

Abstract
Typically, pharmacokinetic–pharmacodynamic (PK/PD) models use plasma concentration as the input that drives the PD model. However, interindividual variability in uptake transporter activity can lead to variable drug concentrations in plasma without discernible impact on the effect site organ concentration. A physiologically based PK/PD model for rosuvastatin was developed that linked the predicted liver concentration to the PD response model. The model was then applied to predict the effect of genotype-dependent uptake by the organic anion-transporting polypeptide 1B1 (OATP1B1) transporter on the pharmacological response. The area under the plasma concentration–time curve (AUC0–∞) was increased by 63 and 111% for the c.521TC and c.521CC genotypes vs. the c.521TT genotype, while the PD response remained relatively unchanged (3.1 and 5.8% reduction). Using local concentration at the effect site to drive the PD response enabled us to explain the observed disconnect between the effect of the OATP1B1 c521T>C polymorphism on rosuvastatin plasma concentration and the cholesterol synthesis response.
Physiologically based pharmacokinetic (PBPK) models are increasingly being used to predict the impact of physiological and pathophysiological patient factors and concomitant medication on drug exposure to support drug development and regulatory submissions.1,2 Typically, the end point of a PBPK model is a prediction of the pharmacokinetics of a drug, while the ultimate success of a drug is dependent on demonstration of efficacy without toxicity. Because PBPK models can predict drug concentration in tissues and in plasma, a natural progression is to link them to pharmacodynamic (PD) models via the concentration at the site of action.3,4 Compared with the traditional approach of pharmacokinetic/pharmacodynamic (PK/PD) modeling that uses plasma concentration to drive the response, this may allow a better understanding of true PD variability vs. variability that results from drug disposition to the site of action.4 This is particularly pertinent where transporters are involved in drug disposition to its effect site. In this case, interindividual variability in transporter activity can result in a lack of correlation between plasma concentration and concentration at the site of action between individuals.5
Rosuvastatin is a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, thus reduces the conversion of acetyl-CoA to mevalonic acid (MVA), which is the rate-limiting step in hepatic cholesterol biosynthesis.6 Rosuvastatin has low passive permeability across biological membranes that limits distribution to tissues and oral absorption.7 However, rosuvastatin is extensively distributed into the liver, its major site of action, through the action of specific uptake transporters, including the organic anion-transporting polypeptides OATP1B1, OATP1B3, and OATP2B1 and the sodium-dependent taurocholate cotransporting polypeptide.8,9 Both liver canalicular and intestinal efflux of rosuvastatin are mediated by the breast cancer resistance protein.10 Multidrug resistance–associated protein-2 also contributes to the liver canalicular efflux of rosuvastatin but plays a more significant role in rats than in humans.11,12 The liver is a site of elimination of rosuvastatin, predominantly through biliary elimination and to a lesser extent metabolic elimination (~10%).13
Genetic variants of OATP1B1 and breast cancer resistance protein have been identified that contribute to interindividual variability in rosuvastatin disposition, exposure, and therapeutic or side effects.14,15 In this study, we focus on the OATP1B1 c.521T>C single-nucleotide polymorphism (SNP). This SNP has been associated with increased exposure to rosuvastatin because of reduced clearance16,17 and a significant increase in the risk of myopathy for the statins simvastatin and atorvastatin, although a statistically significant increase in risk has not been found for rosuvastatin.18,19,20
A PK/PD model describing the effect of rosuvastatin on plasma MVA concentration has been published that uses an indirect response model with a circadian rhythm on the input rate.21 This model is typical of PK/PD models in that it uses the total plasma concentration to drive the PD model. However, the concentration of rosuvastatin at the site of action, i.e., the hepatic unbound intracellular water concentration (CuIW), is a more relevant driving concentration for the PD model. This is supported by a recent publication that showed an improved correlation in the cholesterol-lowering effect between humans and a mouse model when hepatic extraction was accounted for.22 PBPK models have previously been described for rosuvastatin that account for transporter-mediated disposition and allow prediction of hepatic CuIW.11,23,24
The aim of this study was to demonstrate the added utility of linking the PBPK model-predicted concentration at the site of action to the PD response compared with the plasma concentration, using rosuvastatin as an example. PBPK and PD models were integrated, creating a PBPK/PD model that links the unbound hepatocellular concentration, predicted by a permeability-limited liver model within a full PBPK model, to the rate of cholesterol synthesis over time (Figure 1). The developed model was then used to predict the impact of OATP1B1 c.521TT, TC, and CC genotypes on the PK and PD of rosuvastatin, and to compare the predictions with those when plasma concentration was used to drive the PD response and with clinical data. Simulations also investigated the impact of reduced hepatic uptake transporter function on plasma, liver, and muscle concentration, which relates to the myopathy side effect of statins, in addition to the rate of cholesterol synthesis.
Figure 1.

Schematic of the physiologically based pharmacokinetic-pharmacodynamic (PBPK/PD) model used in this study. The unbound concentration of rosuvastatin in the intracellular water of hepatocytes (CuIW) and the impact of liver uptake transporter activity are predicted by a permeability-limited liver model within a full PBPK model. This accounts for the distribution of unbound drug from the blood into the extracellular water and the passive diffusion and active transport (KtEW-in and KtIW-out) across the sinusoidal membrane between the intracellular and extracellular space. The impact of ionization is considered (red circles represent ionized drug and blue circles unionized drug) as well as the association of drug with extracellular proteins and intracellular acidic phospholipids and partitioning into neutral lipids and neutral phospholipids. Elimination of intracellular drug via biliary clearance and metabolism are considered. The PD response model uses a modified indirect response model input, with the drug effect resulting from inhibition of the input rate (Kin) and driven by the predicted liver CuIW.
Results
The OATP1B1 hepatic uptake clearance (CLint,T) for the OATP1B1 c.521TT, TC, and CC genotypes was fit using published plasma concentration–time data for subjects stratified by this genotype to obtain values of 126, 30, and 0 μl/min/106 cells, respectively. Parameter estimates were able to satisfactorily recover the clinical plasma concentration profiles for subjects grouped by genotype (Figure 2). For all three genotype groups, the mean observed maximum concentration (Cmax) and area under the plasma concentration–time curve (AUC0–48h) were within the range of those for the 10 simulated profiles matched in terms of subject age, proportion of females, and study size for each genotype (Supplementary Table S1). However, in all cases, the simulated time after administration to maximum plasma concentration (Tmax) underestimated the observed data (Supplementary Table S1). The simulations predict that the mean AUC0–∞ and Cmax were increased by 86 and 90% for the c.521CC genotype and 62 and 60% for the c.521TC genotype, relative to the c.521TT genotype. The clinically observed increases in AUC0–∞ and Cmax were reported as 62 and 79%, respectively, for the c.521CC genotype group and 57 and 52%, respectively, for the c.521TC genotype group.17
Figure 2.

Simulated and observed plasma rosuvastatin concentration profiles for the (a) wild-type (c.521TT), (b) heterozygous, and (c) homozygous deficient (c.521CC) organic anion-transporting polypeptide 1B1 (OATP1B1) genotypes. Gray lines represent simulated individual trials, and the solid black line represents the simulated mean of 10 trials. Circles represent data extracted from Pasanen et al..15 The simulation study design was matched to that of the clinical study.
The estimated IC50 and Hill coefficient for the drug effect based on rosuvastatin liver CuIW were 0.13 μmol/l and 1.3, respectively. The final model incorporating fitted the parameters allowed adequate recovery of PK and PD profiles (Figure 3). The simulated reduction in MVA AUC relative to baseline was 34 and 27%, respectively, for the evening and morning dose of rosuvastatin.
Figure 3.

(a) Simulated and observed plasma rosuvastatin concentration profile after multiple daily dosing (10 mg) for 14 days. Gray lines represent simulated individual trials, the solid black line represents the simulated mean of 10 trials, and circles are mean observed data. (b) Mean simulated (lines) and observed (markers) plasma MVA profile before rosuvastatin administration (baseline) and after morning (07:00) or evening (18:00) rosuvastatin administration. The time is 06:00 at 312 h. Observed data are from Martin et al..7 The simulation study design was matched to that of the clinical study. MVA, mevalonic acid.
Simulations were performed to predict the effect of the OATP1B1 c.521T>C sequence variation on the PD response using the established PBPK/PD model (Figure 4). Analysis of the simulated data showed that the mean rosuvastatin plasma AUC0–∞ was increased by 63 and 111% for the heterozygote and CC-homozygote genotypes relative to the wild type (Table 1). The liver CuIW AUC0–∞ was reduced by 5.7 and 9.6%, respectively (Table 1). The average MVA AUC relative to baseline corresponding to these sequence variations was increased by 30 and 35% when plasma concentration was used as the input to the PD response model, as in the original model21 (Table 1). However, when liver CuIW was used as the input to the PD response in our modified model, the simulated average MVA AUC relative to baseline was reduced by 3.1 and 5.8%, respectively (Table 1). The latter model is more consistent with clinical data and with mechanistic understanding of statin action (see Discussion), so this was used in further analysis.
Figure 4.

The effect of the organic anion-transporting polypeptide 1B1 (OATP1B1) sequence variation on the simulated mean (a) plasma concentration and (b) liver unbound intracellular water concentration (CuIW) of rosuvastatin and (c) plasma mevalonic acid (MVA) concentration using plasma concentration or (d) liver CuIW as the driving concentration for the response. Data are the mean of 100 simulated individuals based on a population that was 50% female with an age range 20–50 years. Individuals were dosed with 10 mg oral rosuvastatin at 18:00 daily for 5 days. The time is 18:00 at 108 h.
Table 1. Mean simulated plasma and liver exposure and pharmacodynamic response to rosuvastatin for the three OATP1B1 genotypes investigated.
There was a large interindividual variability in both the plasma AUC0–∞ of rosuvastatin and the reduction in MVA AUC from baseline (Figure 5), resulting in overlap between the interquartile ranges for the three OATP1B1 genotype groups. This overlap is most pronounced for the PD response, in which median values for each group are within the interquartile range for all groups.
Figure 5.

Comparison of the interindividual variability of simulated (a) plasma rosuvastatin AUC0–∞ and (b) reduction in MVA AUC from baseline for OATP1B1 c.521TT, TC, and CC genotype groups. Box and whisker plots: horizontal lines from bottom to top are the minimum, 25th percentile, median, 75th percentile, and maximum values. The diamond symbol is the mean. Distributions obtained for 100 simulated healthy volunteer individuals for each genotype. Variability was included in PBPK model parameters but not the PD model parameters.
Further analysis of the impact of the total hepatic uptake transporter CLint,T on the plasma, muscle, and liver CuIW rosuvastatin AUC0–24 h and MVA AUC relative to baseline predicted by the integrated PBPK/PD model was performed using sensitivity analysis for a range between 0 and 250 μl/min/106 cells. Both plasma and muscle exposure to rosuvastatin decreased as the overall hepatic uptake CLint,T increased (Figure 6a,c). In contrast, both liver CuIW and PD response increased as the overall hepatic uptake CLint,T increased (Figure 6b,d). In all cases, sensitivity to the value of CLint,T is greatest when the value of CLint,T is low. The elasticity index (EI) (Figure 6e) is a measure of the relative change in selected variable to the relative change in the CLint,T, normalizing the scale for comparison of the sensitivity of multiple model output parameters. The EI of the muscle rosuvastatin AUC to uptake transporter CLint,T is identical to the plasma AUC and has its greatest absolute value, indicating the greatest proportional change in AUC, when hepatic uptake transporter CLint,T is between 175 and 225 μl/min/106 cells. Changes are in the opposite direction for liver CuIW and PD response (positive rather than negative), with greatest elasticity at overall hepatic CLint,T of 25 μl/min/106 cells. The PD response shows a lower elasticity to changes in CLint,T than the liver CuIW.
Figure 6.

Sensitivity analysis of the influence of total uptake transporter intrinsic clearance (CLint,T) on (a) plasma area under the curve (AUC0–24 h), (b) liver unbound concentration in intracellular water (CuIW) AUC0–24 h, (c) muscle AUC0–24 h of rosuvastatin, and (d) the reduction in plasma mevalonic acid (MVA) AUC relative to baseline. (e) The elasticity index allows direct comparison of the relative change of plasma, liver CuIW, and muscle AUC of rosuvastatin and reduction in MVA AUC ratio to the relative change in total uptake transporter CLint,T. MVA, mevalonic acid.
Discussion
This study aimed to demonstrate the added utility of linking the PBPK model-predicted concentration at the site of action to the PD response compared with that of the use of plasma concentration. Rosuvastatin was selected as an example drug because there are clinical data demonstrating the contrasting effect of hepatic transporter activity, specifically for the OATP1B1 c.521T>C genotype, on the plasma concentration and PD response. In addition, both a PBPK model that includes hepatic transporter activity and a PD model for rosuvastatin have been published. Thus, using these models, a combined PBPK/PD model could be generated with changes to only three parameter values to account for genotype-specific OATP1B1 uptake activity and altered sensitivity of the PD response to the different input concentration used. Compared with using plasma concentration as the driving concentration for the PD response, the model is better able to capture the clinical effect of the OATP1B1 c.521T>C SNP on the therapeutic effect of rosuvastatin.
The genotype-specific OATP1B1 CLint,T for rosuvastatin, considering only the c.521T>C SNP, was estimated using clinical data from Pasanen et al..17 The fitted values of 126, 30, and 0 μl/min/106 cells for the c.521TT, c.521TC, and c.521CC genotypes, respectively, are consistent with an average OATP1B1 CLint,T of 109 μl/min/106 cells determined for rosuvastatin for the north European Caucasian population,13 in which the c.521T allele predominates. Simulations using the parameter estimates for the different OATP1B1 genotype groups and a simulated study design matched to that reported by Pasanen et al.17 were able to recover the clinical data well (Figure 2). The simulated average increase in plasma AUC for the OATP1B1 c.521CC genotype relative to the TT genotype (111%) is also in close agreement with that reported in a separate clinical study for white subjects, in which a 117% increase in activity was observed.25 However, estimation of complete loss of transport activity for the OATP1B1 c.521CC genotype conflicts with in vitro studies that have shown reduced, but not complete loss of, rosuvastatin transport activity for expressed OATP1B1 with the c.521C SNP.8,26 Accounting for residual OATP1B1 activity for the c.521CC genotype would reduce the simulated plasma rosuvastatin concentration, which tends to overestimate mean observed data with current settings (Figure 2).
Although the estimated CLint,T for the OATP1B1 c.521TC genotype group was able to recover clinical data from Pasanen et al.,17 a much smaller increase in plasma AUC of only 6.3% for the c.521TC relative to the c.521TT genotype was reported in another clinical study of white subjects.25 Thus, this study was poorly predicted by our model and the impact of the c.521TC genotype on plasma concentration is controversial. A large interindividual variability in rosuvastatin exposure is predicted for the different genotype groups (Figure 5), indicating the influence of many covariates, most of which remain unknown in clinical studies. Discrepancies between the impact of OATP1B1 c.521T>C sequence variation between clinical studies and between clinical and in vitro data may reflect limitations of the fitting approach, which fixed parameter values for all but the fitted parameter and used average clinical data from a small study. The availability of more clinical plasma concentration data stratified by genotype or data that are required for in vitro–in vivo extrapolation (such as absolute transporter abundance and in vitro activity data with extrapolation factors) would help generate CLint,T values with increased confidence. Recent reports on measurement of transporter protein abundance data in human hepatocytes can improve the in vitro–in vivo extrapolation of transporters.27
The PD model was an adaptation of that reported by Aoyama et al.,21 which describes the change in plasma MVA concentration in healthy subjects receiving 10 mg oral rosuvastatin daily. The model was modified by refitting the drug effect parameters (the IC50 and Hill coefficient) when the hepatic CuIW predicted from the PBPK model was used as the input to the PD model. The estimated IC50 for inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity by rosuvastatin (0.13 µmol/l) is considerably higher than the reported IC50 for the inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity of a purified catalytic fragment (3.5–5.4 × 10−3 µmol/l) and in isolated rat hepatocytes (1.3 × 10−3 µmol/l).28,29 This suggests that the PD model may lack sufficient mechanistic detail to make in vitro–in vivo extrapolation of the IC50 appropriate or that the in vitro methodology fails to reflect the in vivo situation.
The final PBPK/PD model maintained the ability to describe the plasma MVA profile adequately (Figure 3). The simulated reduction in MVA AUC relative to baseline of 34 and 27%, respectively, for the evening and morning dose of rosuvastatin is in reasonable agreement with the reduction of 40 and 32% simulated by Aoyama et al.21 using their PK/PD model with plasma concentration as the PD model input. The study used in fitting of the model7 is the only published clinical study that has reported the change in plasma MVA concentration after rosuvastatin administration. Consequently, the PD model has not been tested against other datasets, including those using a dose other than 10 mg rosuvastatin or in different ethnic or disease populations. Therefore, caution should be taken in generalizing the model to other populations or dosing regimens, particularly in terms of the precise quantitative description of the MVA profile. The model also assumes that there is no influence of OATP1B1 activity on the concentration of MVA at baseline. An increased baseline cholesterol synthesis rate has been reported for the OATP1B1 c.521CC genotype, although there was no effect on the total plasma cholesterol.30 Mechanistically, the increased cholesterol synthesis rate may be related to reduced OATP-mediated hepatic bile acid uptake, leading to reduced hepatic bile acid concentration and removal of the inhibitory effect of hepatic bile acid on cholesterol catabolism.30 However, data are for a small study size (32 subjects, only 4 with the c.521CC genotype), and the biomarker for cholesterol synthesis was not the plasma MVA concentration, so this was not included in the present model.
When rosuvastatin liver CuIW was linked to the PD response, the PBPK/PD model predicted a small reduction in the MVA AUC relative to baseline for individuals with the OATP1B1 c.521T>C SNP (3.1% for c.521TC and 5.8% for c.521CC). In contrast, a larger increase in the MVA AUC relative to baseline (30% for c.521TC and 35% for c.521CC) was predicted if the PD response was linked to the plasma concentration. The former is in agreement with a number of studies that have shown either no effect or a slightly reduced therapeutic response associated with the OATP1B1 c.521T>C SNP for the cholesterol-lowering response to statins.16,30,31 For example, in a genome-wide study of over 3,000 patients allocated to receive rosuvastatin 20 mg daily for a year, the OATP1B1 c.521T>C SNP was associated with a 2.6% reduction in fractional low-density lipoprotein cholesterol reduction per allele.32 Large interindividual variability in the PD response is predicted by the PBPK/PD model with considerable overlap in the distributions (Figure 5), suggesting that a large study size is required to detect the small effect of the OATP1B1 genotype on the PD response to rosuvastatin. A limitation of this study is that interindividual variability was not included for the parameters describing the PD response because the published PD model adapted for this study was estimated from average profiles.7,21 As a result, variability is only introduced by the PBPK model parameters, and PD variability is likely to be underestimated, possibly to a large extent because PD variability can be much greater than PK variability.33
Taken together, the results indicate that reduced OATP1B1 activity results in a relatively large increase in plasma rosuvastatin concentration and a small decrease in PD response. These results are in agreement with the predictions of Watanabe et al.5 for the effect of uptake transporter activity on plasma and liver concentration of pravastatin. Reduced uptake activity may be expected to decrease liver CuIW, but this effect is countered by increased plasma concentration of rosuvastatin because reduced liver exposure leads to reduced hepatic drug elimination. The higher plasma concentration results in increased liver unbound concentration in extracellular water (CuEW), which drives both passive and active uptake into the liver. Because the PD response is driven by the liver CuIW, both show a similar sensitivity to uptake transporter activity, but the relative sensitivity of the PD response is slightly lower because of the nonlinearity in the PD model (Figure 6e). A limitation of the study by Watanabe et al.5 is that predictions were based on sensitivity analysis to probe model behavior that was not confirmed by clinical or preclinical data. Our study provides verification that the modeling approach is able to recover clinical data for the impact of hepatic uptake transporter activity on the pharmacokinetics (plasma concentration) and pharmacodynamics of rosuvastatin for specific OATP1B1 genotypes.
The EI, a normalized measure of sensitivity, for the effect of uptake transporter activity on muscle AUC exactly matched with that of the plasma AUC (Figure 6e). This is expected because prediction of muscle concentration was based on the perfusion-limited model, thus assuming uptake by rapid, passive diffusion into the tissue. The results are in agreement with the association that has been observed between the plasma concentration of statins and the risk of muscle-related side effects, such as myopathy and rhabdomyolysis.34 Thus, in contrast to the results for the cholesterol-lowering effect of rosuvastatin, this supports the use of plasma concentration as a surrogate for the concentration at the site of action (muscle) in assessing risk of statin-induced muscle toxicity in individuals with different hepatic uptake transporter activity. One study has suggested a role for transporter-mediated uptake, by OATP2B1, and efflux in skeletal muscle exposure to rosuvastatin.35 However, at present, the importance of transporter-mediated uptake of rosuvastatin into muscle remains unclear. Expression of mRNA for OATP2B1 was considerably lower in skeletal muscle than in the liver,35 and insufficient data are available to model the impact of specific transporters on rosuvastatin uptake into muscle.
The potential power of linking PBPK to PD models has previously been recognized;3 however, there are few published examples that demonstrate successful application of this approach and the added value it can offer. To our knowledge, this is the first published study to use the liver concentration from a full PBPK model to drive the PD response and demonstrate improved ability to assess the impact of transporters involved in the uptake to the site of action compared with using plasma concentration to drive the PD model. This study also adds to existing knowledge by providing a specific application example validated against clinical data that confirms predictions of the discordant effect of transporter activity on the concentration of the statins in plasma and liver. It is anticipated that the approach used is applicable to other drugs with intracellular sites of action that rely on transporter-mediated processes for distribution to the site of action. It would be useful in the prediction of the impact of transporter-mediated drug–drug interactions on PD response in addition to the effect of genetic variations in transporter activity.
Methods
Development of a PBPK/PD model describing the effect of rosuvastatin on cholesterol synthesis rate
A PBPK/PD model of rosuvastatin in the north European Caucasian healthy volunteer population was constructed in the Simcyp Simulator (version 12 Release 2) as outlined in Figure 1. Detail of the PBPK model inputs for rosuvastatin has been described previously,13 and further details of the calculation of the unbound concentration of a monoprotic acid in the liver extracellular and intracellular water are given in the Supplementary Methods. Briefly, the disposition of rosuvastatin was described using a whole-body PBPK model with tissue partition coefficients predicted by the method of Rodgers and Rowland,36 assuming perfusion-limited distribution for tissues other than the liver and gut. Distribution of rosuvastatin to the liver was described by a permeability-limited liver model that included the transporter-mediated intrinsic clearance (CLint,T) for the sinusoidal uptake transporters OATP1B1, OATP1B3, OATP2B1, and sodium-dependent taurocholate cotransporting polypeptide and the canalicular efflux transporter breast cancer resistance protein. The model assumes that there is no transporter-mediated basolateral efflux from the liver, although recent work has indicated a role of basolateral efflux transporters hepatic efflux.37 Absorption of oral rosuvastatin was modeled using the Advanced Dissolution, Absorption and Metabolism (ADAM) model38 and included active efflux by breast cancer resistance protein.
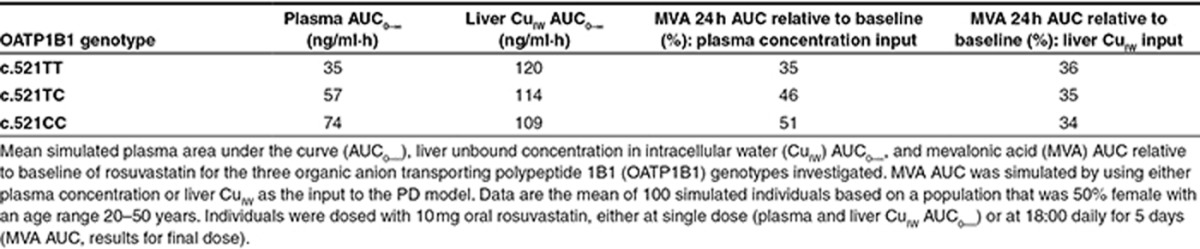
Parameters describing rosuvastatin absorption, distribution, metabolism, and elimination were not changed from the previously published values,13 with the exception of the OATP1B1 hepatic sinusoidal uptake transporter intrinsic clearance for the three OAPT1B1 c.521T>C genotypes. Using published clinically observed concentration–time data,17 the Simcyp Parameter Estimation module was used with the Nelder Mead optimization algorithm to obtain the uptake clearance of rosuvastatin into the liver for OATP1B1 genotypes with c.521TT, TC, and CC sequence variations. To reduce the likelihood of overestimating interindividual variability in liver disposition within the different OATP1B1 genotype groups, the variability in OATP1B1 relative activity in the liver was adjusted to maintain the same overall coefficient of variation (CV) for the OATP1B1 uptake CLint,T in the northern European Caucasian population as when genotype was not considered (CV 69%). Equal variability in OATP1B1 activity was assumed for each genotype, and an adjusted CV of 44% was calculated using Eqs. 1 and 2.
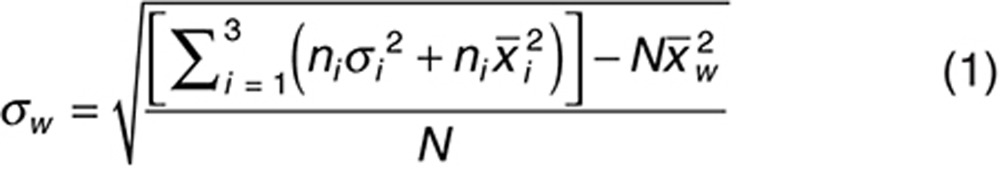 |
 |
where σw is the overall weighted standard deviation for all groups, ni is the fractional frequency of genotype i in the population, σi is the standard deviation of genotype group i, 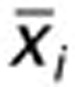 is the mean value of genotype group i, N is the total frequency of all genotypes in a population (N = 1), and
is the mean value of genotype group i, N is the total frequency of all genotypes in a population (N = 1), and 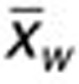 is the weighted mean for the population. The frequency of each genotype was calculated based on a weighted mean for the frequency of the c.521T>C SNP of 17% in the northern European Caucasian population,18,25,39,40,41,42,43,44,45 and assuming Hardy–Weinberg equilibrium.18,40,42,44
is the weighted mean for the population. The frequency of each genotype was calculated based on a weighted mean for the frequency of the c.521T>C SNP of 17% in the northern European Caucasian population,18,25,39,40,41,42,43,44,45 and assuming Hardy–Weinberg equilibrium.18,40,42,44
The structural model for the effect of rosuvastatin on cholesterol synthesis was coded using the custom PD scripting facility within the simulator (see Supplementary Data) and was based on the report by Aoyama et al.21 Equations describing the MVA concentration in plasma were used as reported in this publication, with the exception of the concentration input to the PD model (see Supplementary Methods). The parameters for baseline MVA concentration in plasma were kept as in the original publication. However, the drug effect (inhibition of MVA synthesis) in our model was driven by the unbound intracellular water concentration (CuIW) in liver, as opposed to plasma in the original model. Therefore, associated values (IC50 and the Hill coefficient for the inhibitory sigmoid Emax function) were obtained by refitting the data using the Simcyp Parameter Estimation module with the Nelder–Mead optimization algorithm and clinical data for the change in MVA concentration for morning and evening dosing of rosuvastatin in dominantly wild-type OATP1B1 genotypes (using the compound file default OATP1B1 CLint,T 109 μl/min/106 hepatocytes).13
Although parameter variability to account for interindividual variability in plasma and tissue concentration profiles was incorporated in the PBPK model, this was not possible for the PD model because the published PD model adapted for this work was generated from fitting of average profiles.21
Simulations
All simulations used the Simcyp north European Caucasian healthy volunteer population, and 10 trials were simulated. Where simulations aimed to replicate clinical studies, the trial design was matched as closely as possible to the study population in terms of dosing regimen, trial size, subject age range, and the proportion of females, as summarized in Supplementary Table S2. For predictive simulations, all simulations used a 10 mg oral dose of rosuvastatin dosed daily for 5 days and a trial size of 10 subjects with an age range of 20–50 years and the proportion of females as 50%. A 10 mg dose was selected because this was used by both the PK and PD study used in model development.7,17 The simulated plasma concentration and PD response reached steady state prior to the fifth dose of rosuvastatin (data not shown), thus the results for the final dose reflect predictions at steady state.
The overall PD effect was summarized by calculating the % reduction in 24-h MVA AUC from baseline at steady state using the trapezium rule and as described previously.21
Sensitivity analysis for the impact of total uptake transporter CLint,T on rosuvastatin disposition and PD response
The automated sensitivity analysis option within the Simcyp Simulator was used to investigate the effect of total uptake transporter CLint,T on the plasma, liver, and muscle exposure to rosuvastatin and the change in PD response. The specific output variables investigated were plasma, liver CuIW, and muscle AUC0–24 h and % reduction in MVA AUC from baseline. Sensitivity analysis was selected to investigate overall CLint,T over the range of normal transporter activity for rosuvastatin uptake (250 μl/min/106 cells; approximately the sum of uptake transporter activity as reported in Jamei et al.13) to a complete loss of uptake transporter activity (0 μl/min/106 cells).
The EI is a dimensionless expression of sensitivity that measures the relative change in an output variable Q (e.g., AUC) for a relative change in the input parameter P (in this case, CLint,T).46 EI was calculated as follows for each input parameter value n:
 |
where Q(Pn) is the value of Q when P = Pn and SI is a measure of the change in the output variable Q per unit change in the input parameter value Pn + 1 from its initial value Pn, calculated using Eq. 4.
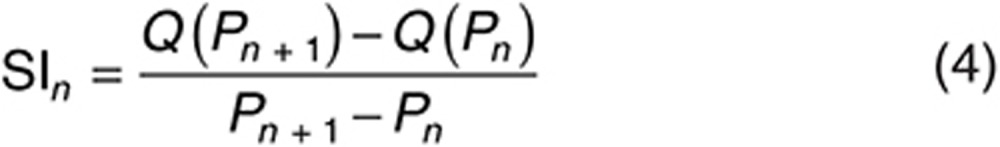 |
For the highest input parameter value, N, SI is approximated as follows:
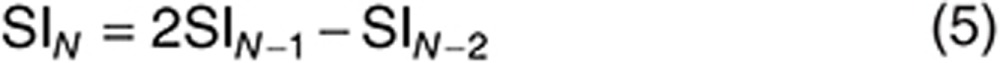 |
Author Contributions
R.H.R., S.N., and M.J. designed the research. R.H.R. performed the research and analyzed the data. R.H.R., S.N., K.A., M.C., A.R.-H., and M.J. wrote the manuscript.
Conflict of Interest
R.H.R., S.N., K.A., M.C., and M.J. are employees of Simcyp Limited (a Certara company). A.R.-H. is an employee of the University of Manchester and part-time secondee to Simcyp Limited (a Certara Company).
Study Highlights

Acknowledgments
This work was funded by Simcyp Limited (a Certara Company). The Simcyp Simulator is freely available, after completion of the training workshop, to approved members of academic institutions and other non-for-profit organizations for research and teaching purposes. The authors thank Eleanor Savill for help in preparing the manuscript and Theresa Cain for advice on statistical analysis.
Supplementary Material
References
- Zhao P., Rowland M., Huang S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 2012;92:17–20. doi: 10.1038/clpt.2012.68. [DOI] [PubMed] [Google Scholar]
- Zhao P., et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 2011;89:259–267. doi: 10.1038/clpt.2010.298. [DOI] [PubMed] [Google Scholar]
- Perera V., Elmeliegy M.A., Rao G., Forrest A. The link between pharmacodynamics and physiologically based pharmacokinetic models. Clin. Pharmacol. Ther. 2013;93:151–152. doi: 10.1038/clpt.2012.178. [DOI] [PubMed] [Google Scholar]
- Rostami-Hodjegan A. Response to “The link between pharmacodynamics and physiologically based pharmacokinetic models.”. Clin. Pharmacol. Ther. 2013;93:152. doi: 10.1038/clpt.2012.216. [DOI] [PubMed] [Google Scholar]
- Watanabe T., Kusuhara H., Maeda K., Shitara Y., Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J. Pharmacol. Exp. Ther. 2009;328:652–662. doi: 10.1124/jpet.108.146647. [DOI] [PubMed] [Google Scholar]
- Ahmad S., et al. (3R,5S,E)-7-(4-(4-fluorophenyl)-6-isopropyl-2-(methyl(1-methyl-1h-1,2,4-triazol-5-yl)amino)pyrimidin-5-yl)-3,5-dihydroxyhept-6-enoic acid (BMS-644950): a rationally designed orally efficacious 3-hydroxy-3-methylglutaryl coenzyme-a reductase inhibitor with reduced myotoxicity potential. J. Med. Chem. 2008;51:2722–2733. doi: 10.1021/jm800001n. [DOI] [PubMed] [Google Scholar]
- Martin P.D., Mitchell P.D., Schneck D.W. Pharmacodynamic effects and pharmacokinetics of a new HMG-CoA reductase inhibitor, rosuvastatin, after morning or evening administration in healthy volunteers. Br. J. Clin. Pharmacol. 2002;54:472–477. doi: 10.1046/j.1365-2125.2002.01688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho R.H., et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–1806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- Kitamura S., Maeda K., Wang Y., Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 2008;36:2014–2023. doi: 10.1124/dmd.108.021410. [DOI] [PubMed] [Google Scholar]
- Hu M., To K.K., Mak V.W., Tomlinson B. The ABCG2 transporter and its relations with the pharmacokinetics, drug interaction and lipid-lowering effects of statins. Expert Opin. Drug Metab. Toxicol. 2011;7:49–62. doi: 10.1517/17425255.2011.538383. [DOI] [PubMed] [Google Scholar]
- Ellis L.C., Hawksworth G.M., Weaver R.J. ATP-dependent transport of statins by human and rat MRP2/Mrp2. Toxicol. Appl. Pharmacol. 2013;269:187–194. doi: 10.1016/j.taap.2013.03.019. [DOI] [PubMed] [Google Scholar]
- Li M., et al. Identification of interspecies difference in efflux transporters of hepatocytes from dog, rat, monkey and human. Eur. J. Pharm. Sci. 2008;35:114–126. doi: 10.1016/j.ejps.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Jamei M., et al. A mechanistic framework for in vitro-in vivo extrapolation of liver membrane transporters: prediction of drug-drug interaction between rosuvastatin and cyclosporine. Clin. Pharmacokinet. 2014;53:73–87. doi: 10.1007/s40262-013-0097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini K.M., et al. International Transporter Consortium International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin. Pharmacol. Ther. 2013;94:23–26. doi: 10.1038/clpt.2013.12. [DOI] [PubMed] [Google Scholar]
- Bailey K.M., et al. SPACE ROCKET Trial Group Hepatic metabolism and transporter gene variants enhance response to rosuvastatin in patients with acute myocardial infarction: the GEOSTAT-1 Study. Circ. Cardiovasc. Genet. 2010;3:276–285. doi: 10.1161/CIRCGENETICS.109.898502. [DOI] [PubMed] [Google Scholar]
- Niemi M., Pasanen M.K., Neuvonen P.J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011;63:157–181. doi: 10.1124/pr.110.002857. [DOI] [PubMed] [Google Scholar]
- Pasanen M.K., Fredrikson H., Neuvonen P.J., Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 2007;82:726–733. doi: 10.1038/sj.clpt.6100220. [DOI] [PubMed] [Google Scholar]
- Link E., et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N. Engl. J. Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- Puccetti L., Ciani F., Auteri A. Genetic involvement in statins induced myopathy. Preliminary data from an observational case-control study. Atherosclerosis. 2010;211:28–29. doi: 10.1016/j.atherosclerosis.2010.02.026. [DOI] [PubMed] [Google Scholar]
- Voora D., et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J. Am. Coll. Cardiol. 2009;54:1609–1616. doi: 10.1016/j.jacc.2009.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T., et al. Pharmacokinetic/pharmacodynamic modeling and simulation of rosuvastatin using an extension of the indirect response model by incorporating a circadian rhythm. Biol. Pharm. Bull. 2010;33:1082–1087. doi: 10.1248/bpb.33.1082. [DOI] [PubMed] [Google Scholar]
- van de Steeg E, et al. Combined analysis of pharmacokinetic and efficacy data of preclinical studies with statins markedly improves translation of drug efficacy to human trials. J. Pharmacol. Exp. Ther. 2013;347:635–644. doi: 10.1124/jpet.113.208595. [DOI] [PubMed] [Google Scholar]
- Jones H.M., et al. Mechanistic pharmacokinetic modeling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab. Dispos. 2012;40:1007–1017. doi: 10.1124/dmd.111.042994. [DOI] [PubMed] [Google Scholar]
- Hobbs M., Parker C., Birch H., Kenworthy K. Understanding the interplay of drug transporters involved in the disposition of rosuvastatin in the isolated perfused rat liver using a physiologically-based pharmacokinetic model. Xenobiotica. 2012;42:327–338. doi: 10.3109/00498254.2011.625452. [DOI] [PubMed] [Google Scholar]
- Lee E., et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin. Pharmacol. Ther. 2005;78:330–341. doi: 10.1016/j.clpt.2005.06.013. [DOI] [PubMed] [Google Scholar]
- van de Steeg E., et al. Drug-drug interactions between rosuvastatin and oral antidiabetic drugs occurring at the level of OATP1B1. Drug Metab Dispos. 2013;41:592–601. doi: 10.1124/dmd.112.049023. [DOI] [PubMed] [Google Scholar]
- Prasad B., et al. Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab. Dispos. 2014;42:78–88. doi: 10.1124/dmd.113.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTaggart F., et al. Preclinical and clinical pharmacology of Rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am. J. Cardiol. 2001;87:28B–32B. doi: 10.1016/s0002-9149(01)01454-0. [DOI] [PubMed] [Google Scholar]
- Holdgate G.A., Ward W.H., McTaggart F. Molecular mechanism for inhibition of 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase by rosuvastatin. Biochem. Soc. Trans. 2003;31:528–531. doi: 10.1042/bst0310528. [DOI] [PubMed] [Google Scholar]
- Pasanen M.K., Miettinen T.A., Gylling H., Neuvonen P.J., Niemi M. Polymorphism of the hepatic influx transporter organic anion transporting polypeptide 1B1 is associated with increased cholesterol synthesis rate. Pharmacogenet. Genomics. 2008;18:921–926. doi: 10.1097/FPC.0b013e32830c1b5f. [DOI] [PubMed] [Google Scholar]
- Tachibana-Iimori R., et al. Effect of genetic polymorphism of OATP-C (SLCO1B1) on lipid-lowering response to HMG-CoA reductase inhibitors. Drug Metab. Pharmacokinet. 2004;19:375–380. doi: 10.2133/dmpk.19.375. [DOI] [PubMed] [Google Scholar]
- Chasman D.I., Giulianini F., MacFadyen J., Barratt B.J., Nyberg F., Ridker P.M. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ. Cardiovasc. Genet. 2012;5:257–264. doi: 10.1161/CIRCGENETICS.111.961144. [DOI] [PubMed] [Google Scholar]
- Levy G. Predicting effective drug concentrations for individual patients. Determinants of pharmacodynamic variability. Clin. Pharmacokinet. 1998;34:323–333. doi: 10.2165/00003088-199834040-00005. [DOI] [PubMed] [Google Scholar]
- Niemi M. Transporter pharmacogenetics and statin toxicity. Clin. Pharmacol. Ther. 2010;87:130–133. doi: 10.1038/clpt.2009.197. [DOI] [PubMed] [Google Scholar]
- Knauer M.J., et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ. Res. 2010;106:297–306. doi: 10.1161/CIRCRESAHA.109.203596. [DOI] [PubMed] [Google Scholar]
- Rodgers T., Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006;95:1238–1257. doi: 10.1002/jps.20502. [DOI] [PubMed] [Google Scholar]
- Pfeifer N.D., Yang K., Brouwer K.L. Hepatic basolateral efflux contributes significantly to rosuvastatin disposition I: characterization of basolateral versus biliary clearance using a novel protocol in sandwich-cultured hepatocytes. J. Pharmacol. Exp. Ther. 2013;347:727–736. doi: 10.1124/jpet.113.207472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamei M., et al. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009;11:225–237. doi: 10.1208/s12248-009-9099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man M., et al. Genetic variation in metabolizing enzyme and transporter genes: comprehensive assessment in 3 major East Asian subpopulations with comparison to Caucasians and Africans. J. Clin. Pharmacol. 2010;50:929–940. doi: 10.1177/0091270009355161. [DOI] [PubMed] [Google Scholar]
- Mwinyi J., Köpke K., Schaefer M., Roots I., Gerloff T. Comparison of SLCO1B1 sequence variability among German, Turkish, and African populations. Eur. J. Clin. Pharmacol. 2008;64:257–266. doi: 10.1007/s00228-007-0409-y. [DOI] [PubMed] [Google Scholar]
- Niemi M., et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1) Pharmacogenetics. 2004;14:429–440. doi: 10.1097/01.fpc.0000114750.08559.32. [DOI] [PubMed] [Google Scholar]
- Pasanen M.K., Backman J.T., Neuvonen P.J., Niemi M. Frequencies of single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide 1B1 SLCO1B1 gene in a Finnish population. Eur. J. Clin. Pharmacol. 2006;62:409–415. doi: 10.1007/s00228-006-0123-1. [DOI] [PubMed] [Google Scholar]
- Pasanen M.K., Neuvonen P.J., Niemi M. Global analysis of genetic variation in SLCO1B1. Pharmacogenomics. 2008;9:19–33. doi: 10.2217/14622416.9.1.19. [DOI] [PubMed] [Google Scholar]
- Thompson J.F., et al. An association study of 43 SNPs in 16 candidate genes with atorvastatin response. Pharmacogenomics J. 2005;5:352–358. doi: 10.1038/sj.tpj.6500328. [DOI] [PubMed] [Google Scholar]
- Tirona R.G., Leake B.F., Merino G., Kim R.B. Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J. Biol. Chem. 2001;276:35669–35675. doi: 10.1074/jbc.M103792200. [DOI] [PubMed] [Google Scholar]
- Loucks D.P., van Beek E., Stedinger J.R., Dijkman J.P.M., Villars M.T. Water Resources Systems Planning and Management: An Introduction to Methods, Models and Applications. UNESCO; Paris; 2005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.