these tissues was assessed using cytokine antibody arrays.
In placental tissue (6.5 h after poly I:C), poly I:C caused a robust increase of several cytokines, including the proinflammatory cytokine IL-6 (Figure 1). An overall comparison of cytokine profiles by three-way ANOVA with genotype and treatment as between-subjects factors and cytokine as within-subjects factor yielded a significant main effect of poly I:C treatment (P = 0.0003). Additional analyses on the level of individual cytokines (using two-way ANOVAs with genotype and treatment as between-subjects factors) showed clear increases of G-CSF, IL-6, IL12 p40/p70, KC, MCP-1, MIP-2, and RANTES in the placental samples of poly I:C treated animals (Figure 1). Poly I:C-induced placental cytokine levels were similar in Tsc2+/− and WT samples (Figure 1).
Figure 1.
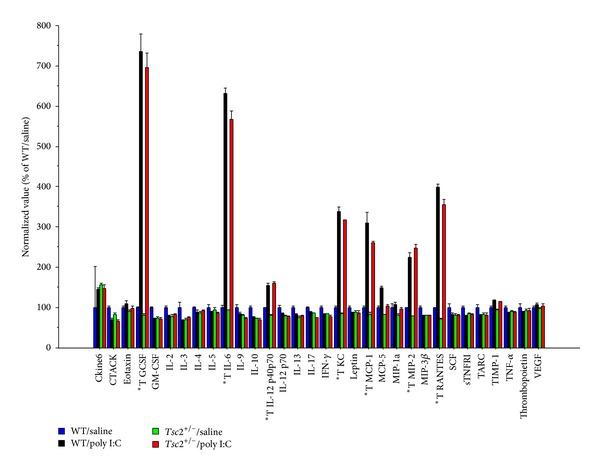
The graph shows relative cytokine abundances in placental tissue lysates at 6.5 h after poly I:C injection as measured by cytokine antibody array. *T denotes individual cytokines with main effect of treatment at P < 0.01 and a fold change > 1.5. Plotted are means +/− SEM.
Poly I:C effects on cytokines in fetal whole brain extracts were more moderate than those seen in the placenta: At 2 h after poly I:C, the levels of several cytokines appeared to be decreased in the treated group compared to saline controls (Figures 2(a) and 2(b)). An overall comparison of cytokine profiles (by three-way ANOVA with genotype and treatment as between-subjects factors and cytokine as within-subjects factor) showed a nonsignificant trend towards an effect of poly I:C (P = 0.09), while there was no effect of genotype (P = 0.34) and no genotype x treatment interaction (P = 0.95). Exploratory statistical analyses focusing on individual cytokines (using two-way ANOVAs with genotype and treatment as between-subjects factors) revealed a number of molecules with significantly altered abundances in the poly I:C groups (Figures 2(a) and 2(b)), such as Fas ligand, fractalkine, IL-3, IL-4, IL-5, lymphotactin, MCP-5, M-CSF, MIG, MIP-1alpha, TARC, TECK, TNF-alpha, and VCAM-1. These analyses, however, showed few effects of Tsc2 haploinsufficiency on fetal cytokine levels (Figures 2(a) and 2(b)).
Figure 2.
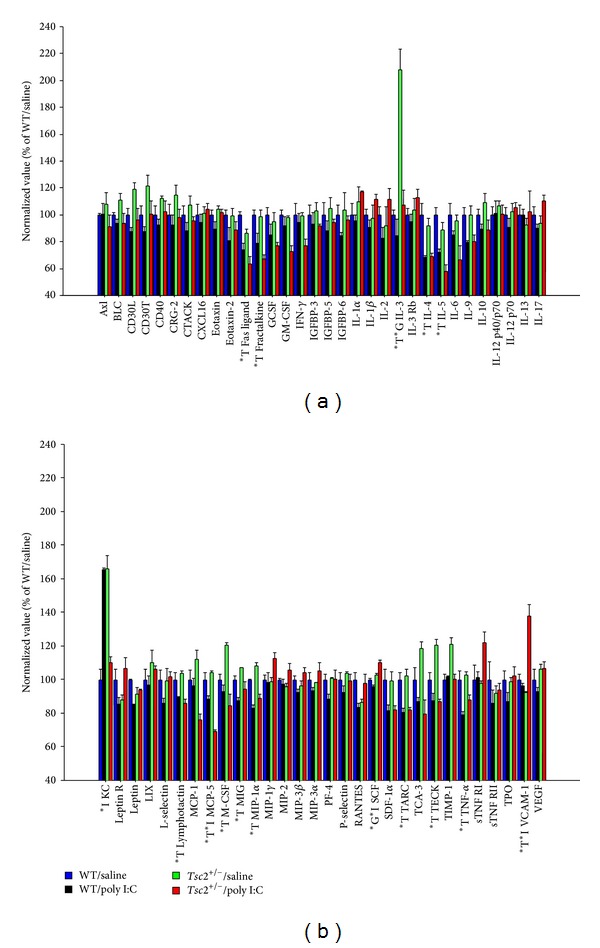
(a) and (b) The figure shows relative cytokine abundances as measured by cytokine antibody arrays in fetal brain at 2 h after gestational poly I:C. Individual cytokines with a main effect of treatment (*T; P < 0.01), genotype (*G; P < 0.01), or a genotype x treatment interaction (*I; P < 0.01) are highlighted. Shown are means +/− SEM.
At 6.5 h after poly I:C, cytokines appeared to be, overall, modestly elevated in poly I:C fetal brains relative to controls, although this finding did not reach statistical significance (Figures 3(a) and 3(b); two-way ANOVA, WT/saline versus poly I:C, treatment as between-subjects factor and cytokine as within-subjects factor: effect of treatment, P = 0.09). Tsc2 haploinsufficiency did not seem to modulate the cytokine response induced by gestational poly I:C in any obvious way (Figures 3(a) and 3(b); two-way ANOVA, WT/poly I:C versus Tsc2+/−/poly I:C, genotype as between-subjects factor and cytokine as within-subjects factor: effect of genotype, P = 0.89; measure x genotype interaction, P = 0.34).
Figure 3.
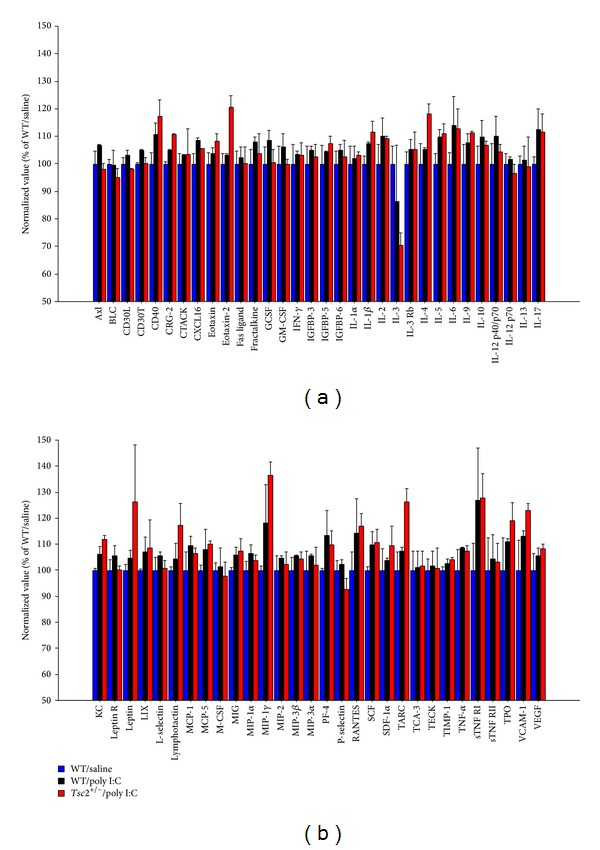
(a) and (b) This graph shows relative cytokine abundances as measured by cytokine antibody arrays in fetal brain at 6.5 h after gestational poly I:C. Shown are means +/− SEM.
Taken together, these provisional data indicate that alterations in fetal brain cytokine abundance during MIA are less pronounced than placental changes and they suggest that cytokine responses during MIA are not substantially influenced by a heterozygous Tsc2 mutation.
4. Discussion
In a previous study, we have found an interactive influence of Tsc2 haploinsufficiency and gestational poly I:C on fetal development and adult behavioral traits in mice [24]: fewer Tsc2+/− than WT pups were born to poly I:C-injected mothers, while the genotype ratio was balanced in vehicle-treated mothers, which suggested, together with the presence of fetal resorption, that gestational poly I:C triggered the preferential elimination of Tsc2+/− fetuses in utero. Surviving poly I:C-exposed Tsc2+/− animals, but not Tsc2+/−/saline and WT/poly I:C mice, showed abnormal social approach behavior when tested in adulthood [24]. Collectively, these data indicated that poly I:C-related gestational immune activation and Tsc2 haploinsufficiency interact to perturb fetal development.
Gestational immune activation may, at least to some extent, interfere with normal fetal development because of adverse effects of cytokines on the developing organism [19, 22, 25–27]. TSC-related cell signaling regulates immunological and inflammatory processes [28, 29] and, therefore, it is conceivable that an exacerbated immune response in the Tsc2+/− fetal brain to gestational poly I:C contributes to the interactive effects of Tsc2 haploinsufficiency and maternal immune activation.
The present analysis of poly I:C-triggered cytokine responses suggests that Tsc2+/− and WT fetal brains are exposed to qualitatively and quantitatively similar cytokine responses upon midgestational poly I:C administration. Cytokine antibody arrays revealed a pronounced increase in the abundance of specific cytokines in the placenta at 6.5 h after poly I:C, representing evidence for the mounted maternal immune activation triggered by poly I:C. Poly I:C caused a complex alteration in cytokine levels in the fetal brain that comprised an initial phase of decreased cytokine abundance (at 2 h after poly I:C) and a subsequent phase of modestly elevated tissue cytokine levels (at 6.5 h after poly I:C), which is consistent with prior data that show that the fetal brain cytokine response during maternal immune activation is a complex function of various parameters, such as time after poly I:C injection and gestational age [30]. Cytokine levels were similar in fetal Tsc2+/− and WT brains, suggesting that genotype-dependent differences in cytokine exposure are unlikely to account for the interactive effects of Tsc2 haploinsufficiency and gestational immune activation [24].
As TSC/mTOR signaling not only regulates immune responses, but also is a downstream signaling mediator of many cytokines and other factors induced by poly I:C, it remains to be explored if altered TSC-related cell signaling downstream of cytokines/other poly I:C-induced factors contributes to the genotype-dependent effects of poly I:C [24]. In addition, it is also possible that the disruptive effects of Tsc2 haploinsufficiency and gestational immune activation converge on certain developmental processes ongoing at the time of poly I:C injection (E12.5). For instance, both gestational poly I:C and TSC gene mutations interfere with and disrupt neurogenesis, neuroblast migration, and proper cortical layering [31–33], which are important features of the developing CNS at E12.5. Accordingly, gestational immune activation and Tsc2 haploinsufficiency may display synergistic effects on these processes and interactively lead to the phenotypic precipitation of otherwise subthreshold neurobiological changes.
5. Conclusion
The present study revealed a similar cytokine induction profile in Tsc2 mutant and WT control brains in the context of a midgestational immune activation paradigm. These findings indicate that an exacerbated cytokine response in Tsc2 mutants is unlikely to account for the interactive effects of Tsc2 haploinsufficiency and maternal immune activation identified in a previous study [24]. Future experiments will address if Tsc2 haploinsufficiency and gestational immune activation may possibly interact by converging on cytokine-induced cell signaling and/or by jointly disrupting midgestational neurodevelopmental processes, such as neurogenesis and/or cell migration.
Conflict of Interests
The author declares that there is no conflict of interests regarding the publication of this paper.
References
- 1.Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480(7375):63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehninger D, Silva AJ. Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends in Molecular Medicine. 2011;17(2):78–87. doi: 10.1016/j.molmed.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelleher RJ, III, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135(3):401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 4.Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. Journal of Autism and Developmental Disorders. 2003;33(4):365–382. doi: 10.1023/a:1025054610557. [DOI] [PubMed] [Google Scholar]
- 5.Smalley SL. Autism and tuberous sclerosis. Journal of Autism and Developmental Disorders. 1998;28(5):407–414. doi: 10.1023/a:1026052421693. [DOI] [PubMed] [Google Scholar]
- 6.Smalley SL, Tanguay PE, Smith M, Gutierrez G. Autism and tuberous sclerosis. Journal of Autism and Developmental Disorders. 1992;22(3):339–355. doi: 10.1007/BF01048239. [DOI] [PubMed] [Google Scholar]
- 7.Butler MG, Dazouki MJ, Zhou X, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. Journal of Medical Genetics. 2005;42(4):318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neves-Pereira M, Müller B, Massie D, et al. Deregulation of EIF4E: a novel mechanism for autism. Journal of Medical Genetics. 2009;46(11):759–765. doi: 10.1136/jmg.2009.066852. [DOI] [PubMed] [Google Scholar]
- 9.Williams PG, Hersh JH. Brief report: the association of neurofibromatosis type 1 and autism. Journal of Autism and Developmental Disorders. 1998;28(6):567–571. doi: 10.1023/a:1026012414193. [DOI] [PubMed] [Google Scholar]
- 10.Atladóttir HÓ, Thorsen P, Østergaard L, et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. Journal of Autism and Developmental Disorders. 2010;40(12):1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- 11.Hagerman RJ, Jackson AW, III, Levitas A, Rimland B, Braden M. An analysis of autism in fifty males with the fragile X syndrome. American Journal of Medical Genetics. 1986;23(1-2):359–374. doi: 10.1002/ajmg.1320230128. [DOI] [PubMed] [Google Scholar]
- 12.Chess S. Autism in children with congenital rubella. Journal of Autism and Childhood Schizophrenia. 1971;1(1):33–47. doi: 10.1007/BF01537741. [DOI] [PubMed] [Google Scholar]
- 13.Chess S. Follow up report on autism in congenital rubella. Journal of Autism and Childhood Schizophrenia. 1977;7(1):69–81. doi: 10.1007/BF01531116. [DOI] [PubMed] [Google Scholar]
- 14.Chess S, Fernandez P, Korn S. Behavioral consequences of congenital rubella. Journal of Pediatrics. 1978;93(4):699–703. doi: 10.1016/s0022-3476(78)80921-4. [DOI] [PubMed] [Google Scholar]
- 15.Sweeten TL, Posey DJ, McDougle CJ. Brief report: autistic disorder in three children with cytomegalovirus infection. Journal of Autism and Developmental Disorders. 2004;34(5):583–586. doi: 10.1007/s10803-004-2552-y. [DOI] [PubMed] [Google Scholar]
- 16.Yamashita Y, Fujimoto C, Nakajima E, Isagai T, Matsuishi T. Possible association between congenital cytomegalovirus infection and autistic disorder. Journal of Autism and Developmental Disorders. 2003;33(4):455–459. doi: 10.1023/a:1025023131029. [DOI] [PubMed] [Google Scholar]
- 17.Garbett K, Ebert PJ, Mitchell A, et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiology of Disease. 2008;30(3):303–311. doi: 10.1016/j.nbd.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgan JT, Chana G, Pardo CA, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biological Psychiatry. 2010;68(4):368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 19.Patterson PH. Immune involvement in Schizophrenia and Autism: etiology, pathology and animal models. Behavioural Brain Research. 2009;204(2):313–321. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 20.Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of Neurology. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- 21.Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. Journal of Neuroscience. 2007;27(40):10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuckerman L, Weiner I. Post-pubertal emergence of disrupted latent inhibition following prenatal immune activation. Psychopharmacology. 2003;169(3-4):308–313. doi: 10.1007/s00213-003-1461-7. [DOI] [PubMed] [Google Scholar]
- 24.Ehninger D, Sano Y, De Vries PJ, et al. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Molecular Psychiatry. 2012;17(1):62–70. doi: 10.1038/mp.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64(1):61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 26.Nawa H, Takahashi M, Patterson PH. Cytokine and growth factor involvement in schizophrenia—support for the developmental model. Molecular Psychiatry. 2000;5(6):594–603. doi: 10.1038/sj.mp.4000730. [DOI] [PubMed] [Google Scholar]
- 27.Shi L, Tu N, Patterson PH. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. International Journal of Developmental Neuroscience. 2005;23(2-3):299–305. doi: 10.1016/j.ijdevneu.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Cao W, Manicassamy S, Tang H, et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nature Immunology. 2008;9(10):1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colina R, Costa-Mattioli M, Dowling RJO, et al. Translational control of the innate immune response through IRF-7. Nature. 2008;452(7185):323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 30.Meyer U, Nyffeler M, Engler A, et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. Journal of Neuroscience. 2006;26(18):4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Miranda J, Yaddanapudi K, Hornig M, Villar G, Serge R, Lipkin WI. Induction of toll-like receptor 3-mediated immunity during gestation inhibits cortical neurogenesis and causes behavioral disturbances. mBio. 2010;1(4) doi: 10.1128/mBio.00176-10.e00176-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magri L, Cominelli M, Cambiaghi M, et al. Timing of mTOR activation affects tuberous sclerosis complex neuropathology in mouse models. Disease Models & Mechanisms. 2013;6:1185–1197. doi: 10.1242/dmm.012096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soumiya H, Fukumitsu H, Furukawa S. Prenatal immune challenge compromises the normal course of neurogenesis during development of the mouse cerebral cortex. Journal of Neuroscience Research. 2011;89(10):1575–1585. doi: 10.1002/jnr.22704. [DOI] [PubMed] [Google Scholar]