

Abstract
While aspirin is generally effective for prevention of cardiovascular disease, considerable variation in drug response exists, resulting in some individuals displaying high on-treatment platelet reactivity. We used pharmacometabolomics to define pathways implicated in variation of response to treatment. We profiled serum samples from healthy subjects pre- and postaspirin (14 days, 81 mg/day) using mass spectrometry. We established a strong signature of aspirin exposure independent of response (15/34 metabolites changed). In our discovery (N = 80) and replication (N = 125) cohorts, higher serotonin levels pre- and postaspirin correlated with high, postaspirin, collagen-induced platelet aggregation. In a third cohort, platelets from subjects with the highest levels of serotonin preaspirin retained higher reactivity after incubation with aspirin than platelets from subjects with the lowest serotonin levels preaspirin (72 ± 8 vs. 61 ± 11%, P = 0.02, N = 20). Finally, ex vivo, serotonin strongly increased platelet reactivity after platelet incubation with aspirin (+20%, P = 4.9 × 10−4, N = 12). These results suggest that serotonin is implicated in aspirin response variability.
Aspirin is the most commonly used drug worldwide and has known analgesic, antipyretic, anti-inflammatory, and antiplatelet effects. Despite its wide use, some molecular and metabolic mechanisms by which aspirin exerts this diverse range of therapeutic effects remain unclear.1 As an antiplatelet agent, aspirin is employed for both primary prevention of cardiovascular disease and secondary prevention of recurrent cardiovascular events following a myocardial infarction.2 Aspirin significantly reduces the risk of cardiovascular death, but ~25% of high-risk patients show persistent platelet reactivity3,4 (i.e., laboratory aspirin resistance) and atherothrombotic events (i.e., clinical aspirin resistance) are relatively common2 while on aspirin therapy. Aspirin exerts its antiplatelet action by irreversibly inhibiting cyclooxygenase-1 (COX-1), thereby preventing the conversion of arachidonic acid (AA) to the potent platelet agonist thromboxane A2.5 The mechanisms underlying variability in aspirin response are poorly understood. Incomplete COX-1 inhibition has been observed in several settings.6,7 However, poor response despite complete COX-1 inhibition has also been reported,6,8 suggesting non–COX-1-mediated mechanisms. To better understand these non–COX-1-mediated mechanisms, it is important to probe pathways beyond traditional measures of aspirin response (e.g., thromboxane B2 levels and AA-stimulated platelet aggregation). Understanding these non–COX-1-related mechanisms is critical in order to help identify patients who will fail to respond to aspirin so that alternative medications may be used to improve clinical outcomes.3,9
Pharmacometabolomics is an emerging field that aims to use metabolomics tools to define the mechanisms of action for drugs and the biochemical basis for variation in response to treatment.10,11,12,13,14,15 The low-molecular-weight metabolites measured with metabolomics in biofluids such as urine or blood are the end result of the entire chain of regulatory changes that occur in response to treatment. Metabolic profiles therefore integrate genetic and environmental influences and provide unique information that can help explain the drug–response phenotype. Several recent studies have demonstrated that metabolic profiles can indeed contribute to predicting treatment outcomes.16,17,18
In the present study, we hypothesized that pharmacometabolomics might help identify metabolites influencing response to aspirin. Given that amino acids and urea cycle amines have previously been associated with cardiovascular disease,19,20 we used metabolic profiling targeted for the measurement of metabolites containing an amine functional group in serum samples from healthy subjects before and after aspirin therapy. Our specific aims were to characterize the metabolic signature of aspirin exposure and to identify metabolites implicated in response variation to aspirin antiplatelet therapy.
Results
Population characteristics and platelet aggregation
Healthy volunteers enrolled in the Heredity and Phenotype Intervention (HAPI) Heart Trial were selected for metabolic profiling based on their response to low-dose aspirin treatment. Supplementary Figure S1 describes the procedure for sample selection. Briefly, all 745 subjects who underwent a 2-week aspirin intervention study (81 mg/day) as part of the initial HAPI study were divided into sex-specific quartiles of collagen-stimulated ex vivo platelet aggregation. To maximize the probability of identifying candidate metabolites implicated in aspirin response variability, we first selected samples from 42 subjects from the first quartile and 38 subjects from the fourth quartile of postaspirin collagen-stimulated platelet aggregation (discovery cohort). The two groups were matched for sex and age and had similar body mass index. Table 1 presents subject characteristics and platelet aggregation.
Table 1. Subject characteristics in the discovery cohort.
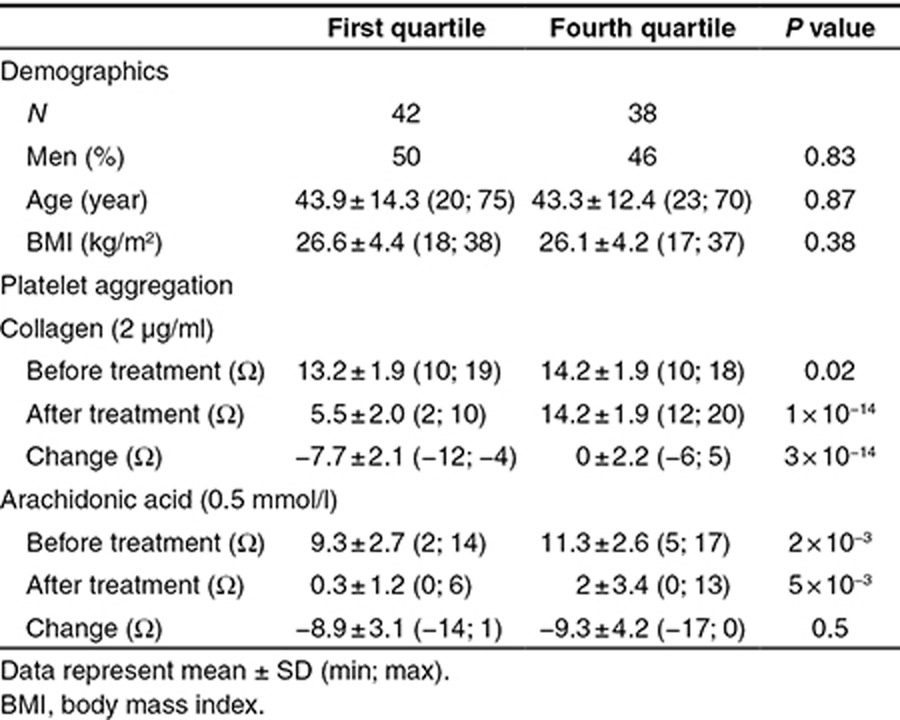
Before treatment, collagen-induced aggregation was higher in subjects from the fourth quartile than in those from the first quartile of aspirin response (P = 0.02). By design, postaspirin collagen-induced aggregation was significantly lower in subjects from the first quartile than in those from the fourth quartile (5.5 ± 2.0 vs. 14.2 ± 1.9 Ω, P = 10−14).
Metabolic signature of aspirin exposure
Multilevel principal component analysis21 was used to investigate the primary sources of variation in serum metabolic profiles of subjects from the discovery cohort, using the 35 metabolites measured. Multilevel principal component analysis plots revealed a trend toward a separation between metabolic profiles before and after aspirin on principal components 1 and 2, irrespective of gender or quartile (Supplementary Figure S2A). This demonstrates that aspirin treatment was one of the largest sources of variation in the metabolic profiles. As described in the Methods, we subsequently used linear mixed models to evaluate simultaneously the effects of aspirin exposure, gender, quartile, and their interaction on each metabolite level. We identified 19 individual metabolites significantly different post- compared to preaspirin exposure in those 80 subjects (Table 2).
Table 2. Metabolites significantly changed during aspirin exposure.
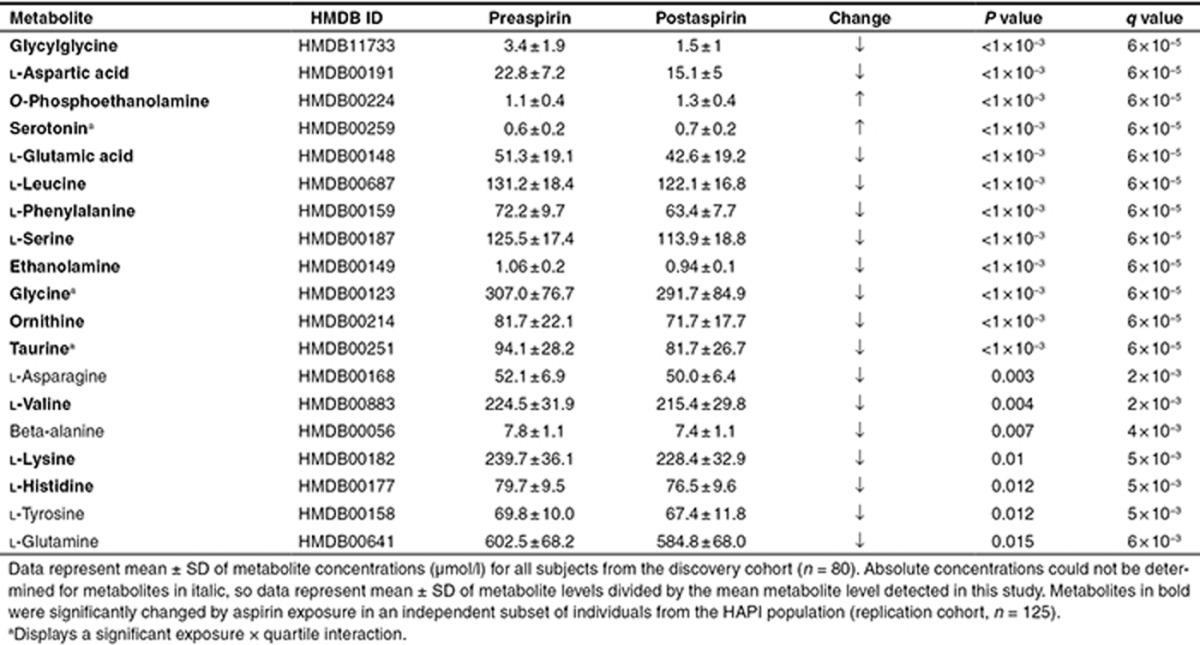
Significant differences in metabolite levels were observed between men and women both pre- and postaspirin (Supplementary Table S1). However, no significant interaction between aspirin treatment and gender was observed, demonstrating that aspirin affected metabolite levels similarly in men and women.
Differential metabolic signature of aspirin exposure in the two extreme quartiles of aspirin response
We evaluated the differences in metabolic signatures in the two extreme quartiles of aspirin response and identified four metabolites with significant treatment × quartile interaction: alanine (P = 0.003, q = 0.02), taurine (P = 0.01, q = 0.04), glycine (P = 0.03, q = 0.05), and serotonin (P = 0.03, q = 0.05).
Alanine was significantly decreased in subjects from the first quartile (P < 10−3, q < 10−3) but not in those from the fourth quartile upon aspirin treatment, resulting in lower levels in subjects from the first quartile postaspirin (P = 0.006, q < 10−3). Subjects from the fourth quartile had higher levels of taurine before aspirin (P = 0.005, q = 0.002) that significantly decreased upon treatment (P < 10−3, q < 10−3), resulting in similar levels in both groups posttreatment. Glycine was significantly decreased in subjects from the first quartile only upon aspirin (P < 10−3, q < 10−3), resulting in lower postaspirin levels in those subjects (P < 10−3, q < 10−3) (Supplementary Figure S3).
Interestingly, subjects from the fourth quartile had higher levels of serotonin preaspirin (0.66 ± 0.25 vs. 0.53 ± 0.19 µmol/l, P = 0.02, q = 0.05) and the difference in serotonin levels between the two groups increased further postaspirin (0.76 ± 0.28 vs. 0.55 ± 0.18 µmol/l, P < 10−3, q < 10−3) (Figure 1a).
Figure 1.

Serotonin in the two extreme quartiles of aspirin response. Data represent means and 95% confidence intervals of serotonin levels before (blue) and after (red) aspirin exposure in the discovery cohort (a: first quartile n = 42, fourth quartile n = 38) and in the replication cohort (b, first quartile n = 19, fourth quartile n = 19). *P < 0.05, ***P < 0.001.
Replication cohort
To validate our findings, we profiled serum samples in an independent group of 125 HAPI subjects across the entire distribution of drug response. This replication set was matched for sex, age, and body mass index with our discovery cohort (Supplementary Table S2).
Consistent with our discovery sample, multilevel principal component analysis showed a trend toward a separation between metabolic profiles pre- and postaspirin exposure in all subjects (Supplementary Figure S2B) and 15 individual metabolites changed significantly post- compared to preaspirin (Table 2, highlighted in bold), confirming the strong effect of aspirin on amine metabolites.
To provide a direct replication of our previous results in our discovery cohort, the four metabolites that were related to drug response in our discovery sample (i.e., serotonin, taurine, glycine, and l-alanine) were first investigated in the subjects from the two extreme quartiles in this replication cohort. Again, serotonin levels were significantly higher in subjects from the fourth quartile (n = 19) compared to those from the first quartile (n = 19) both pre- and postaspirin (P < 0.001 for both, Figure 1b). No significant differences in taurine, glycine, or l-alanine levels were observed (Supplementary Figure S3).
We next investigated whether metabolite levels were correlated to ex vivo agonist-induced platelet aggregation across the continuum of aspirin response in all replication subjects (n = 125). Pre- and postaspirin serotonin levels correlated with postaspirin collagen-induced platelet aggregation (ρ = 0.39 and P = 9 × 10−6; ρ = 0.38 and P = 1 × 10−5, respectively) (Figure 2). Pre- and postaspirin serotonin levels were highly correlated to each other (Supplementary Figure S4). Importantly, serotonin levels did not correlate with AA-induced platelet aggregation. Finally, taurine, glycine, l-alanine, or any other metabolite did not correlate significantly with AA- or collagen-induced platelet aggregation pre- or posttreatment (Supplementary Figure S5).
Figure 2.

Correlation between serotonin and ex vivo agonist-induced platelet aggregation in the replication cohort (N = 125). (a–c) Platelet aggregation after stimulation with collagen (2 µg/ml). (d–f) Platelet aggregation after stimulation with arachidonic acid (0.5 mmol/l).
Serotonin as a predictor of on-aspirin platelet reactivity
Because we consistently found that serotonin was associated with aspirin response evaluated by ex vivo collagen-induced platelet aggregation in Amish subjects from the HAPI study, a genetically and culturally homogeneous group, we next sought to investigate whether baseline serotonin levels could help predict which individuals would retain high platelet reactivity postaspirin in an independent cohort of 38 unrelated healthy subjects from the general population (functional cohort 1). We first compared serotonin levels in platelet-rich plasma (PRP), platelet-poor plasma (PPP), and serum. As expected, PPP serotonin levels were very low (Figure 3a), whereas serum and PRP serotonin levels were highly correlated (R2 = 0.7, P = 1 × 10−6, Figure 3b), demonstrating that serum serotonin mostly reflected platelet-derived serotonin. However, because serotonin can bind to proteins during the coagulation process,22 thereby reducing the amount of serotonin in serum, we used PRP in the following experiments. Subjects were divided into quartiles of preaspirin PRP serotonin levels, which ranged from 0.37 to 2.22 nmol/l/106 platelets in subjects from the lowest quartile (n = 10) and from 3.26 to 6.56 nmol/l/106 platelets in subjects from the highest quartile (n = 10). Platelet aggregation was measured at baseline and after ex vivo aspirin exposure (53 µmol/l for 10 min).
Figure 3.

Ex vivo functional studies of serotonin. Thirty-eight healthy subjects (functional cohort 1) independent from the Heredity and Phenotype Intervention study were selected to participate in functional studies. (a,b) Serotonin was measured at fasting in platelet-rich plasma (PRP), serum, and platelet-poor plasma (PPP). (c,d) Subjects were separated into four quartiles of PRP serotonin levels. Arachidonic acid–stimulated (c) and collagen-stimulated (d) platelet aggregation were measured before (blue) and after (red) incubation of PRP with aspirin in subjects with low (N = 10) vs. high (N = 10) baseline serotonin level. (e) Platelets from 12 additional healthy subjects (functional cohort 2) were exposed ex vivo to serotonin (1 µmol/l) alone or to serotonin plus aspirin. Data represent means and 95% confidence intervals of collagen-stimulated platelet aggregation. *P < 0.05, ***P < 0.001.
Before aspirin, no difference was observed between the subjects with the highest and the lowest serotonin levels preaspirin in AA- or collagen-induced platelet aggregation (Figure 3c,d). Postaspirin, AA-induced platelet aggregation was completely inhibited and no difference was found between the two groups (Figure 3c), whereas collagen-stimulated platelet aggregation was inhibited to a lesser extent in individuals with higher serotonin levels preaspirin (61 ± 11 vs. 72 ± 8% respectively, P = 0.02) (Figure 3d).
Using all 38 subjects from this additional cohort, we also investigated the correlations between PRP serotonin levels and platelet aggregation measures. We observed a positive correlation between PRP serotonin levels and postaspirin collagen-induced platelet aggregation (ρ = 0.30, P = 0.06; Supplementary Figure S6). No significant correlation was observed between serotonin and AA-induced platelet aggregation (data not shown).
Effect of serotonin on on-aspirin platelet aggregation
Finally, although serotonin is a well-described weak platelet activator,23,24 little is known regarding its action in the context of aspirin therapy. Therefore, we evaluated how ex vivo addition of serotonin to platelets could modify collagen-induced platelet aggregation pre- and postaspirin incubation in a fourth independent group of 12 healthy subjects (functional cohort 2). As expected, in the absence of aspirin, addition of serotonin resulted in a slight increase in collagen-induced platelet aggregation (+2%, P = 0.01) (Figure 3e). After aspirin incubation (16.7 µmol/l or 30 min), the effect of serotonin on platelet aggregation was significantly and substantially larger (+20%, P = 4.9 × 10−4) (Figure 3e). These results demonstrate that serotonin potentiated the agonist effects of collagen on platelet aggregation more strongly upon aspirin exposure.
Discussion
We investigated the metabolic signature of aspirin exposure in healthy volunteers using a quantitative mass spectrometry-based metabolomics platform targeted to the measurement of metabolites containing an amine functional group. Our first finding was that aspirin induced strong changes in the serum metabolic profiles of all subjects, independently of their gender or their response to treatment. Indeed, we observed a change in the concentrations of 15 amine metabolites upon aspirin exposure in all subjects. These results are in accordance with our previous findings.25 In our previous work, the same serum samples were profiled using an untargeted gas chromatography–mass spectrometry platform, which provided a broader coverage of the metabolome than the targeted method used here. Using gas chromatography–mass spectrometry, we also observed a strong general signature of aspirin exposure on the metabolic profiles of all subjects, including significant changes in the level of metabolites involved in purine metabolism. Our past and present results therefore consistently demonstrate systemic metabolic effects of aspirin that are not directly attributed to prostaglandin inhibition. These findings are of utmost importance to unveil new biology regarding the mechanisms of action of aspirin. Oral bioavailability of aspirin is ~50–60% because a fraction of the absorbed dose is deacetylated to salicylic acid in plasma and liver before entering the systemic circulation. The serum half-life of aspirin is ~20 min and the fall in aspirin concentration is associated with a rapid rise in salicylic acid concentration, which is itself renally excreted or further metabolized.26 Our present findings highlight the importance of systemic non–COX-1 mediated pathways in aspirin's general mechanisms of action. Whether aspirin itself or its metabolites disrupt these pathways remains to be determined.
Our second major finding was that increased levels of serotonin correlated with higher postaspirin platelet reactivity. In humans, serotonin is synthesized only in neurons27 and in enterochromaffin (intestine) cells28 and platelets are the only blood cells capable of transporting serotonin when it is released into plasma. In platelets, serotonin is stored at high concentrations into dense granules and is secreted upon activation. Serotonin is considered to be only a weak agonist, which per se does not activate platelets, but dose dependently enhances platelet activation induced by other agonists,23 an effect mediated through the binding of secreted serotonin to the 5HT2A receptor on the surface of surrounding platelets23,29 and through a receptor-independent signaling pathway.24 Mice with elevated blood serotonin in the absence of other cardiovascular risk factors had a platelet phenotype of hyperreactivity.30 In humans, the importance of serotonergic mechanisms in platelet aggregation has been highlighted by several studies on selective serotonin reuptake inhibitors (SSRIs). SSRIs block the serotonin transporter, thereby inhibiting serotonin uptake into neurons as well as into platelets,31 leading to lower platelet serotonin content and reduced platelet aggregation.32 These mechanisms might participate to the protective role of SSRIs against cardiovascular disease.33,34 Moreover, epidemiological studies have shown that the concomitant use of SSRIs and aspirin increases the risk of bleeding compared to each treatment alone.35 Our data add to these epidemiological data by suggesting that serotonin may mediate the increased bleeding risk of aspirin plus SSRIs, rather than the interaction being purely an additive pharmacodynamics effect.
The role of serotonin in cardiovascular disease is not completely understood, however, it seems that plasma serotonin is elevated in a subset a patients with cardiovascular disorders. Vikenes et al.36 have shown for example that plasma serotonin was associated with the extent of coronary artery disease and the occurrence of cardiac event in patients undergoing coronary angiography. Our metabolomics results suggest that high serotonin levels are also associated with poor response to aspirin antiplatelet therapy, as assessed by ex vivo collagen-stimulated platelet aggregation. Moreover, we show that serotonin potentiates collagen-induced platelet aggregation significantly more after platelets were incubated with aspirin than before aspirin incubation. These ex vivo findings deserve in vivo confirmation but highlight the fact that the effect of serotonin on platelets might be further enhanced by aspirin.
In our metabolomics cohorts, baseline (preaspirin) serotonin levels were higher in those subjects from the fourth quartile of aspirin response. Using a more heterogeneous group of healthy volunteers (functional cohort 1), we confirmed that platelets from subjects that have high baseline serotonin levels retain higher ex vivo collagen-induced reactivity after incubation with aspirin (Figure 3d). Together, our results provide preliminary evidence that serotonin might help predict those individuals that retain high on-aspirin platelet reactivity. It should be noted that we measured serotonin in serum. The measurements of serotonin in serum reflect both intraplatelet serotonin and the plasma circulating serotonin mostly bound to proteins. Plasma serotonin levels are usually very low (0.07 µmol/l, Figure 3a), accounting for <15% of serotonin in serum. Thus, the ~20–25% difference observed in our study between serum serotonin levels of subjects from the first and the fourth quartiles of aspirin response is most probably driven by differences in intraplatelet serotonin. Whether plasma serotonin also correlates with aspirin response is unknown.
There are many pathways of platelet activation, besides stimulation of the thromboxane A2 receptor, including stimulation of the platelet glycoprotein receptors for collagen, ADP, van Willebrand factor, thrombin, epinephrine, and serotonin.37 Constitutive or drug-induced enhanced sensitivity of platelets to pathways that are independent from COX-1 and not blocked by aspirin might represent one mechanism of poor response to aspirin. In poor responders to low-dose aspirin in vitro, enhanced sensitivity of platelets to pathways of aggregation that do not depend on thromboxane A2, such as ADP38 and collagen,39 has already been reported. Here, we found that serotonin levels were highly correlated with COX-1–independent platelet aggregation (namely collagen-induced aggregation) but not with AA-induced aggregation, therefore supporting the hypothesis that in our population, subjects that retain high collagen-induced platelet aggregation on aspirin also have enhanced sensitivity to serotonergic mechanisms and that this phenomenon is independent from COX-1 inhibition by aspirin. Consistent with our present findings, Duerschmied et al.40 recently demonstrated that serotonin receptor antagonism improved platelet inhibition in low responders to clopidogrel, another widely used antiplatelet drug that acts by inhibiting the P2Y12-ADP receptor on the platelet surface.
We used healthy volunteers in this study, which is one limitation. However, this is advantageous because the HAPI subjects underwent a strict drug and supplement washout period before treatment, leading to a thorough evaluation of aspirin intervention alone. A second limitation is that we investigated the interaction between serotonin and aspirin ex vivo in our functional cohort 2. Since aspirin is not metabolized within platelets, we therefore do not take into account the possible role of aspirin metabolites. Finally, we evaluated aspirin response using agonist-induced platelet aggregation. Although these tests are widely used surrogate markers that evaluate response to antiplatelet therapy41 and have been correlated to clinical outcomes,6,42,43 we aim to ultimately predict the occurrence of cardiovascular events while on therapy. Our current findings therefore need to be validated in patients with cardiovascular disease.
In conclusion, we successfully demonstrate the use of pharmacometabolomics in establishing new metabolic signature of drug exposure. We demonstrate that low-dose aspirin treatment induces profound changes in the metabolic profiles of healthy individuals that cannot be directly attributed to COX-1 inhibition. We then show that correlating metabolic profiles to clinically relevant surrogate endpoints can identify metabolites implicated in variation in drug response. Using this approach, we previously demonstrated that serum levels of purine metabolites correlated with on-aspirin platelet reactivity.25 Here, we show that intraplatelet levels of serotonin correlate with on-aspirin platelet reactivity. Together, our results suggest that certain metabolite levels, together with other factors, could contribute to predicting aspirin response.
Methods
HAPI study design and platelet aggregation measures
Samples for metabolomics profiling were obtained from subjects enrolled in the HAPI study, which has been described previously.44 Briefly, participants were adult members of the Old Order Amish population from Lancaster County, PA, and considered healthy. A total of 745 subjects participated in a short-term aspirin intervention where they were given 81 mg of aspirin for 14 consecutive days.45 Blood samples were obtained after an overnight fast and ex vivo platelet aggregometry was performed before aspirin therapy and again the morning after the last dose by the same technician as previously described.25,44 Aggregation was induced with collagen (2 µg/ml) or AA (0.5 mmol/l). Blood samples for serum preparation were allowed to clot at room temperature for 15 min, centrifuged at 3,000 rpm for 10 min then immediately frozen at −80 °C.
Metabolomics
Sample selection. All 745 HAPI subjects were grouped into sex-specific quartiles of aspirin response as assessed by postaspirin collagen-induced platelet aggregation adjusted for age and preaspirin collagen-induced platelet aggregation. We specifically used collagen-stimulated platelet aggregation instead of AA-stimulated platelet aggregation as a surrogate measure of drug response in order to preferentially identify non–COX-mediated changes to the metabolome. Only non–first-degree relatives were selected for metabolic profiling. In the discovery cohort, samples originated from 42 subjects from the first drug–response quartile and from 38 subjects from the fourth drug–response quartile (Supplementary Figure S1). In the replication cohort, samples originated from 19, 46, 41, and 19 subjects from the first, second, third, and fourth quartile, respectively.
Mass spectrometry. The liquid chromatography–mass spectrometry method used is targeted to the analysis of primary and secondary amines. Detailed procedures for metabolomics measurement are provided in the Supplementary Methods, but briefly, analysis was performed using AccQ-Tag derivatization and samples were analyzed by ultra performance liquid chromatography–tandem mass spectrometry. Thirty metabolites were quantified and four metabolites were semiquantified because calibration standards were not available (Supplementary Table S3).
Follow-up functional studies
Two independent cohorts that were not part of the HAPI Heart study, referred to as functional cohorts 1 and 2, were evaluated to assess the effects of serotonin on aspirin response.
Functional cohort 1. Forty healthy participants were recruited from Duke University Medical Center. They completed questionnaires, which assessed for factors that could interfere with endogenous serotonin levels (dietary information, medication history, and smoking). Two subjects were not included in the study because one was pregnant and one was on aspirin therapy. Subject characteristics are presented in Supplementary Table S4.
Whole blood was drawn after an overnight fast using standard venipuncture technique into sodium citrate (0.105 mol/l, 3.2%) Vacutainer tubes. These tubes were spun immediately at 135 g for 15 min to separate PRP from red cells. Tubes were centrifuged further at 2,100 g to obtain PPP. Platelet counts of the pooled PRP were determined using a Sysmex KX-21N Hematology Analyzer (Sysmex America, Lincolnshire, IL) and PRP was adjusted with PPP to 250,000 platelets/µl. In addition to the sodium citrate tubes, a Vacutainer serum collection tube was obtained during venipuncture. The tube was allowed to clot for at least 30 min and centrifuged at 2,100 g to obtain serum.
Serotonin was quantified using reverse phase high-pressure liquid chromatography with electrochemical detection. The mobile phase contained 0.1 mol/l sodium phosphate, 0.8 mmol/l octanesulfonic acid, 0.1 mmol/l sodium ethylenediaminetetraacetic acid, and 18% methanol, the pH was adjusted to 3.1 and flow rate was 0.7 ml/min. Samples were quantified with a BAS Epsilon electrochemical detector (Bioanalytical Systems, W. Lafayette, IN) with dual 3-mm glassy carbon electrodes (MF-1000) set to 0.7 V.
Subjects were divided into quartiles of PRP serotonin concentration. Platelet function was evaluated in PRP by the same technician using a 470-VS Chrono-Log aggregometer pre- and postaspirin incubation (53 µmo/l for 10 min at room temperature) after stimulation with collagen (5 µg/ml) or AA (0.5 mmol/l), using PPP as a referent. Measures of platelet aggregation were compared between the first and the fourth quartiles of preaspirin serotonin levels.
Functional cohort 2. PRP was isolated from blood samples drawn into 3.2% citrate-anticoagulated tubes from 12 healthy individuals who were not on antiplatelet medication. Blood samples were deidentified and thus no other clinical information was available. Platelet aggregation was measured in PRP by the same technician using a PAP8E aggregometer (Becton-Dickinson, Franklin Lakes, NJ) before and after ex vivo aspirin exposure (16.5 µmol/l for 30 min at 37 °C) following stimulation with collagen (2 µg/ml) ± serotonin (1 µM) using PPP as a referent. Collagen and serotonin were purchased from Chrono-Log and Sigma-Aldrich (St Louis, MO), respectively.
Statistical analyses. Missing ion intensity values (maximum 2.5%) were assumed to result from areas falling below the limits of detection and were imputed with the observed minimum for that metabolite divided by 2.46 For the four metabolites for which absolute concentrations were not obtained, we report metabolite response value divided by the mean metabolite response value for the metabolite, a scaling that does not affect our conclusions. Metabolite response values were log-transformed before statistical analysis and back-transformed for presentation. We assessed the significance of the effects of aspirin exposure, gender, and quartile on metabolite level using linear modeling and linear mixed modeling47 in GenStat 14th edition (VSN International, Hemel Hempstead, UK). Three models were fitted for each metabolite.
The first global linear mixed modeling included all data for each metabolite:
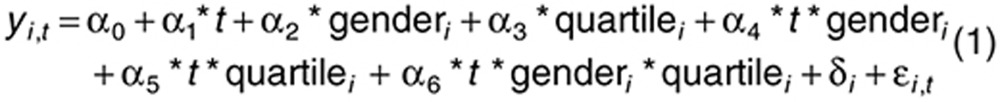 |
in which i = 1,2…81, yi,t = vector of metabolite concentrations for the ith individual at time t; t = time (pre- or postaspirin exposure), gender = man or woman, quartile = first or fourth quartile of aspirin response, fixed effects; α0…α6 = regression coefficients; δi = random effect associated with the ith individual; ɛi,t = error term.
The second and third linear models included data before and after treatment, respectively:
 |
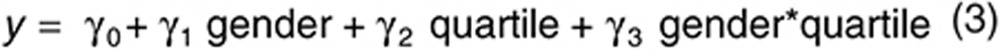 |
The effect of aspirin therapy on metabolite level was considered significant when P <0.05 for α1. The effect of quartile was considered significant before, after and during treatment when P <0.05 for β2, γ2, or α5, respectively. We computed a false discovery rate value for each P value using the mafdr function in Matlab version R2009a (MathWorks, Natick, MA), which uses a procedure introduced by Storey.48 q <0.05 was considered significant.
Additional analyses are described in the Supplementary Methods.
Informed consents. The investigations were approved by the Institutional Review Boards of the University of Maryland, Baltimore and Duke University, Durham. Informed consents were obtained before participation. The HAPI Heart Study was monitored by an external Data Safety and Monitoring Board.
Author Contributions
S.E.-S., J.P.L., A.G., L.M.Y.-A., A.L.B., R.B.H., A.R.S., T.H., and R.K.-D. designed the research; A.D., A.C.H., R.R., R.J.V., C.G.P., C.L.S., C.K., and T.L.O. performed the research; S.E.-S., A.G., A.D., and H.Z. analyzed the data; S.E.-S., J.P.L., A.G., L.M.Y.-A., A.L.B., R.B.H., T.H., and R.K.-D. wrote the manuscript.
Conflict of Interest
R.K.-D. is an inventor on patents in the metabolomics field. The other authors declare no conflict of interest.
Study Highlights

Acknowledgments
We would like to thank Margriet Hendriks and Fred van Eeuwijk for their helpful advice on linear mixed modeling and Keith Tanner for his help with sample preparation. The National Institutes of Health supported this study through (RC2GM092729) as part of the American Recovery and Reinvestment Act (ARRA). The work was implemented by the Pharmacometabolomics Research Network. The effort of S.E.-S. was supported by the research program of the Netherlands Metabolomics Centre (NMC), part of the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research; HAPI was supported by grants (U01-HL72515), the University of Maryland General Clinical Research Center (GCRC; M01-RR-16500), the Johns Hopkins University GCRC (M01-RR-000052), National Center for Research Resources, and the Clinical Nutrition Research Unit of Maryland (P30-DK072488). The effort of A.L.B. was supported by NIH grant K23-HL091120 and J.P.L. was supported by NIH grants T32-HL72751 and K23-GM102678.
Supplementary Material
References
- Amin A.R., Attur M.G., Pillinger M., Abramson S.B. The pleiotropic functions of aspirin: mechanisms of action. Cell. Mol. Life Sci. 1999;56:305–312. doi: 10.1007/s000180050432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovens M.M., Snoep J.D., Eikenboom J.C., van der Bom J.G., Mertens B.J., Huisman M.V. Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review. Am. Heart J. 2007;153:175–181. doi: 10.1016/j.ahj.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Gum P.A., Kottke-Marchant K., Welsh P.A., White J., Topol E.J. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J. Am. Coll. Cardiol. 2003;41:961–965. doi: 10.1016/s0735-1097(02)03014-0. [DOI] [PubMed] [Google Scholar]
- Patrignani P., Filabozzi P., Patrono C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J. Clin. Invest. 1982;69:1366–1372. doi: 10.1172/JCI110576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frelinger A.L., 3rd, et al. Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirin-treated patients presenting for cardiac catheterization. Circulation. 2009;120:2586–2596. doi: 10.1161/CIRCULATIONAHA.109.900589. [DOI] [PubMed] [Google Scholar]
- Eikelboom J.W., et al. Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management and Avoidance (CHARISMA) Investigators Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: determinants and effect on cardiovascular risk. Circulation. 2008;118:1705–1712. doi: 10.1161/CIRCULATIONAHA.108.768283. [DOI] [PubMed] [Google Scholar]
- Ohmori T., et al. Aspirin resistance detected with aggregometry cannot be explained by cyclooxygenase activity: involvement of other signaling pathway(s) in cardiovascular events of aspirin-treated patients. J. Thromb. Haemost. 2006;4:1271–1278. doi: 10.1111/j.1538-7836.2006.01958.x. [DOI] [PubMed] [Google Scholar]
- Hankey G.J., Eikelboom J.W. Aspirin resistance. Lancet. 2006;367:606–617. doi: 10.1016/S0140-6736(06)68040-9. [DOI] [PubMed] [Google Scholar]
- Clayton T.A., et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature. 2006;440:1073–1077. doi: 10.1038/nature04648. [DOI] [PubMed] [Google Scholar]
- Nicholson J.K., Wilson I.D., Lindon J.C. Pharmacometabonomics as an effector for personalized medicine. Pharmacogenomics. 2011;12:103–111. doi: 10.2217/pgs.10.157. [DOI] [PubMed] [Google Scholar]
- Kaddurah-Daouk R., et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One. 2011;6:e25482. doi: 10.1371/journal.pone.0025482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddurah-Daouk R., et al. Lipidomic analysis of variation in response to simvastatin in the Cholesterol and Pharmacogenetics Study. Metabolomics. 2010;6:191–201. doi: 10.1007/s11306-010-0207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trupp M., et al. Metabolomics reveals amino acids contribute to variation in response to simvastatin treatment. PLoS One. 2012;7:e38386. doi: 10.1371/journal.pone.0038386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff W.R., et al. Pharmacometabolomics Research Network Pharmacometabolomics reveals racial differences in response to atenolol treatment. PLoS One. 2013;8:e57639. doi: 10.1371/journal.pone.0057639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y., et al. Glycine and a glycine dehydrogenase (GLDC) SNP as citalopram/escitalopram response biomarkers in depression: pharmacometabolomics-informed pharmacogenomics. Clin. Pharmacol. Ther. 2011;89:97–104. doi: 10.1038/clpt.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddurah-Daouk R., et al. Pretreatment metabotype as a predictor of response to sertraline or placebo in depressed outpatients: a proof of concept. Transl. Psychiatry. 2011;1 doi: 10.1038/tp.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton T.A., Baker D., Lindon J.C., Everett J.R., Nicholson J.K. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatine M.S., et al. Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation. 2005;112:3868–3875. doi: 10.1161/CIRCULATIONAHA.105.569137. [DOI] [PubMed] [Google Scholar]
- Shah S.H., et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ. Cardiovasc. Genet. 2010;3:207–214. doi: 10.1161/CIRCGENETICS.109.852814. [DOI] [PubMed] [Google Scholar]
- van Velzen E.J., et al. Multilevel data analysis of a crossover designed human nutritional intervention study. J. Proteome Res. 2008;7:4483–4491. doi: 10.1021/pr800145j. [DOI] [PubMed] [Google Scholar]
- Ingebretsen O.C., Bakken A.M., Farstad M. Liquid chromatography of serotonin and adenine nucleotides in blood platelets, illustrated by evaluation of functional integrity of platelet preparations. Clin. Chem. 1985;31:695–698. [PubMed] [Google Scholar]
- Li N., Wallén N.H., Ladjevardi M., Hjemdahl P. Effects of serotonin on platelet activation in whole blood. Blood Coagul. Fibrinolysis. 1997;8:517–523. doi: 10.1097/00001721-199711000-00006. [DOI] [PubMed] [Google Scholar]
- Walther D.J., et al. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell. 2003;115:851–862. doi: 10.1016/s0092-8674(03)01014-6. [DOI] [PubMed] [Google Scholar]
- Yerges-Armstrong L.M., et al. Purine pathway implicated in mechanism of resistance to aspirin therapy: pharmacometabolomics-informed pharmacogenomics. Clin. Pharmacol. Ther. 2013;94:525–532. doi: 10.1038/clpt.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needs C.J., Brooks P.M. Clinical pharmacokinetics of the salicylates. Clin. Pharmacokinet. 1985;10:164–177. doi: 10.2165/00003088-198510020-00004. [DOI] [PubMed] [Google Scholar]
- Walther D.J., Bader M. A unique central tryptophan hydroxylase isoform. Biochem. Pharmacol. 2003;66:1673–1680. doi: 10.1016/s0006-2952(03)00556-2. [DOI] [PubMed] [Google Scholar]
- Furness J.B., Costa M. Neurons with 5-hydroxytryptamine-like immunoreactivity in the enteric nervous system: their projections in the guinea-pig small intestine. Neuroscience. 1982;7:341–349. doi: 10.1016/0306-4522(82)90271-8. [DOI] [PubMed] [Google Scholar]
- Killam A.L., Cohen M.L. Characterization of rat platelet serotonin receptors with tryptamine agonists and the antagonists: ketanserin and SCH 23390. Thromb. Res. 1991;64:331–340. doi: 10.1016/0049-3848(91)90004-g. [DOI] [PubMed] [Google Scholar]
- Ziu E., et al. Down-regulation of the serotonin transporter in hyperreactive platelets counteracts the pro-thrombotic effect of serotonin. J. Mol. Cell. Cardiol. 2012;52:1112–1121. doi: 10.1016/j.yjmcc.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich N., Aigner M., Eichler H.G., Entlicher J., Drucker C., Jilma B. Paroxetine decreases platelet serotonin storage and platelet function in human beings. Clin. Pharmacol. Ther. 2000;68:435–442. doi: 10.1067/mcp.2000.110456. [DOI] [PubMed] [Google Scholar]
- Bismuth-Evenzal Y., Gonopolsky Y., Gurwitz D., Iancu I., Weizman A., Rehavi M. Decreased serotonin content and reduced agonist-induced aggregation in platelets of patients chronically medicated with SSRI drugs. J. Affect. Disord. 2012;136:99–103. doi: 10.1016/j.jad.2011.08.013. [DOI] [PubMed] [Google Scholar]
- Sauer W.H., Berlin J.A., Kimmel S.E. Selective serotonin reuptake inhibitors and myocardial infarction. Circulation. 2001;104:1894–1898. doi: 10.1161/hc4101.097519. [DOI] [PubMed] [Google Scholar]
- Schlienger R.G., Meier C.R. Effect of selective serotonin reuptake inhibitors on platelet activation: can they prevent acute myocardial infarction. Am. J. Cardiovasc. Drugs. 2003;3:149–162. doi: 10.2165/00129784-200303030-00001. [DOI] [PubMed] [Google Scholar]
- Labos C., Dasgupta K., Nedjar H., Turecki G., Rahme E. Risk of bleeding associated with combined use of selective serotonin reuptake inhibitors and antiplatelet therapy following acute myocardial infarction. CMAJ. 2011;183:1835–1843. doi: 10.1503/cmaj.100912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikenes K., Farstad M., Nordrehaug J.E. Serotonin is associated with coronary artery disease and cardiac events. Circulation. 1999;100:483–489. doi: 10.1161/01.cir.100.5.483. [DOI] [PubMed] [Google Scholar]
- Ueno M., Kodali M., Tello-Montoliu A., Angiolillo D.J. Role of platelets and antiplatelet therapy in cardiovascular disease. J. Atheroscler. Thromb. 2011;18:431–442. doi: 10.5551/jat.7633. [DOI] [PubMed] [Google Scholar]
- Macchi L., et al. Resistance to aspirin in vitro is associated with increased platelet sensitivity to adenosine diphosphate. Thromb. Res. 2002;107:45–49. doi: 10.1016/s0049-3848(02)00210-4. [DOI] [PubMed] [Google Scholar]
- Kawasaki T., Ozeki Y., Igawa T., Kambayashi J. Increased platelet sensitivity to collagen in individuals resistant to low-dose aspirin. Stroke. 2000;31:591–595. doi: 10.1161/01.str.31.3.591. [DOI] [PubMed] [Google Scholar]
- Duerschmied D., et al. Serotonin antagonism improves platelet inhibition in clopidogrel low-responders after coronary stent placement: an in vitro pilot study. PLoS One. 2012;7:e32656. doi: 10.1371/journal.pone.0032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison P., Frelinger A.L., 3rd, Furman M.I., Michelson A.D. Measuring antiplatelet drug effects in the laboratory. Thromb. Res. 2007;120:323–336. doi: 10.1016/j.thromres.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Snoep J.D., Hovens M.M., Eikenboom J.C., van der Bom J.G., Huisman M.V. Association of laboratory-defined aspirin resistance with a higher risk of recurrent cardiovascular events: a systematic review and meta-analysis. Arch. Intern. Med. 2007;167:1593–1599. doi: 10.1001/archinte.167.15.1593. [DOI] [PubMed] [Google Scholar]
- Reny J.L., De Moerloose P., Dauzat M., Fontana P. Use of the PFA-100 closure time to predict cardiovascular events in aspirin-treated cardiovascular patients: a systematic review and meta-analysis. J. Thromb. Haemost. 2008;6:444–450. doi: 10.1111/j.1538-7836.2008.02897.x. [DOI] [PubMed] [Google Scholar]
- Mitchell B.D., et al. The genetic response to short-term interventions affecting cardiovascular function: rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. Am. Heart J. 2008;155:823–828. doi: 10.1016/j.ahj.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H., et al. Aspirin Resistance in healthy drug-naive men versus women (from the Heredity and Phenotype Intervention Heart Study) Am. J. Cardiol. 2009;104:606–612. doi: 10.1016/j.amjcard.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J., Psychogios N., Young N., Wishart D.S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37:W652–W660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro J., Bates D. Mixed-Effects Models in S and S-PLUS. Springer-Verlag, New York,; 2000. [Google Scholar]
- Storey J.D. A direct approach to false discovery rates. J Roy Stat Soc. B. 2002;64:479–498. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.