

Abstract
Neutropenia is a lethal dose-limiting toxicity of docetaxel. Our previous report indicated that the prevalence of severe docetaxel-induced neutropenia is significantly associated with genetic polymorphisms in solute carrier organic anion transporter 1B3 (SLCO1B3) (encoding organic anion–transporting polypeptide 1B3 (OATP1B3)) and ATP-binding cassette subfamily C2 (ABCC2) (encoding multidrug-resistant–associated protein 2 (MRP2)). Therefore, we investigated their significance in docetaxel-induced neutropenia. In vitro experiments suggested their possible involvement in the hepatic uptake of docetaxel and its efflux from bone marrow cells. To further characterize a quantitative impact of OATP1B3 and MRP2 on neutropenia, we used an in silico simulation of the neutrophil count in docetaxel-treated subjects with functional changes in OATP1B3 and MRP2 in a pharmacokinetic/pharmacodynamic model. The clinically reported odds ratios for docetaxel-induced neutropenia risk were explained by the decreased function of OATP1B3 and MRP2 to 41 and 32%, respectively. These results suggest that reduced activities of OATP1B3 and MRP2 associated with systemic exposure and local accumulation in bone marrow cells, respectively, account for the docetaxel-induced neutropenia observed clinically.
Introduction
Docetaxel exerts an antitumor activity by stabilizing and inhibiting the depolymerization of tubulin,1 and is used as a first-line therapy in the treatment of several kinds of cancers. Neutropenia is a dose-limiting toxicity of docetaxel, and it sometimes restricts the clinical use. However, the determining factors for docetaxel-induced hematopoietic toxicity are not understood well.
When infused intravenously in humans, docetaxel is metabolized mainly by cytochrome P450 (CYP) 3A in the liver, and only a small fraction of the parent docetaxel is excreted into the bile and urine.2 Because the metabolites of docetaxel have less biological activity than the parent form,3 systemic exposure to docetaxel is thought to be an important causal factor of docetaxel-induced toxicity. Previous reports suggested that the area under the plasma concentration–time curve of docetaxel correlated with a decrease in neutrophil count.4,5
Kiyotani et al.6 reported that the risk of severe neutropenia induced by docetaxel chemotherapy was significantly associated with genetic polymorphisms of solute carrier organic anion transporter 1B3 (SLCO1B3), encoding organic anion–transporting polypeptide 1B3 (OATP1B3), and of ATP-binding cassette subfamily C2 (ABCC2), encoding multidrug-resistant–associated protein 2 (MRP2). It was reported earlier that docetaxel was a substrate of OATP1B3 and MRP2.7,8,9,10 OATP1B3 is expressed almost exclusively on the basolateral side of human hepatocytes and mediates the hepatic uptake of a wide variety of drugs such as telmisartan.11,12,13 Although docetaxel is reported to be a substrate of OATP1B3, the relative contribution of OATP1B3 to hepatic uptake of docetaxel remains to be clarified. MRP2 is expressed on the bile canalicular membrane and is responsible for the biliary excretion of various organic anions including glucuronide and glutathione conjugates.14,15,16 However, because docetaxel is eliminated predominantly by cytochrome P450 3A–mediated metabolism, it is unlikely that hepatic MRP2 controls its systemic exposure. Instead, MRP2 in hematopoietic cells or their precursor cells is likely to suppress the entry of docetaxel into cells and protects them against its cytotoxicity.
The pharmacokinetic/pharmacodynamic (PK/PD) model is a powerful tool for predicting both drug efficacy and toxicity. A population PK/PD model was built to predict the time profile of blood neutrophil count after treatment with anticancer drugs including docetaxel, and this model could explain well the quantitative risk of neutropenia.17 Because the mutations in SLCO1B3 and ABCC2 reported previously are not located in their coding regions, it is difficult to estimate the functional alterations in these transporters from in vitro experimental data. Alternatively, we decided to use the reported PK/PD model describing the drug-induced neutropenia for that purpose.
In this study, we tried to investigate the possible roles of OATP1B3 and MRP2 in the systemic pharmacokinetics and local exposure of docetaxel in bone marrow cells, which finally links to the hematopoietic toxicity. Contribution of OATP1B3 to the overall uptake of docetaxel in human hepatocytes and the protective role of MRP2 in the cytotoxicity induced by docetaxel were investigated by in vitro experiments. Then, the impact of functional changes in OATP1B3 and MRP2 on docetaxel-induced toxicity was also estimated quantitatively using Monte Carlo simulation approach.
Results
Uptake of docetaxel in hepatic uptake transporter expression systems
Among hepatic uptake transporters in humans (OATP1B1, OATP1B3, OATP2B1, OAT2, Na+-taurocholate cotransporting polypeptide (NTCP), and organic cation transporter (OCT)1), docetaxel is taken up significantly only into OATP1B3-expressing HEK293 cells compared with control cells (Figure 1a). Transport activity of each expression system was confirmed as the uptake of typical substrates, estrone-3-sulfate (E1S) for OATP1B1 and OATP2B1 (49 and 14 µl/0.5 min/mg protein), cholecystokinin octapeptide (CCK-8) for OATP1B3 (35 µl/5 min/mg protein), xanthine for OAT2 (18 µl/15 min/mg protein), taurocholate for NTCP (14 µl/2 min/mg protein), and tetraethylammonium for OCT1 (90 µl/10 min/mg protein). The saturation kinetics of OATP1B3-mediated uptake of docetaxel was evaluated by its transport for 5 min because the time-dependent linear uptake was maintained (data not shown). The protein-unbound fraction of docetaxel (0.001–100 µmol/l) in the transport buffer with 3% human serum albumin was measured to estimate the transport clearance with regard to the unbound concentration of docetaxel. The protein-unbound fraction was constant within the range of 0.001–1 µmol/l, but increased gradually in the concentration range of 3–100 µmol/l, suggesting a saturation of protein binding (Supplementary Figure S1). The uptake clearance was calculated with regard to the unbound concentration and is shown as Eadie–Hofstee plots (Figure 1b). The concentration-dependent uptake could be explained by one saturable component with a Km value of 0.325 ± 0.064 µmol/l (mean ± computer-calculated SD).
Figure 1.

In vitro uptake of docetaxel into HEK293 cells expressing solute carrier transporters expressed in human hepatocytes and into human hepatocytes. (a) The uptake of docetaxel (1 µmol/l) into OATP-, OAT-, OCT1-, and NTCP–expressing HEK293 cells and vector-transfected control cells (vector) was measured at 5 min. (b) Saturation of the docetaxel uptake in OATP1B3-expressing HEK293 cells was also investigated. The solid line is a fitted curve calculated by nonlinear regression analysis based on Eq. 1, as described in the Methods. (c) Concentration (0.1 µmol/l and 2 mmol/l)-dependent uptake of estrone-3-sulfate (E1S) in OATP1B1-expressing HEK293 cells, (d) inhibitory effects of 2 mmol/l estradiol-17β-glucuronide (E217βG) and E1S on the uptake of cholecystokinin octapeptide (CCK-8) in OATP1B3-expressing HEK293 cells, and (e) docetaxel uptake into cryopreserved human hepatocytes in the presence of E1S and E217βG were investigated. These uptake assays were carried out in the presence of 3% human serum albumin. Each bar represents the mean ± SE (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Inhibitory effects of E1S and estradiol-17β-glucuronide on docetaxel uptake
The in vitro uptake study with OATP1B1- and OATP1B3-expressing HEK293 cells confirmed that 2 mmol/l E1S with 3% human serum albumin specifically inhibited OATP1B1-mediated transport but not OATP1B3-mediated transport (Figure 1c,d). By contrast, 2 mmol/l estradiol-17β-glucuronide (E217βG) inhibited both OATP1B1- and OATP1B3-mediated transport. The uptake of docetaxel in human hepatocytes was inhibited by 100 µmol/l unlabeled docetaxel and 2 mmol/l E217βG, but was not inhibited by 2 mmol/l E1S (Figure 1e).
Cytotoxicity and intracellular accumulation of docetaxel in MRP2-expressing MDCKII cells
The LC50 values in MRP2-expressing and vector-transfected MDCKII cells were calculated to be 12.0 ± 5.4 and 3.27 ± 0.91 nmol/l, respectively (mean ± computer-calculated SD; Figure 2a). The accumulation of docetaxel for 24 h in these cell lines was also measured. The distribution volume in MRP2-expressing cells (271 ± 9 µl/mg protein) was significantly smaller than that in vector-transfected cells (665 ± 20 µl/mg protein) (Figure 2b).
Figure 2.

Effect of exogenous expression of MRP2 on the cytotoxicity (a) and intracellular accumulation (b) of docetaxel. (a) Vector-transfected MDCKII cells (closed circles) or MRP2-expressing MDCKII cells (open circles) were incubated for 96 h with various concentrations (0.01–100 nmol/l) of docetaxel. Cell survival was quantified by the MTT assay as described in the Methods. (b) Cell lines were incubated for 24 h with 1 nmol/l of docetaxel, and the volume uptake of docetaxel into these cells was determined. Each point and bar represents the mean ± SE (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Colony-forming assay
A colony-forming assay was performed to investigate the inhibitory effect of docetaxel on the proliferation and maturation of bone marrow cells induced by granulocyte colony–stimulating factor. Docetaxel decreased the number of colonies in a concentration-dependent manner. The inhibitory effect of docetaxel on colony formation was significantly more potent in bone marrow cells derived from the MRP2-deficient rats (Eisai hyperbilirubinemic rats), compared with cells from normal Sprague Dawley rats (Figure 3a). The inhibition potency of docetaxel was also significantly increased in the presence of 50 µmol/l of the MRP inhibitor, MK571 (Figure 3b).
Figure 3.

Inhibitory effect of docetaxel on granulocyte colony–stimulating factor (G-CSF)-induced colony formation. (a) Bone marrow cells obtained from Sprague Dawley rats (open circles) and Eisai hyperbilirubinemic rats (closed circles) were incubated for 6 days with various concentrations (0–5 ng/ml) of docetaxel. (b) Bone marrow cells derived from Sprague Dawley rats were also incubated for 6 days with various concentrations (0–5 ng/ml) of docetaxel in the presence (closed circles) or absence (open circles) of 50 µmol/l MK571. The number of colonies induced by G-CSF was counted. Each point represents the mean ± SE (n = 9).*P < 0.05, **P < 0.01, ***P < 0.001.
Estimation of EC50 values for subjects with no targeted mutations in SLCO1B3 and ABCC2 for an in silico simulation using the PK/PD model
We initially performed an in silico simulation by using a reported PK/PD model describing the time course of docetaxel pharmacokinetics and neutrophil count (Figure 5; Supplementary Material online).17,18 However, in subjects with wild-type (wt) SLCO1B3 and ABCC2 alleles, the ratio of the number of patients with grade 3–4 neutropenia to those without neutropenia calculated from an in silico simulation with reported PK/PD parameters (1.71) largely differed from the clinically observed data (0.0968) (data not shown). We considered that this difference might reflect the difference in the EC50 value in this model because the very frequent ABCC2 genotype, which can affect the transport function of MRP2 and subsequently the EC50 value, was not taken into consideration in the previous parameter estimation.17 Therefore, we re-evaluated the EC50 value that best fitted our clinical data. Setting the EC50 values to 10, 20, 22, 24, 26, 28, or 30 µmol/l gave the ratios of the number of patients with grade 3–4 neutropenia to those without neutropenia of 1.09, 0.178, 0.146, 0.146, 0.106, 0.0880, and 0.0864, respectively (Figure 4a). This estimation gave an optimum EC50 value that would best account for our clinical data of about 28 µmol/l.
Figure 4.

Estimation of EC50 values in the control group and the functional changes to MRP2, which best explained the clinical observations reported previously.6 (a) Ratio of the number of subjects with severe neutropenia (grade 3 or 4) to those without any hematopoietic toxicity (non-ADR) was calculated with EC50 values of 10, 20, 22, 24, 26, 28, or 30 µmol/l, respectively. The broken line shows the clinically reported value (0.0968). (b) The impact of MRP2 function on the OR for the expression of severe (grade 3 or 4) neutropenia was simulated. The upper, middle, and lower broken lines represent the ORs for OATP1B3 (wt/wt)/MRP2 (mut/mut) (11.8), OATP1B3 (wt/mut)/MRP2 (wt/mut) (9.04), and OATP1B3 (wt/wt)/MRP2 (wt/mut) (3.72), respectively, as observed clinically. ADR, adverse drug reaction; OR, odds ratio.
Simulated odds ratios in subjects with mutations in SLCO1B3 and ABCC2
In the heterozygotes of SLCO1B3 rs11045585 (A>G), the intrinsic hepatic clearance was expected to decrease to 70.5% because this was estimated from the difference in the intrinsic hepatic clearance of telmisartan between homozygotes of wt SLCO1B3 alleles (wt/wt) and heterozygotes of the mutated (mut) SLCO1B3 allele (wt/mut) in our previous pharmacogenetic study.19 On the basis of this information, the hepatic clearances of docetaxel were estimated to be 36.7, 29.5, and 20.0 l/h in subjects with wt/wt, wt/mut, and homozygotes of mutated alleles (mut/mut) of SLCO1B3, respectively, assuming that the change in the intrinsic clearance of telmisartan was the same as that of docetaxel.
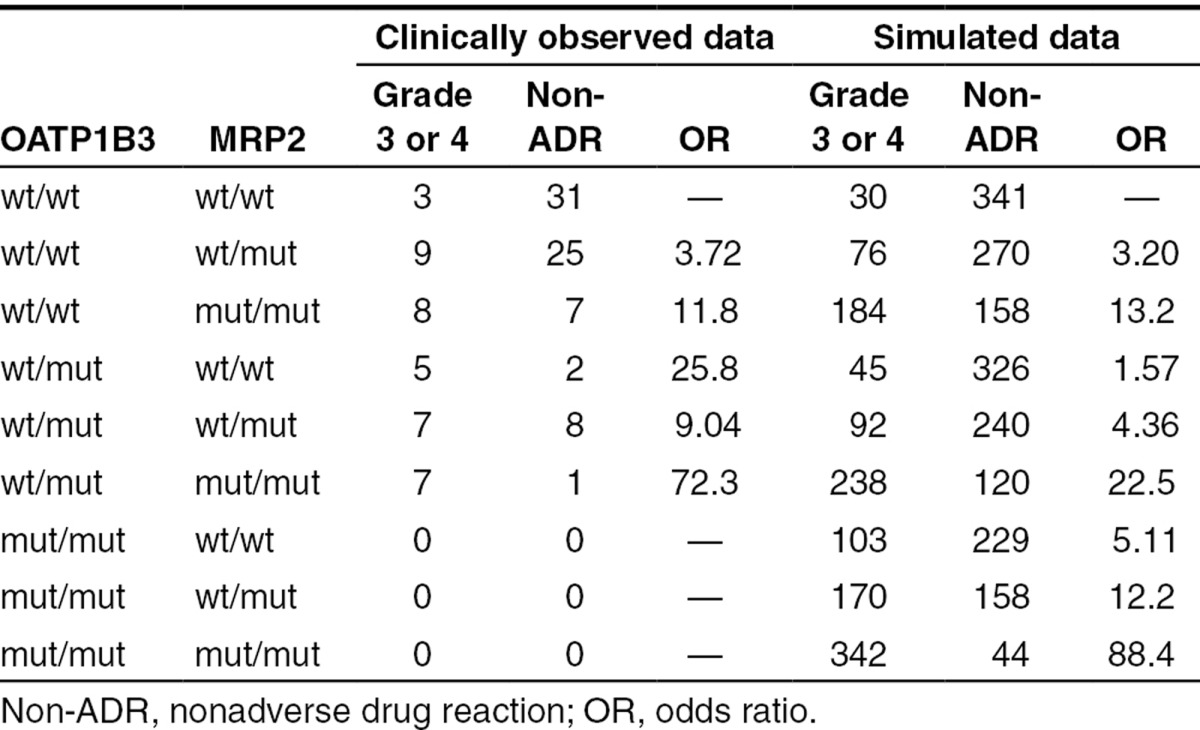
Regarding the functional change of MRP2 caused by the ABCC2 rs12762549 (G>C) mutation, we estimated the odds ratio (OR) in subjects with mutated alleles of ABCC2 by changing the EC50 value when the in vivo transport activity of MRP2 from mutated ABCC2 was assumed to decrease to 10–90% (Figure 4b). By minimizing the sum of the square of differences normalized by observed ORs between clinically observed ORs in subjects with SLCO1B3 and ABCC2 diplotypes of wt/wt and wt/mut (ORwt/wt+wt/mut; 3.72), wt/wt and mut/mut (ORwt/wt+mut/mut; 11.8), and wt/mut and wt/mut (ORwt/mut+wt/mut; 9.04) and calculated ORs with changing EC50 value, we conclude that the decrease in the MRP2 function to 32% would best explain our clinical data (Table 1).
Table 1. Comparison of the number of patients who received docetaxel and the ORs for docetaxel-induced neutropenia between clinically observed data and simulated data (each genotype: n = 500) when the mean EC50 value was set at 28 µmol/l.
Discussion
Although docetaxel is used in the treatment of several kinds of cancers, neutropenia sometimes impedes its continuous clinical use. The severity of neutropenia is thought to be determined by the exposure to docetaxel in the bone marrow, which is dominated by both its systemic concentration and local accumulation in hematopoietic cells or their precursors. In the present study, we tested our hypothesis that hepatic uptake of docetaxel is controlled mainly by OATP1B3 and that MRP2 in hematopoietic cells acts as a barrier to the entry of docetaxel into hematopoietic cells.
Among the hepatic uptake transporters, only OATP1B3 recognizes docetaxel as a substrate, as reported previously7,8; however, its relative contribution to the hepatic uptake of docetaxel is unknown. The uptake of docetaxel into human hepatocytes was inhibited by E217βG (an inhibitor of both OATP1B1 and OATP1B3), but was not inhibited by E1S (an OATP1B1-selective inhibitor).11 This suggests that the hepatic uptake of docetaxel is mediated mainly by OATP1B3, as is the case for telmisartan. These results support our hypothesis that the increased risk of docetaxel-induced neutropenia by the SLCO1B3 rs11045585 mutation is caused by the decreased expression of OATP1B3 because this mutation is not located in the exon of SLCO1B3.
Because the fraction of biliary excretion of parent docetaxel is thought to be very small, we hypothesized that MRP2 might play an important role in bone marrow cells. Previous reports showed that MRP2 expression was not detected in CD34−KSL cells (hematopoietic stem cells), but was detected in CD34+KSL cells (immature hematopoietic cells).20 In human MRP2-expressing cells, the intracellular accumulation of docetaxel decreased significantly, and sensitivity to the cytotoxicity defined based on its medium concentration subsequently decreased. Moreover, bone marrow cells from Mrp2-deficient Eisai hyperbilirubinemic rats were more sensitive to docetaxel-induced cytotoxicity compared with cells from normal Sprague Dawley rats. The presence of MK571, an MRP inhibitor, also sensitized bone marrow cells to docetaxel. These results support our hypothesis that MRP2 in bone marrow cells works as a barrier to docetaxel-induced hematopoietic cytotoxicity.
The single-nucleotide polymorphisms in SLCO1B3 and ABCC2 linked to the increased risk of docetaxel-induced neutropenia are located in the noncoding region of each gene (intron 11 and the 3′-untranslated region, respectively).6 Thus, it is difficult to estimate the quantitative functional changes of these transporters from an in vitro study, although we cannot neglect the possibility that unknown causal mutations are linked to these single-nucleotide polymorphisms. Telmisartan is expected to be an in vivo probe substrate for OATP1B3.11,21 We demonstrated recently that the intrinsic clearance of telmisartan decreased to 70.5% in heterozygotes with the SLCO1B3 rs11045585 mutation compared with homozygotes of wt alleles.19 That result fits our hypothesis, explaining why the risk of docetaxel-induced hematopoietic toxicity is increased by the SLCO1B3 mutation. On the other hand, because MRP2 is not involved in the biliary excretion of docetaxel, decreased expression of MRP2 might occur in certain types of bone marrow cells.
A mathematical PK/PD model for predicting neutropenia induced by anticancer drugs including docetaxel has been proposed.17 In this model, a multicompartment PK model is connected to the PD model that is used to describe the maturation, feedback regulation, and turnover of hematopoietic cells and their precursor cells. The elimination rate constant (K10) of docetaxel is affected directly by a functional change in OATP1B3. By contrast, the EC50 value was defined with regard to the intracellular concentration of docetaxel in hematopoietic cells in this model, and so the decreased function of MRP2 corresponds to the decrease in EC50 values.
To understand the distribution of the clinical outcomes in a certain population, a set of parameters was generated randomly in the PK/PD model for each subject (a so-called “virtual person”) while retaining the distribution of parameters in one population using Monte Carlo simulation (Figure 5c). Kato et al. used this approach to demonstrate that the interindividual variability of cytochrome P450 3A4 function could be estimated from the variation in the clinical pharmacokinetics of cytochrome P450 3A4 substrates.22 In our simulation, assuming that the variability of each parameter was unchanged when the parameter (K10 or EC50) reflecting the transporter function was changed by genetic polymorphisms of SLCO1B1 and ABCC2, the model could predict the distribution of neutrophil counts in subjects with different genotypes.
Figure 5.

Schematic diagram of the PK/PD model describing docetaxel-induced neutropenia. (a) A three-compartment model was used to describe the pharmacokinetics of docetaxel. (b) The model of Friberg et al. was used to estimate the neutrophil count.17 (c) Neutrophil concentration profile after docetaxel administration in five individuals selected from subjects with SLCO1B3 and ABCC2 diplotypes of wt/wt and wt/wt. Broken lines show borders between non-ADR, neutropenia grade 1/2, and neutropenia grade 3/4. Non-ADR, nonadverse drug reaction; PD, pharmacodynamic; PK, pharmacokinetic.
Initially, using the default parameters reported,17 the calculated ratio of the number of patients with severe (grade 3–4) neutropenia to that without any hematopoietic toxicity in the control group was 1.71, which differed considerably from the reported value of 0.0968 (data not shown). One possible reason is the high frequency of the ABCC2 rs12762549 genotype in the Japanese population (0.636). To fit the real situation, we first optimized the EC50 value to adjust it to the reported value. When the EC50 value in subjects with wt SLCO1B3 and ABCC2 alleles was set at 28 µmol/l, the calculated ratio was 0.0880, which was close to the reported value.
The decrease of oral clearance in heterozygotes of SLCO1B3 rs11045585 could explain the clinically observed OR for the risk of docetaxel-induced neutropenia. A metabolomic study revealed that the plasma concentrations of some MRP2 substrates were increased in carriers of ABCC2 rs12762549,23 suggesting a decreased function of MRP2. However, it is difficult to determine the extent of its functional change. Therefore, we decided to estimate the function in mutated MRP2 by comparing the reported ORs for neutropenia with the calculated ones in subjects with SLCO1B3 and ABCC2 genotypes of wt/mut and wt/mut, wt/wt and wt/mut, and wt/wt and mut/mut, with numbers of more than 15 in each group. When the MRP2 function was assumed to decrease to 32% by this mutation, the calculated OR was similar to the reported value. Our sensitivity analysis showed that even a small change in MRP2 function has a large effect on the OR. Previous reports suggested that many factors, such as environmental and pathophysiological conditions, can change the expression of MRP224 and that the large interindividual differences in the risk of neutropenia partly reflect the differences in the accumulation of docetaxel into certain types of bone marrow cells, which are partly determined by MRP2.
In conclusion, we demonstrated that the hepatic uptake of docetaxel is mediated mainly by OATP1B3 and that MRP2 plays an important role as a barrier against the entry of docetaxel into hematopoietic cells. The in silico PK/PD analyses showed that the increased risk of docetaxel-induced neutropenia could be explained by functional decreases in OATP1B3 and MRP2. Our model could explain the clinically observed ORs for docetaxel-induced neutropenia when the function of OATP1B3 and MRP2 decreased to 41 and 32%, respectively, because of their single-nucleotide polymorphisms.
Methods
Materials
[3H]-docetaxel (60 Ci/mmol) and [3H]-xanthine (19.4 Ci/mmol) were purchased from American Radiolabeled Chemicals (St Louis, MO). [3H]-E217βG (45 Ci/mmol), [3H]-E1S (46 Ci/mmol), [3H]-taurocholic acid (5.0 Ci/mmol), and [14C]-tetraethylammonium (3.2 mCi/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). [3H]-CCK-8 (77 Ci/mmol) was purchased from Amersham Pharmacia Biotech (Buckinghamshire, UK). Unlabeled E217βG, E1S, and CCK-8 were purchased from Sigma-Aldrich (St Louis, MO), and docetaxel was purchased from LC Laboratories (Woburn, MA). All other chemicals were of analytical grade and are commercially available.
Animals
Male Sprague Dawley rats and Eisai hyperbilirubinemic rats (7–8 weeks old) were used in this study. All animals were maintained under standard conditions with a reversed dark–light cycle and were treated humanely. Food and water were available ad libitum. The studies were carried out in accordance with the guidelines provided by the Institutional Animal Care Committee (Graduate School of Pharmaceutical Sciences, the University of Tokyo, Tokyo, Japan).
Transport studies using transporter expression systems
Human OATP-, OAT-, or NTCP–expressing HEK293 cells have been constructed previously.25,26,27 Human OCT1-transfected HEK293 cells were constructed as follows. Human OCT1 (GenBank Accession No. NM_153187) transcripts were amplified and subcloned into pcDNA3.1 vector (Invitrogen, Carlsbad, CA). The expression vector carrying human OCT1 cDNA was then transfected into HEK293 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol and stably transfected cells were selected by adding G418 (0.8 mg/ml) (Invitrogen) to the culture medium. The transport study was carried out as described previously.27
Transport study using cryopreserved human hepatocytes
Cryopreserved human hepatocytes (Lot. 109) were purchased from BD Gentest (Franklin Lakes, NJ). Immediately before the study, the hepatocytes in 1-ml suspension were thawed at 37°C and then quickly suspended in thawing medium (Celsis In Vitro Technologies, Baltimore, MD). The cells were centrifuged, the medium was replaced with plating medium, and the hepatocytes were transferred to collagen-precoated 24-well plates (Becton Dickinson Labware, Bedford, MA) at a cell density of 1.8 × 105 viable cells/well in 0.5 ml of plating medium and then incubated for 24 h. The uptake study was performed as described previously.27
Kinetic analysis of OATP1B3-mediated transport
Docetaxel uptake was expressed as volume (µl/mg protein) and given as the amount of radioactivity associated with the cells (dpm/mg protein) divided by its initial concentration in the incubation medium (dpm/µl). Specific uptake was calculated by subtracting uptake in the vector-transfected cells from that in the transporter cDNA-transfected cells. The kinetic parameters were calculated using the following equation:
 |
where v, S, Km, Vmax, and Pdif represent the uptake velocity of the substrate (pmol/min/mg protein), substrate concentration in the medium (µmol/l), Michaelis constant (µmol/l), maximum uptake rate (pmol/min/mg protein), and nonsaturable uptake clearance (µl/min/mg protein), respectively. Fitting was performed using a nonlinear least-squares method using the MULTI program,28 and the Damping Gauss–Newton algorithm was used for curve fitting. These input data were weighted as the reciprocals of the observed values.
To measure the saturable hepatic uptake clearance in human hepatocytes, we first determined the hepatic uptake clearance (CL(2–0.5 min)) (µl/min/mg protein) by calculating the slope of the uptake volume (Vd) (µl/mg protein) between 0.5 and 2 min (Eq. 2). The saturable component of the hepatic uptake clearance (CLhep) was determined by subtracting CL(2−0.5 min) in the presence of 100 µmol/l substrate (excess) from that in the presence of 10 nmol/l (E1S and CCK-8) or 10.9 nmol/l (docetaxel) substrate (tracer) (Eq. 3):
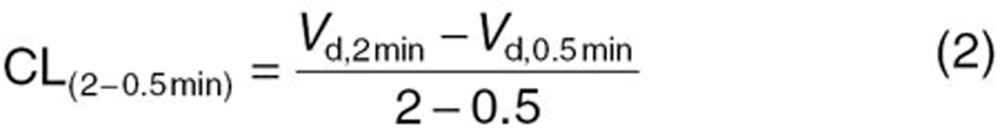 |
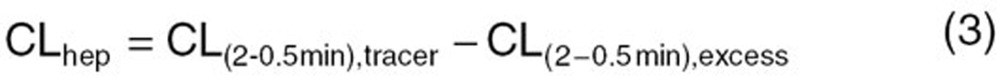 |
MTT viability assays
Human MRP2-expressing and vector-transfected MDCKII cells were seeded in 96-well plates at 1 × 103 cells/well. After 12 h incubation, the medium was replaced with Dulbecco's modified Eagle's medium containing docetaxel at concentrations of 0.01–100 nmol/l. After 96 h, the medium was removed and the cells were washed with phosphate-buffered saline twice. Fresh Dulbecco's modified Eagle's medium (without phenol red) containing 100 µl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to each well. After 3 h of incubation, the medium was replaced, 100 µl of dimethyl sulfoxide was added to each well to dissolve the formazan, and the absorbance at 560 nm was measured. The LC50 value was calculated using the following equation:
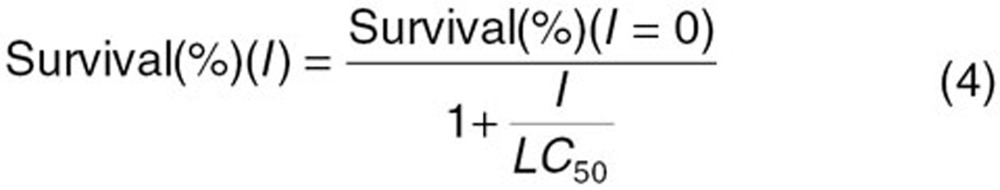 |
where I represents the concentration of docetaxel. Fitting was performed with a nonlinear least-squares method using the MULTI program,28 and the Damping Gauss–Newton algorithm was used for curve fitting.
Accumulation study
MRP2-expressing and vector-transfected MDCKII cells were seeded in 12-well plates at 1.25 × 105 cells/well. After 48-h incubation, the medium was replaced with fresh medium containing [3H]-docetaxel and the cells were incubated for 24 h. The cells were then washed three times with 1 ml of ice-cold phosphate-buffered saline, solubilized with 500 µl of 0.2 N NaOH and stored overnight at 4 °C. After 250 µl of 0.4 N HCl was added to solubilized cells, aliquots (600 µl) were transferred to scintillation vials. The radioactivity associated with the cells and the incubation buffer was measured with a liquid scintillation counter after the addition of 3 ml of scintillation fluid (Clear-sol I; Nacalai Tesque, Kyoto, Japan) to the scintillation vials.
Colony-forming assay
The inhibitory effect of docetaxel on the colony formation of bone marrow cells induced by granulocyte colony–stimulating factor was performed as described.29 Bone marrow cells were collected from the femurs of rats by flushing dissected and cleaned femurs with α-medium (GIBCO BRL, Gaithersburg, MD) containing 10% fetal bovine serum. The cells were collected and then incubated for 2 h. The nonadherent cells were passed through a 40-µm cell strainer (Becton Dickinson Labware, Franklin Lakes, NJ) to prepare single cell suspensions. The methylcellulose colony-forming assay was performed in 35-mm dishes. The culture medium consisted of α-medium, 1.2% 1500-cps methylcellulose, 30% fetal bovine serum, 10% bovine serum albumin, 96 µmol/l 2-mercaptoethanol, 0.1 mmol/l 1-thioglycerol, and 0.1% Neutrogin (Chugai Pharmaceuticals, Tokyo, Japan). Cells were plated in 35-mm dishes at a concentration of 1 × 106 cells and incubated for 6 days. Aggregates comprising more than 50 cells were counted as colonies and quantified by observation with an inverted microscope (Diaphot; Nikon, Tokyo, Japan).
In silico simulation of PK/PD model for predicting neutrophil count after the treatment of docetaxel.
The reported PK/PD model shown in Figure 5 was used to predict the neutrophil count after docetaxel administration.17,18 The differential equations used in the PK/PD model were as follows:
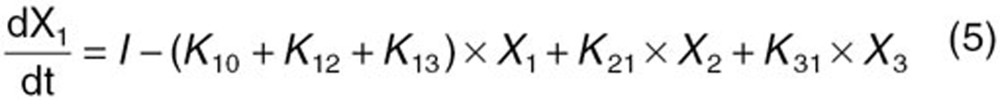 |
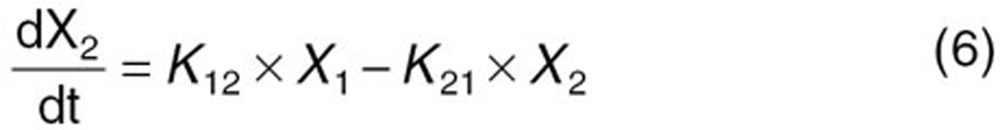 |
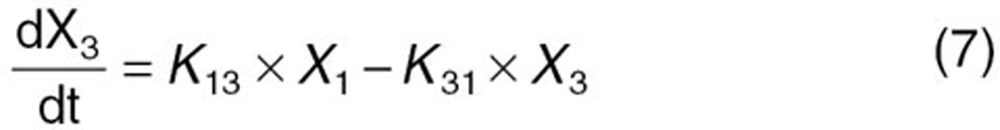 |
 |
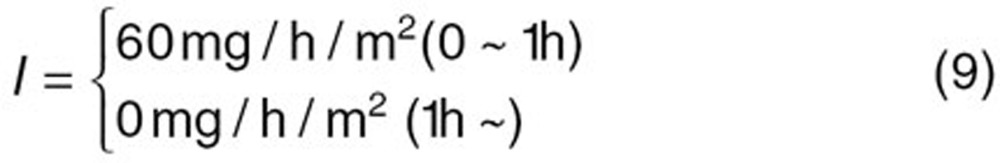 |
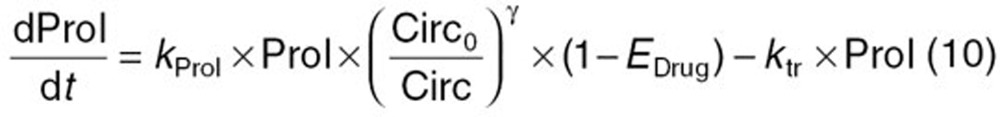 |
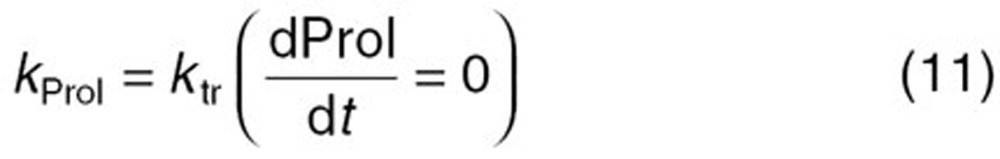 |
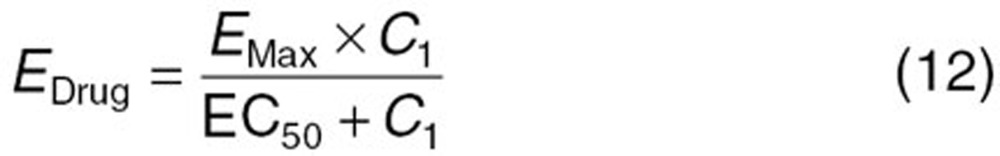 |
 |
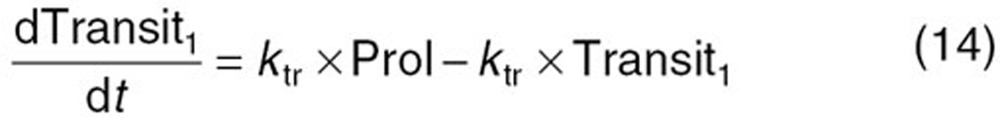 |
 |
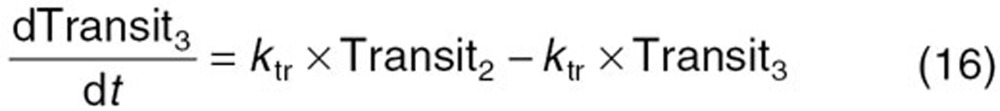 |
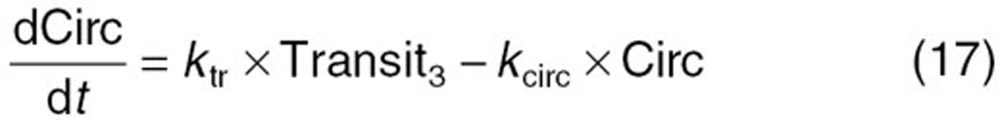 |
 |
 |
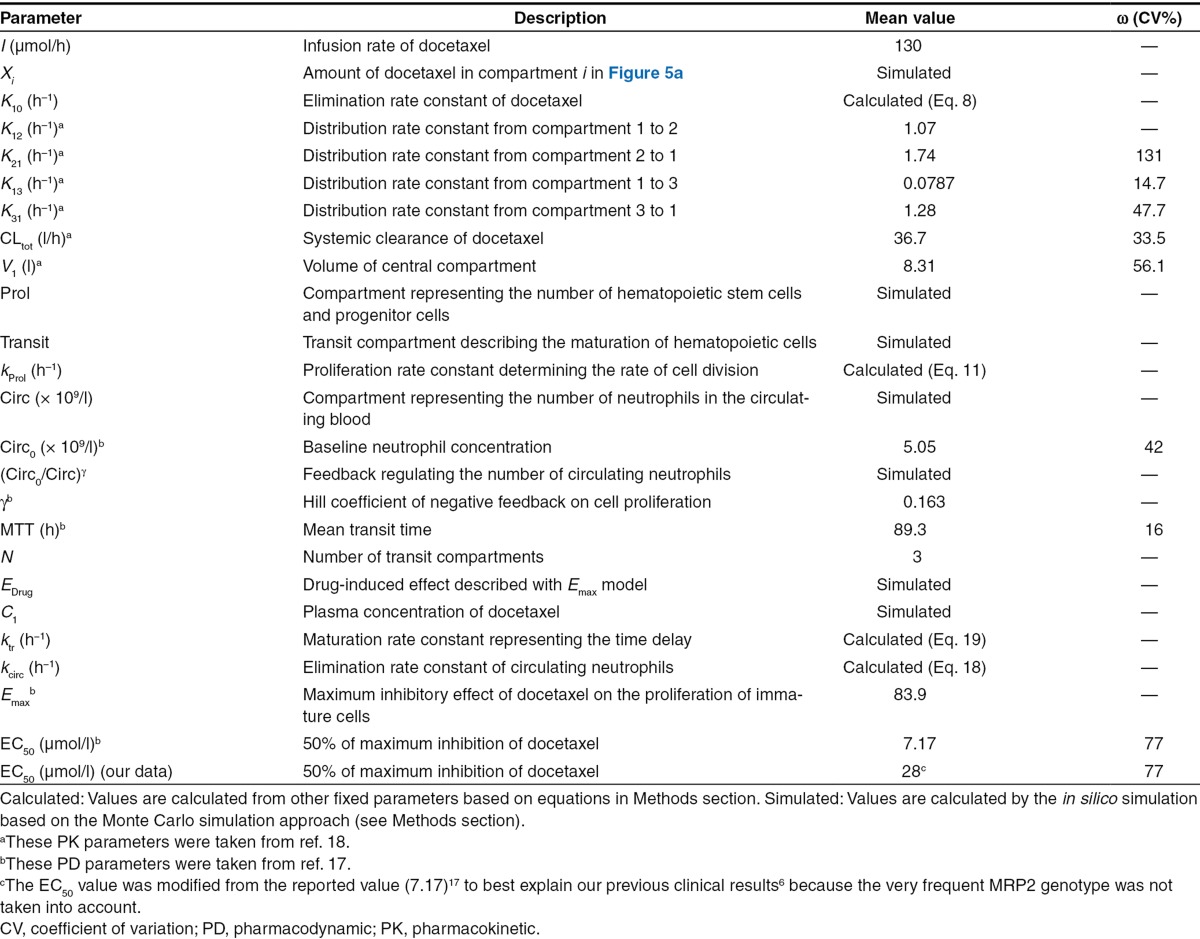
Detailed descriptions of all parameters are summarized in Table 2. The means and coefficients of variation for each parameter are summarized in Table 2.
Table 2. Descriptions for all the parameters and mean values and their CV of kinetic parameters in the PK/PD model for the in silico simulation of docetaxel-induced neutropenia.17,18.
To determine the prevalence of severe neutropenia in specific populations by in silico simulation, each parameter in the PK/PD model for virtual subjects was generated as described.22 The distribution of all parameters was assumed to be lognormal. The number of virtual subjects generated in one simulation was set at 500. After entering the parameters for each individual into the PK/PD model, the time course of neutrophil concentration in blood was simulated based on the differential equations described above using the Runge–Kutta–Gill method (Figure 4c). The calculation was performed with MULTI-RUNGE program coded by Visual Basic in Excel 2007 (Microsoft Corporation, Redmond, WA) as described.22
The grade of neutropenia was judged from the nadir based on the National Cancer Institute Common Toxicity Criteria version 2.0 (nonadverse drug reaction, >2.0 × 109/l; grade 1, 1.5–2.0 × 109/l; grade 2, 1.0–1.5 × 109/l; grade 3, 0.5–1.0 × 109/l; and grade 4, <0.5 × 109/l).
Our previous pharmacogenetic study showed that the intrinsic clearance of telmisartan in heterozygotes with the SLCO1B3 rs11045585 mutation decreased to 70.5%.19 On the basis of this observation, we calculated the systemic clearance of docetaxel in homozygotes of SLCO1B3 rs11045585 using the following equation:
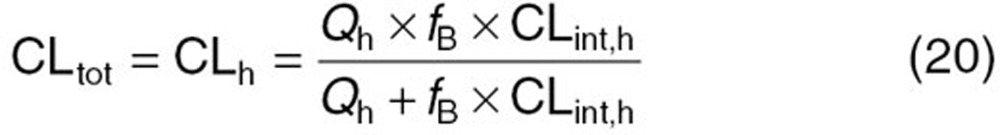 |
where CLtot and CLh represent the systemic and hepatic clearances, respectively. Because docetaxel is eliminated predominantly by hepatic metabolism, CLtot can be regarded as CLh. Qh represents the hepatic blood flow rate (20.7 ml/min/kg), fB represents the protein-unbound fraction of docetaxel in blood, and CLint,h represents the intrinsic hepatic clearance. As for MRP2, because there is no information about the functional alteration in MRP2 caused by ABCC2 rs12762549 mutation, we optimized the EC50 values by minimizing the following objective function (Obj).
 |
where ORobs and ORcalc represent the clinically observed OR and calculated OR, respectively.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
A.Y., K.M., K.K., T.M., Y.N., and Y.S. wrote the manuscript and designed the research. A.Y., K.M., and Y.S. performed the research and analyzed the data. A. Y. and K. M. contributed new reagents/analytical tools.
Study Highlights

Acknowledgments
This study was partly supported by the Research Project for the “Establishment of Evolutional Drug Development With the Use of Microdose Clinical Trial” sponsored by the New Energy and Industrial Technology Development Organization (to Y.S.) and a Grant-in-Aid for Young Scientists (B) (21790145) from the Ministry of Education, Culture, Sports, Science, and Technology (to K.M.). We thank Dr. Motohiro Kato (Chugai Pharmaceutical) for his technical support of the Monte Carlo simulation.
Supplementary Material
References
- Rowinsky E.K., Donehower R.C. The clinical pharmacology and use of antimicrotubule agents in cancer chemotherapeutics. Pharmacol. Ther. 1991;52:35–84. doi: 10.1016/0163-7258(91)90086-2. [DOI] [PubMed] [Google Scholar]
- Clarke S.J., Rivory L.P. Clinical pharmacokinetics of docetaxel. Clin. Pharmacokinet. 1999;36:99–114. doi: 10.2165/00003088-199936020-00002. [DOI] [PubMed] [Google Scholar]
- Sparreboom A., et al. Isolation, purification and biological activity of major docetaxel metabolites from human feces. Drug Metab. Dispos. 1996;24:655–658. [PubMed] [Google Scholar]
- Extra J.M., Rousseau F., Bruno R., Clavel M., Le Bail N., Marty M. Phase I and pharmacokinetic study of Taxotere (RP 56976; NSC 628503) given as a short intravenous infusion. Cancer Res. 1993;53:1037–1042. [PubMed] [Google Scholar]
- Baker S.D., et al. Relationship of systemic exposure to unbound docetaxel and neutropenia. Clin. Pharmacol. Ther. 2005;77:43–53. doi: 10.1016/j.clpt.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Kiyotani K., Mushiroda T., Kubo M., Zembutsu H., Sugiyama Y., Nakamura Y. Association of genetic polymorphisms in SLCO1B3 and ABCC2 with docetaxel-induced leukopenia. Cancer Sci. 2008;99:967–972. doi: 10.1111/j.1349-7006.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith N.F., Acharya M.R., Desai N., Figg W.D., Sparreboom A. Identification of OATP1B3 as a high-affinity hepatocellular transporter of paclitaxel. Cancer Biol. Ther. 2005;4:815–818. doi: 10.4161/cbt.4.8.1867. [DOI] [PubMed] [Google Scholar]
- Baker S.D., et al. Pharmacogenetic pathway analysis of docetaxel elimination. Clin. Pharmacol. Ther. 2009;85:155–163. doi: 10.1038/clpt.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman M.T., Chhatta A.A., van Tellingen O., Beijnen J.H., Schinkel A.H. MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int. J. Cancer. 2005;116:824–829. doi: 10.1002/ijc.21013. [DOI] [PubMed] [Google Scholar]
- Zimmermann C., van de Wetering K., van de Steeg E., Wagenaar E., Vens C., Schinkel A.H. Species-dependent transport and modulation properties of human and mouse multidrug resistance protein 2 (MRP2/Mrp2, ABCC2/Abcc2) Drug Metab. Dispos. 2008;36:631–640. doi: 10.1124/dmd.107.019620. [DOI] [PubMed] [Google Scholar]
- Ishiguro N., et al. Predominant contribution of OATP1B3 to the hepatic uptake of telmisartan, an angiotensin II receptor antagonist, in humans. Drug Metab. Dispos. 2006;34:1109–1115. doi: 10.1124/dmd.105.009175. [DOI] [PubMed] [Google Scholar]
- Shimizu M., et al. Contribution of OATP (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metab. Dispos. 2005;33:1477–1481. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- Matsushima S., Maeda K., Ishiguro N., Igarashi T., Sugiyama Y. Investigation of the inhibitory effects of various drugs on the hepatic uptake of fexofenadine in humans. Drug Metab. Dispos. 2008;36:663–669. doi: 10.1124/dmd.107.017814. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Sugiyama Y. Excretion of GSSG and glutathione conjugates mediated by MRP1 and cMOAT/MRP2. Semin. Liver Dis. 1998;18:359–376. doi: 10.1055/s-2007-1007170. [DOI] [PubMed] [Google Scholar]
- König J., Nies A.T., Cui Y., Leier I., Keppler D. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim. Biophys. Acta. 1999;1461:377–394. doi: 10.1016/s0005-2736(99)00169-8. [DOI] [PubMed] [Google Scholar]
- Keppler D., König J., Büchler M. The canalicular multidrug resistance protein, cMRP/MRP2, a novel conjugate export pump expressed in the apical membrane of hepatocytes. Adv. Enzyme Regul. 1997;37:321–333. doi: 10.1016/s0065-2571(96)00013-1. [DOI] [PubMed] [Google Scholar]
- Friberg L.E., Henningsson A., Maas H., Nguyen L., Karlsson M.O. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J. Clin. Oncol. 2002;20:4713–4721. doi: 10.1200/JCO.2002.02.140. [DOI] [PubMed] [Google Scholar]
- Bruno R., Vivier N., Vergniol J.C., De Phillips S.L., Montay G., Sheiner L.B. A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J. Pharmacokinet. Biopharm. 1996;24:153–172. doi: 10.1007/BF02353487. [DOI] [PubMed] [Google Scholar]
- Yamada A., et al. The impact of pharmacogenetics of metabolic enzymes and transporters on the pharmacokinetics of telmisartan in healthy volunteers. Pharmacogenet. Genomics. 2011;21:523–530. doi: 10.1097/FPC.0b013e3283482502. [DOI] [PubMed] [Google Scholar]
- Zhou S., et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- Shimizu K., et al. Whole-body distribution and radiation dosimetry of [11C]telmisartan as a biomarker for hepatic organic anion transporting polypeptide (OATP) 1B3. Nucl Med Biol. 2012;39:847–53. doi: 10.1016/j.nucmedbio.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Kato M., Tachibana T., Ito K., Sugiyama Y. Evaluation of methods for predicting drug-drug interactions by Monte Carlo simulation. Drug Metab. Pharmacokinet. 2003;18:121–127. doi: 10.2133/dmpk.18.121. [DOI] [PubMed] [Google Scholar]
- Kato K., et al. Association of multidrug resistance-associated protein 2 single nucleotide polymorphism rs12762549 with the basal plasma levels of phase II metabolites of isoflavonoids in healthy Japanese individuals. Pharmacogenet. Genomics. 2012;22:344–354. doi: 10.1097/FPC.0b013e3283517012. [DOI] [PubMed] [Google Scholar]
- Payen L., Sparfel L., Courtois A., Vernhet L., Guillouzo A., Fardel O. The drug efflux pump MRP2: regulation of expression in physiopathological situations and by endogenous and exogenous compounds. Cell Biol. Toxicol. 2002;18:221–233. doi: 10.1023/a:1016020626941. [DOI] [PubMed] [Google Scholar]
- Maeda K., Kambara M., Tian Y., Hofmann A.F., Sugiyama Y. Uptake of ursodeoxycholate and its conjugates by human hepatocytes: role of Na(+)-taurocholate cotransporting polypeptide (NTCP), organic anion transporting polypeptide (OATP) 1B1 (OATP-C), and oatp1B3 (OATP8) Mol. Pharm. 2006;3:70–77. doi: 10.1021/mp050063u. [DOI] [PubMed] [Google Scholar]
- Tahara H., et al. Molecular cloning and functional analyses of OAT1 and OAT3 from cynomolgus monkey kidney. Pharm. Res. 2005;22:647–660. doi: 10.1007/s11095-005-2503-0. [DOI] [PubMed] [Google Scholar]
- Hirano M., Maeda K., Shitara Y., Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J. Pharmacol. Exp. Ther. 2004;311:139–146. doi: 10.1124/jpet.104.068056. [DOI] [PubMed] [Google Scholar]
- Yamaoka K., Tanigawara Y., Nakagawa T., Uno T. A pharmacokinetic analysis program (multi) for microcomputer. J. Pharmacobio-dyn. 1981;4:879–885. doi: 10.1248/bpb1978.4.879. [DOI] [PubMed] [Google Scholar]
- Matsumura-Takeda K., Kotosai K., Ozaki A., Hara H., Yamashita S. Rat granulocyte colony-forming unit (CFU-G) assay for the assessment of drug-induced hematotoxicity. Toxicol. In Vitro. 2002;16:281–288. doi: 10.1016/s0278-6915(02)00048-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.