

Abstract
Background
Drug resistance has become a severe challenge for treatment of HIV infections. Mutations accumulate in the HIV genome and make certain drugs ineffective. Prediction of resistance from genotype data is a valuable guide in choice of drugs for effective therapy.
Results
In order to improve the computational prediction of resistance from genotype data we have developed a unified encoding of the protein sequence and three-dimensional protein structure of the drug target for classification and regression analysis. The method was tested on genotype-resistance data for mutants of HIV protease and reverse transcriptase. Our graph based sequence-structure approach gives high accuracy with a new sparse dictionary classification method, as well as support vector machine and artificial neural networks classifiers. Cross-validated regression analysis with the sparse dictionary gave excellent correlation between predicted and observed resistance.
Conclusion
The approach of encoding the protein structure and sequence as a 210-dimensional vector, based on Delaunay triangulation, has promise as an accurate method for predicting resistance from sequence for drugs inhibiting HIV protease and reverse transcriptase.
Background
HIV/AIDS is a pandemic disease and more than 30 million people are infected worldwide [1]. There is no effective vaccine or medicine to completely cure AIDS; however, the long-term survival of many patients has been enabled by drug therapy. Highly Active Antiretroviral Therapy (HAART) using three or four different drugs with different viral targets is very effective in stabilizing the infection [2]. These antiviral drugs target different stages in the viral life-cycle. Two important drug targets are the HIV protease (PR) and reverse transcriptase (RT), which have essential roles in viral replication. HIV RT converts the viral RNA genome into DNA, which is translated by the host cell machinery into the viral precursor proteins. HIV PR functions to cleave the large viral precursor proteins into individual enzymes and structural proteins, which produces infectious viral particles. Among the 23 approved drugs in current clinical use, there are seven nucleoside RT inhibitors (NRTIs), four non-nucleoside RT inhibitors (NNRTIs), and eight PR inhibitors (PIs) [3]. The approved PIs were designed to bind in the active site of HIV PR, and prevent the processing of viral precursor proteins (Figure 1). NRTIs are chemical analogs of the natural nucleoside substrates of the HIV RT that bind to the protein active site and block its activity in synthesizing DNA from viral RNA. The inhibitors in the NNRTI class also decrease the enzymatic activities of RT, however, they bind in an allosteric site in the palm domain of the p66 subunit instead of the active site of RT (Figure 1).
Figure 1.
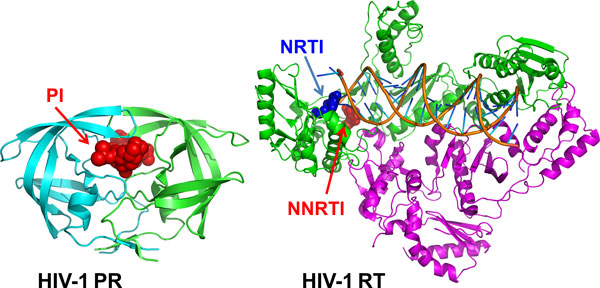
Crystal structures of HIV-1 PR and RT. The structure of HIV-1 PR dimer in complex with the inhibitor (PI) saquinavir is shown from50. The two subunits of HIV-1 PR are shown in green and cyan. The PI is colored red. The structure of HIV-1 RT dimer is shown in complex with DNA and bound NNRTI nevirapine (NVP) and NRTI zidovudine (AZT) from 51-52. The p66 subunit is shown in green and the p51 subunit is shown in purple. NRTI is shown in blue, and NNRTI is red. Double stranded DNA is indicated in orange.
Despite the success of HAART, current therapy is limited by the rapid emergence of drug resistance [3]. The virus can mutate to acquire resistance during therapy due to the lack of proofreading by RT [4] and high replication rate [5]. These resistance mutations alter the drug targets such as PR and RT [6]. Some of the 35 mutations associated with resistance to PRIs alter amino acids located in the active site of PR while the majority alter residues in distal regions of the enzyme structure [7]. Similarly for RT, several of the mutations associated with resistance to NRTIs alter amino acids in the active site of the enzyme while others are located in more distal regions. The amino acid mutations occurring in association with resistance to the NNRTIs tend to cluster around the inhibitor binding site [8,9]. The molecular mechanisms for these antiviral drugs are described in the review [10].
These resistance mutations lower the effectiveness of specific drugs and may cause failure of the treatment. Infections with resistant HIV are prevalent; surveys in North America and Europe show that 8-20% of HIV infections in untreated people contain primary drug resistance mutations [10]. Over time, multiple mutations can accumulate giving a huge number of possible combinations of mutations in each protein. This persistent problem led to the recommendation for resistance testing to guide the choice of drugs in AIDS therapy [11-13]. Fast sequencing of the genome of the infecting virus can be combined with computational predictions of resistance to guide the choice of effective antiviral drugs [13]. Accurate and fast computational predictions are desirable to avoid the expense, limited availability and time involved for performing an experimental cell-based assay for resistance where results can take four weeks.
Accurate predictions can be valuable for prescribing the most effective drugs for infections with resistant HIV. Most genotype interpretation algorithms in clinical use are knowledge based [14]. These interpretation algorithms apply a set of rules or scores for each mutation and drug. The performance of several commonly used interpretation algorithms: Stanford HIVdb [15], HIV-grade [16], REGA and ANRS (http://www.hivfrenchresistance.org/) has been compared [16]. In addition, many computational classification techniques have been evaluated for predicting drug resistance from the genotype data. The standard classification techniques of artificial neural networks (ANN) [17-21], decision tree [19-22], random forests [21], support vector machine (SVM) [21-23] and regression analysis [19] have been applied in HIV drug resistance predictions. Statistical methods can also be applied to analyze the relationship between genotype and phenotype. The association of mutations with resistance to the PIs saquinavir (SQV) and indinavir (IDV) was determined using cluster analysis, recursive partitioning, and linear discriminant analysis [24]. These methods are limited by the high dimensionality of the genotype data, hence non-parametric methods have been proposed and tested on resistance data for the PI amprenavir [25,26]. Protein structural information has been used to generate statistical potentials of mutants for training with SVM or random forest learning algorithms and tested in predicting resistance to the RT inhibitor nevirapine (NVP) [27].
We have evaluated an efficient encoding of information from the three-dimensional protein structure for the prediction of resistance from genotype. The structural encoding via Delaunay triangulation improves the quality of the predictions by representing interactions between amino acid neighbours in the three-dimensional structure unlike the linear sequence representation of other methods. This unified sequence-structure representation was used in supervised training with SVM, ANN, and a new sparse dictionary classification method. The compressive sensing/sparse dictionary representation [28,29] has been applied successfully in image analysis to enhance learning capacity and efficiency. Sparse representation has been employed for image restoration [30,31], denoising [32], deblurring [33], signal processing [34], and face detection [35]. Initial tests of this procedure for classifying resistance to four PIs was presented in [36]. Here, the structural encoding has been expanded to regression analysis and classification of genotype-phenotype data for seven PIs, six NRTIs and three NNRTIs.
Results
We combined structural information with genotype for regression analysis and supervised learning on resistance data. The new graph based sequence-structure encoding was tested with the Genotype-Phenotype Data from the Stanford HIV drug resistance database [37] (http://hivdb.stanford.edu/cgi-bin/GenoPhenoDS.cgi). Data were available for two different protein targets: HIV-1 PR and HIV-1 RT. For HIV-1 PR, eight PR inhibitors atazanavir (ATV), IDV, nelfinavir (NFV), ritonavir (RTV), lopinavir (LPV), tipranavir (TPV) and SQV were tested. While for the study of HIV RT inhibitor resistance, NNRTIs nevirapine (NPV), delaviridine (DLV), efavirenz (EFV), and NRTIs lamivudine (3TC), abacavir (ABC), zidovudine (AZT), stavudine (D4T), didanosine (DDI) and tenofovir (TDF) were tested. The data include the protein sequence (genotype) and resistance value (phenotype) from the PhenoSense (ViroLogic™) assay for each virus isolate. Genotype-phenotype data were available for 744 to 1674 isolates for different inhibitors of HIV PR, while RT was represented by 353 to 746 records for the 9 different NRTIs and NNRTIs. The preprocessing of the sequence and resistance data is detailed in Methods. Genotypes were expanded to unique protein sequences due to the presence of more than one amino acid at some positions. This expansion resulted in a total of 10,228 to 17,545 unique sequences of HIV PR mutants and 2,004 to 11,367 RT mutants for the various inhibitor resistance values.
Graph based protein sequence/structure representation using Delaunay triangulation
The sequences were combined with information from the three-dimensional protein structures by employing a graph generated by Delaunay triangulation as described in [38]. Two structure templates were used: 3OXC [4] for HIV-1 PR, and 2WOM [39] (from http://www.pdb.org). Only one structure vector is needed for each protein. In other words, all PR mutant sequences are combined with a single 210-dimensional vector derived from one PR structure, and similarly, a single structure vector is used for the RT mutants in subsequent regression and classification of resistance data. As a result, all mutants are represented as vectors of constant dimensionality, which is a desirable property for most of the pattern recognition algorithms. This structure vector was combined with sequences in regression analysis and classification for resistance.
Multiple regression on HIV protease inhibitor resistance
After each of the mutated sequences was represented by a 210-dimensional vector, we performed the regression analysis for the drug resistance data. We performed k-fold (k = 5) regression analysis on the sequence and resistance data. The predicted values for relative resistance are plotted against the experimental values as shown in (Figure 2) for the PR inhibitors ATV, NFV, RTV, IDV, LPV, TPV and SQV.
Figure 2.
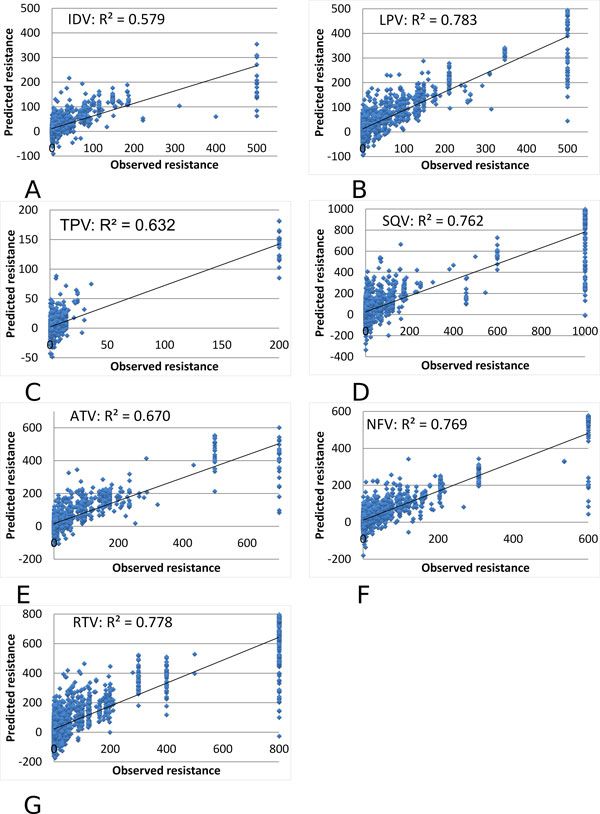
Multiple regression on the predicted and observed resistance for HIV-1 PR inhibitors. The predicted resistance is plotted against the observed value as blue dots. The observed resistance is measured relative to a value of zero for the standard non-resistant virus. The trend line is shown. The regression results are shown for resistance to PIs: (A) IDV, (B) LPV, (C) TPV, (D) SQV, (E) ATV, (F) NFV, and (G) RTV.
The multiple regression gave high R2 values of 0.579-0.783 and very low standard deviations as listed in Table 1. The values are the average of all the R2 values from k-fold regression. The high variance seen for high values of resistance is likely due to limitations of the experimental assay such that the measured resistance value has a cutoff at the upper limit, while the viral strains may have an effective resistance above this cutoff. The excellent correlations demonstrate that relative resistance to PIs can be predicted successfully from genotype by the new sequence/structure encoding method. In order to avoid training to an "optimal" n-fold set for cross validation, cross validation sets are chosen independently for each training run. Therefore, there is always a small variation in the results.
Table 1.
Multiple regression on predicted relative resistance to HIV-1 PR inhibitors.
| IDV | LPV | TPV | SQV | ATV | NFV | RTV | |
|---|---|---|---|---|---|---|---|
| R2 values, mean | 0.579 | 0.783 | 0.632 | 0.762 | 0.670 | 0.769 | 0.778 |
| R2 values, stddev | 0.037 | 0.014 | 0.045 | 0.018 | 0.035 | 0.029 | 0.016 |
Multiple regression on HIV reverse transcriptase inhibitor resistance
Multiple regression analysis was performed similarly on genotype-phenotype data for drugs inhibiting HIV-1 RT. The predicted and observed values are compared for resistance to NRTIs: 3TC, ABC, D4T, DDI, TDF and AZT in Figure 3; and NNRTIs: NPV, DLV and EFV in Figure 4.
Figure 3.
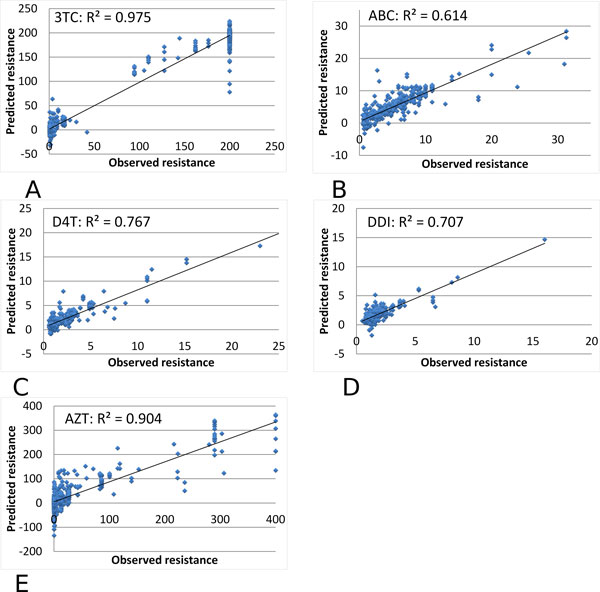
Multiple regression on the predicted and observed resistance for HIV-1 NRTIs. The predicted resistance is plotted against the observed value as blue dots. Observed resistance is measured relative to a value of zero for the standard non-resistant virus. The trend line is shown. The regression results are shown for resistance to NRTIs: (A) 3TC, (2) ABC, (3) D4T, (4) DDI, and (5) AZT.
Figure 4.
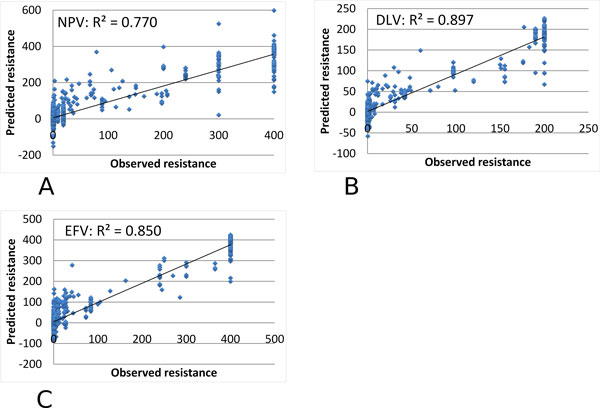
Multiple regression on the predicted and observed resistance for HIV-1 NNRTIs. The predicted resistance is plotted against the observed value as blue dots. Observed resistance is measured relative to a value of zero for the standard non-resistant virus. The trend line is shown. The regression results are shown for resistance to NNRTIs: (A) NPV, (B) DLV, and (3) EFV.
The regression results gave high R2 values of 0.614-0.975 for the different RT inhibitors, as shown in Tables 2 and 3. The resistance to NRTIs was predicted with excellent R2 values of 0.85-0.90 and very low standard deviations, while resistance predictions for NRTIs gave R2 values in the larger range of 0.61-0.98. Larger standard deviations were obtained for analysis of ABC and DDI possibly because the range of values in the dataset was smaller than for the others. Therefore, graph based encoding had excellent success in predicting resistance to RT inhibitors.
Table 2.
Multiple regression on predicted relative resistance for NNRTIs.
| DLV | EFV | NPV | |
|---|---|---|---|
| R2 values, mean | 0.904 | 0.897 | 0.850 |
| R2 values, stddev | 0.015 | 0.012 | 0.015 |
Table 3.
Multiple regression on predicted relative resistance for NRTIs.
| AZT | 3TC | ABC | D4T | DDI | |
|---|---|---|---|---|---|
| R2 values, mean | 0.770 | 0.975 | 0.614 | 0.767 | 0.707 |
| R2 values, stddev | 0.023 | 0.004 | 0.253 | 0.061 | 0.146 |
Classification of resistance with support vector machine
The support vector machine (SVM) was proposed by Vapnik [40], and is widely used as a supervised learning classifier in the machine learning classification area. In this experiment, 5-fold cross validation tests were performed by implementing in MATLAB SVM toolbox [41,42] and the linear kernel was used. The results are shown in Tables 3, 4, 5 for HIV-1 PR inhibitors (PIs), HIV-1 RT NRTIs and HIV-1 RT NNRTIs. This classification shows high accuracy, sensitivity and specificity for all inhibitors. For PIs the accuracy values range from a low of 0.93 to a high of 0.96, while sensitivity and specificity range from 0.92-0.96 and 0.94-0.98, respectively. Resistance to NRTIs is classified with even higher accuracies of 0.97-0.99, sensitivities of greater than 0.98 and specificities of 0.95-0.99, while for NNRTIs the classification performance was superior with all values of over 0.97 for accuracy, sensitivity and specificity. The excellent performance with the linear SVM kernel demonstrates conclusively that the novel encoding using Delaunay triangulation separates the resistant and non-resistant data into two distinct categories.
Table 4.
Classification using SVM for Resistance to PIs.
| ATV | IDV | NFV | RTV | LPV | SQV | TPV | |
|---|---|---|---|---|---|---|---|
| Accuracy | 0.955 | 0.960 | 0.933 | 0.946 | 0.962 | 0.946 | 0.961 |
| Stddev (×102) | 0.400 | 0.510 | 0.350 | 0.580 | 0.220 | 0.580 | 0.290 |
| Sensitivity | 0.943 | 0.951 | 0.923 | 0.945 | 0.952 | 0.945 | 0.957 |
| Stddev (×102) | 0.600 | 1.00 | 0.400 | 0.910 | 0.270 | 0.910 | 0.410 |
| Specificity | 0.968 | 0.970 | 0.943 | 0.947 | 0.972 | 0.947 | 0.965 |
| Stddev (×102) | 0.450 | 0.290 | 0.820 | 0.890 | 0.280 | 0.890 | 0.410 |
Table 5.
Classification using SVM for Resistance to NRTIs.
| 3TC | ABC | AZT | D4T | DDI | TDF | |
|---|---|---|---|---|---|---|
| Accuracy | 0.987 | 0.981 | 0.984 | 0.992 | 0.965 | 0.975 |
| Stddev (×102) | 0.484 | 0.234 | 0.390 | 0.371 | 0.289 | 0.914 |
| Sensitivity | 0.984 | 0.981 | 0.984 | 0.991 | 0.977 | 0.979 |
| Stddev (×102) | 0.613 | 0.379 | 0.627 | 0.417 | 0.436 | 1.21 |
| Specificity | 0.991 | 0.982 | 0.984 | 0.993 | 0.954 | 0.970 |
| Stddev (×102) | 0.510 | 0.397 | 0.470 | 0.505 | 0.625 | 1.76 |
Classification with Artificial Neural Networks
As in the SVM experiment, the 5-cross validation test was applied to the Artificial Neural Networks (ANN) to classify genotype-phenotype data for resistance. Specifically, the three-layer feedforward network was used in Matlab [42-44]. The network had one hidden layer of 20 nodes and was trained with backpropagation with a maximum of 50 training epochs. The results are shown in Tables 6, 7, 8 for HIV-1 PR inhibitors, and RT inhibitors NRTIs and NNRTIs. The values calculated for accuracy, sensitivity and specificity for resistance to PIs have a low of 0.91 and reach 0.97. Improved performance was achieved for classifying resistance to RT inhibitors compared with PIs. Results for NRTIs gave values of accuracy, sensitivity and specificity of 0.96-0.99, while for NNRTIs all values were greater than 0.98.
Table 6.
Classification using SVM for Resistance to NNRTIs.
| NPV | DLV | EFV | |
|---|---|---|---|
| Accuracy | 0.982 | 0.983 | 0.991 |
| Stddev (×102) | 0.254 | 0.473 | 0.316 |
| Sensitivity | 0.972 | 0.976 | 0.986 |
| Stddev (×102) | 0.490 | 0.600 | 0.618 |
| Specificity | 0.992 | 0.991 | 0.996 |
| Stddev (×102) | 0.397 | 0.787 | 0.301 |
Table 7.
Classification using ANN for Resistance to PIs.
| ATV | IDV | NFV | RTV | LPV | SQV | TPV | |
|---|---|---|---|---|---|---|---|
| Accuracy | 0.958 | 0.944 | 0.917 | 0.934 | 0.963 | 0.957 | 0.951 |
| Stddev (×102) | 0.320 | 1.25 | 1.38 | 1.44 | 0.641 | 0. 723 | 1.27 |
| Sensitivity | 0.959 | 0.940 | 0.913 | 0.935 | 0.965 | 0.958 | 0.953 |
| Stddev (×102) | 0.460 | 1.56 | 2.46 | 1.13 | 0.741 | 0.483 | 1.89 |
| Specificity | 0.957 | 0.947 | 0.922 | 0.933 | 0.961 | 0.956 | 0.950 |
| Stddev (×102) | 0.440 | 0.944 | 1.05 | 1.97 | 0.598 | 1.06 | 0. 672 |
Table 8.
Classification using ANN for Resistance to NRTIs.
| 3TC | ABC | AZT | D4T | DDI | TDF | |
|---|---|---|---|---|---|---|
| Accuracy | 0.982 | 0.984 | 0.987 | 0.983 | 0.965 | 0.970 |
| Stddev (×102) | 0.469 | 0.525 | 0.164 | 0.452 | 0.176 | 1.21 |
| Sensitivity | 0.984 | 0.978 | 0.988 | 0.980 | 0.973 | 0.965 |
| Stddev (×102) | 0.994 | 0.700 | 0.428 | 0.983 | 0.434 | 1.67 |
| Specificity | 0.980 | 0.991 | 0.986 | 0.986 | 0.957 | 0.975 |
| Stddev (×102) | 0.835 | 0.474 | 0.490 | 0.687 | 0.168 | 1.00 |
Classification using sparse dictionary
The sparse dictionary classifier was also implemented using the 5-fold cross validation tests using the approach described in [36]. The results are shown in Tables 7, 8, 9 for HIV-1 PR inhibitors, HIV-1 RT NRTIs and NNRTIs. High values were obtained for accuracy, sensitivity, and specificity. Accuracies ranged from 0.95-0.99 for resistance to PIs, 0.82-0.92 for NRTIs and 0.81-0.84 for NNRTIs. The sensitivities were all greater than 0.93 for the calculations on resistance to PIs, and specificities were greater than 0.96. Lower values were obtained for calculations on some of the RT inhibitors where values for sensitivity ranged from 0.75 to 0.96, while high specificity values from 0.86 to 1.00 was calculated. These performance measures are somewhat poorer than for the standard SVM and ANN classifiers. It is not surprising; however, that more development may be necessary for applying the new sparse dictionary as a classifier since previously it has been employed primarily for image processing.
Table 9.
Classification using ANN for Resistance to NNRTIs.
| NPV | DLV | EFV | |
|---|---|---|---|
| Accuracy | 0.983 | 0.986 | 0.986 |
| Stddev (×102) | 0.524 | 0.488 | 0.503 |
| Sensitivity | 0.979 | 0.985 | 0.982 |
| Stddev (×102) | 0.507 | 1.24 | 0.955 |
| Specificity | 0.987 | 0.987 | 0.990 |
| Stddev (×102) | 0.554 | 0.448 | 0.462 |
Comparison with standard genotype interpretation methods
Finally, we compared our methods with the standard drug resistance prediction methods HIV-GRADE, ANRS-rules, Stanford HIVdb, and Rega, which are available at http://www.hiv-grade.de/cms/grade/, using the same genotype-phenotype datasets described in Methods. The procedure discussed in [36] was used to convert the protein sequences into nucleotide sequences. Other methods usually give resistance interpretations in three categories of "resistance, "intermediate" and "susceptible". Since multiple classification is difficult with SVM and ANN, only two classes were considered for calculating the accuracy. Both "resistant" and "intermediate" are considered as "resistant"; while "susceptible" is considered as "non-resistant". The results are shown in Tables 10, 11, 12 for HIV-1 PR inhibitors, HIV-1 RT NRTIs and NNRTIs. N/A means that no output was obtained from the server for this dataset.
Table 10.
Classification using sparse dictionary for resistance to PIs.
| ATV | NFV | RTV | IDV | LPV | SQV | TPV | |
|---|---|---|---|---|---|---|---|
| Accuracy | 0.973 | 0.946 | 0.962 | 0.969 | 0.974 | 0.970 | 0.990 |
| Stddev (×102) | 0.262 | 0.602 | 0.269 | 0.151 | 0. 292 | 0.139 | 0.277 |
| Sensitivity | 0.961 | 0.927 | 0.968 | 0.951 | 0.957 | 0.959 | 0.984 |
| Stddev (×102) | 0.244 | 0.635 | 0.976 | 0.529 | 0.494 | 0.604 | 0.423 |
| Specificity | 0.986 | 0.967 | 0.958 | 0.989 | 0.992 | 0.981 | 0.995 |
| Stddev (×102) | 0.661 | 1.44 | 1.23 | 0.297 | 0.361 | 0.692 | 0.199 |
Table 11.
Classification using sparse dictionary for resistance to NRTIs.
| 3TC | ABC | AZT | D4T | DDI | TDF | |
|---|---|---|---|---|---|---|
| Accuracy | 0.918 | 0.915 | 0.932 | 0.879 | 0.816 | 0.852 |
| Stddev (×102) | 3.44 | 3.14 | 4.20 | 5.06 | 7.63 | 7.20 |
| Sensitivity | 0.963 | 0.872 | 0.947 | 0.814 | 0.801 | 0.789 |
| Stddev (×102) | 2.60 | 5.08 | 4.73 | 6.81 | 6.11 | 8.45 |
| Specificity | 0.888 | 0.973 | 0.933 | 0.987 | 0.860 | 0.972 |
| Stddev (×102) | 6.78 | 0.185 | 8.75 | 1.02 | 12.1 | 4.19 |
Table 12.
Classification using sparse dictionary for resistance to NNRTIs.
| NPV | DLV | EFV | |
|---|---|---|---|
| Accuracy | 0.826 | 0.844 | 0.811 |
| Stddev (×102) | 2.46 | 2.49 | 6.43 |
| Sensitivity | 0.761 | 0.773 | 0.753 |
| Stddev (×102) | 3.48 | 3.82 | 8.43 |
| Specificity | 0.938 | 0.973 | 0.935 |
| Stddev (×102) | 2.87 | 2.11 | 3.55 |
The accuracies demonstrate that classification with our structural encoding significantly outperforms other state of the art methods for predicting resistance to PIs for the three tested classifiers SVM, ANN and the sparse dictionary. Accuracies of 93.4-99.0% were obtained with structural encoding compared to 59.7-87.0% for the standard methods. The highest accuracies of greater than 95% were achieved with the sparse dictionary method. The prediction accuracy for resistance to the NRTI class of RT inhibitors also showed the advantages of our structural encoding with values of 81.6-99.2% compared with 72.7-95.9% for standard methods. In this case, the SVM and ANN classifiers performed better than the new sparse dictionary giving accuracies of at least 97%. For the NNRTIs, the structural encoding with SVM or ANN gave higher accuracies of 98.3-99.1% compared with 94.8-98.7% for standard methods. The sparse dictionary, however, showed lower performance with accuracies of 81.1-84.4% for NNRTI resistance, indicating some improvements may be needed for the new classifier.
Discussion
The serious problem of drug resistance arising during therapy of HIV-infected individuals can be tackled by sequencing the HIV drug targets to identify mutations followed by computational prediction of resistance to guide the choice of effective therapy. Computational predictions of the most effective drugs for the mutated HIV provide a major advantage of low cost and speed relative to experimental assays for resistance. Most standard prediction methods are knowledge based methods, such as the genotype interpretation algorithms. These algorithms either use a set of rules, for example, the Visible Genetics/Bayer Diagnostics genotype interpretation rules [45], to generate the susceptibility of the infecting virus for each drug; or apply a score or 'penalty' for each drug such as the Stanford HIV database [46] and mutation rate based score [47]. Also, a combined rule-based and penalty-based method has been proposed and applied to both HIV-1 PR and RT inhibitors [48]. Although these methods are fast, they suffer from the major disadvantage of relying on specific known mutations strongly associated with resistance and cannot identify newly appearing resistance mutations, or assess the effects of many mutations more weakly associated with resistance.
Various machine learning and statistical methods have been applied to this problem, including the widely used classifiers, ANN [17,18], decision tree [22], and SVM [23]. Statistical methods such as cluster analysis, recursive partitioning, and linear discriminant analysis have been evaluated [24], and non-parametric methods proposed for high dimensionality data [25,26]. Most of these methods are based on the linear protein sequence and omit potentially valuable information from the three-dimensional protein structure. Additional information has been introduced in the form of 544 physicochemical descriptors for the amino acid mutations leading to correlation coefficients of 0.75-0.94 [20]. Other groups have included structural features such as PR-drug contacts in the binding site with majority voting [18]. In another example, Delaunay triangulation of the RT structure was combined with a four-body statistical potential derived from 1200 protein structures in predictions for resistance to NVP and gave cross-validated accuracies of 85% with SVM and 92% with random forest classifiers [27]. Molecular mechanics calculations on the PR-drug structure have been used to predict resistance of mutants, and gave high correlation (R2 of 0.76-0.85) between caclulation and IC50 from the experimental assay [51]. However, these calculations must be performed for each mutant-drug combination and will be slow for assessing large numbers of mutants for resistance.
We have developed a simple graph representation of protein structure for fast classification. The protein structure is a three-dimensional object that has many physical and chemical factors potentially effecting stability and activity. Previously, we showed that Delaunay triangulation was the best of several tested graph-based encodings of protein structure and sequence [37]. The graph-based encoding algorithm condenses a complicated three-dimensional object, a protein structure, into a relatively small hash function with 210 unique values per sequence and structure. One critical outcome is that the graph-based encoding results in a linearly separable data set that can be used readily by several different machine learning algorithms. Similarly, the encoding is sufficiently linear that straightforward multiple linear regression can be performed on the training data. The hash value maintains enough information about the complicated object to provide useful information for machine learning and regression.
This unified sequence-structure encoding gave high accuracy in initial tests on four PIs [36]. Here, we demonstrate successful application of the structure vector in multiple regression analysis and classification on resistance data for seven inhibitors of HIV PR and nine inhibitors of RT. The 5-fold validated regression analysis gave excellent correlation between predicted and observed resistance with excellent R2 values of 0.58-0.78 for PIs, 0.61-0.98 for NRTIs and 0.85-0.90 for NNRTIs. Classification with SVM, ANN or a new sparse dictionary method gave high accuracies for predicting the resistance for PR and RT inhibitors. The structure vector encoding had superior accuracy to predictions on the same sequences using standard interpretation algorithms. The sparse dictionary classifier was the best of tested classifiers for prediction of resistance to PIs, whereas SVM classification gave the best performance on resistance prediction for RT inhibitors. This structure vector encoding of genotype data has the advantage of using a single 210-dimensional vector for each protein target. The algorithm has one slow step for preparing the encoding from a single protein structure that can be applied to all genotypes in a fast calculation, in contrast to molecular mechanics calculations that must be set up in a non-trivial manner for each individual protein sequence. The entire protein sequence is combined with the structure vector, so there is the potential for accommodating new mutations or combinations of mutations with weak but concerted effects on resistance. The procedure can be extended easily in future calculations for resistant mutants with insertions in the protein sequence, which occur commonly in RT [3]. The new sparse dictionary classification approach can be extended to multiple classifiers by using more than two dictionaries, which is a significant advantage over the tested standard SVM or ANN classifiers, and may permit accurate predictions for different levels of resistance.
Conclusions
The simple unified encoding of structural information with genotype gives high accuracy for prediction of resistance to HIV PR and RT inhibitors as well as excellent correlation coefficients in regression analysis. The improvement over algorithms using only linear sequence information suggests the importance of local interactions between mutated residues in the protein structure, which is consistent with the correlated local changes observed in the crystal structures of a highly resistant PR mutant with 20 substitutions [49]. Graph-based encoding of sequence and structure holds promise for fast and accurate predictions of resistance from sequence in order to guide the choice of effective drugs for treatment of HIV infections. In future, this approach can be expanded to predict resistance for other drugs and more diverse types of data.
Materials and methods
Data sets and data preparation
All the datasets were retrieved from Genotype-Phenotype Data on the Stanford HIV drug resistance database [37] (http://hivdb.stanford.edu/cgi-bin/GenoPhenoDS.cgi). In this experiment, the proposed algorithm was tested on two different systems: HIV-1 PR and HIV-1 RT resistance data. For HIV-1 PR, eight PR inhibitors atazanavir (ATV), indinavir (IDV), nelfinavir (NFV), ritonavir (RTV), IDV, LPV, TPV and SQV were tested. While for the study of HIV RT inhibitor resistance, NNRTIs nevirapine (NPV), delaviridine (DLV), efavirenz (EFV), and NRTIs lamivudine (3TC), abacavir (ABC), zidovudine (AZT), stavudine (D4T), didanosine (DDI) and tenofovir (TDF) were tested.
For the drug resistance study on the HIV PR and HIV RT inhibitors, all the genotypes were expanded to individual unique amino acid sequences using the method discussed in [36]. This expansion was needed since the genotyping experiment resulted in more than one possible amino acid at several positions in each genotype, due to potential experimental error or existence of multiple viral sequences infecting one patient. For each of the HIV-1 PR inhibitors the results were: for the inhibitor IDV, a total of 16846 sequences were obtained from 1622 isolates; for the inhibitor LPV, a total of 16269 sequences were obtained from 1322 isolates; for the inhibitor TPV, a total of 10228 sequences were obtained from 744 isolates; for the inhibitor SQV, a total of 17118 sequences were obtained from 1640 isolates; for the inhibitor ATV, a total of 12084 sequences were obtained from 1012 isolates; for the inhibitor IDV, a total of 16846 sequences were obtained from 1621 isolates; for the inhibitor NFV, a total of 17545 sequences were obtained from 1674 isolates; and for the inhibitor RTV, a total of 16652 sequences were obtained from 1589 isolates.
For each of the HIV-1 RT inhibitors the results were: for the inhibitor NPV, a total of 11367 sequences were obtained from 746 isolates; for the inhibitor DLV, a total of 11299 sequences were obtained from 732 isolates; and for the inhibitor EFV, a total of 11354 sequences were obtained from 734 isolates; for the inhibitor 3TC, a total of 4850 sequences were obtained from 633 isolates; for the inhibitor ABC, a total of 4846 sequences were obtained from 628 isolates; for the inhibitor AZT, a total of 4847 sequences were obtained from 630 isolates; for the inhibitor D4T, a total of 4845 sequences were obtained from 630 isolates; for the inhibitor DDI, a total of 4849 sequences were obtained from 632 isolates; for the inhibitor TDF, a total of 2004 sequences were obtained from 353 isolates.
All positive and negative instances of a given mutant were removed from either training or testing dataset before the cross-validation. This may avoid the potential problem of having negative instances associated with a positive test item or positive instances associated with a negative test item, and thus assure the training accuracy.
Pre-processing of the datasets
In order to unify the data in the original datasets, those sequences with an insertion, deletion, or containing a stop codon relative to the consensus have been removed so that the data represent proteases of 99 amino acids.
Many of the sequence records in the dataset have multiple mutations at the same sites yet share the same drug-resistance value, which may be due to sequencing limitations or to the existence of multiple viral strains in the same isolate. In order to represent a single amino acids sequence for each mutant protein, we need to expand the data to multiple sequences with single amino acids at each location. For instance, in one 99-amino acid mutant of HIV PR, at one site there are two different types of amino-acids, and another site has three. In this case, this record must be expanded to a total of 6 = (2 × 3) different sequences, each of which has only one amino-acid for each of its 99 residues, sharing the same drug resistance. We designed a fast way to perform this expansion as detailed in [36], which significantly enriches the test data.)
Cutoffs for resistance/susceptibility for each drug
For the HIV-1 PR inhibitors: ATV, IDV, NFV, and RTV, among all these genotype sequences, those mutants with the relative resistant fold < 3.0 were classified as non- resistant (susceptible), denoted as 0; while those with the relative resistant fold ≥ 3.0 were classified as resistant, denoted as 1 [19].
With the HIV-1 RT inhibitors: for ABC and TPV, those mutants with the relative resistant fold < 2.0 were classified as non-resistant, denoted as 0; while those with the relative resistant fold ≥ 2.0 were classified as resistant, denoted as 1; for 3TC, AZT, NPV, DLV, EFV, SQV, IDV and LPV those mutants with the relative resistant fold < 3.0 were classified as non-resistant, denoted as 0; while those with the relative resistant fold ≥ 3.0 were classified as resistant, denoted as 1; for D4T, DDI and TDF, those mutants with the relative resistant fold < 1.5 were classified as non-resistant, denoted as 0; while those with the relative resistant fold ≥ 1.5 were classified as resistant, denoted as 1 [19].
Encoding structure and sequence with Delaunay triangulation
The sequence and structure of the protein were represented using a graph-based encoding as described in [36]. Delaunay triangulation was used to define a graph which spanned the protein structure and defined adjacent pairs of amino acid residues. Adjacent pairs of amino acids were summarized into a vector of the 210 unique kinds of amino acid pairs by calculating the distance for each adjacent pair in the structure and tabulating by the types of amino acids in that adjacent pair. Only the sequences of the mutated proteins are needed and only one protein structure is necessary. As a result, all mutants are represented as vectors of the same dimensionality, which is a desired property for most of the pattern recognition algorithms. The structures 3OXC [4] for HIV-1 PR, and 2WOM [39] for HIV-1 RT (from http://www.pdb.org) were used as templates for Delaunay triangulation.
k-fold validation
In order to fully use all the data, a k-fold cross-validation was performed in all the experiments for all the drugs. Specifically, we randomly choose (k-1)/k of all the sequences (some are drug resistant, while others are non-drug resistant) for training the classifier and the remaining 1/k data are used for testing. These tests used k = 5. Independent randomly selected k-folds were chosen throughout the study to avoid bias in the results.
Regression analysis for drug resistance prediction
The Genotype-Phenotype Datasets provide a drug resistance value, with respect to a certain type of drug, with each genotype. The mutations relative to a standard sequence are denoted as where N is the total number of mutations and is the structure vector. Also the corresponding drug resistance values are denoted as the real numbers including 0 for the resistance value of the wild type virus. We then seek a linear model between the xi's and yi's by minimizing the cost function E :
| (1) |
with respect to the 210 dimensional vector A and scalar b.
Furthermore, in order to better utilize the available data set, we performed a k-fold cross-validation (in this work, k = 5). Specifically, the training set of size N is randomly divided into k groups. Among them, k - 1 groups are utilized for constructing the linear model as in Equation (1). Then, the linear model is used to predict the drug resistance for the remaining group with N/k mutations. The predicted resistances are compared with the measured ones and the R2 values are recorded. Finally, the average and standard deviation of the k R2 values are computed.
Sparse dictionary classification
In this experiment, we applied our newly proposed method described in [36] on both HIV-1 PR and HIV-1 RT data sets. In this case, the sequences of the mutants are considered as the group of signals, and given these signals, we would like to construct a dictionary to represent them sparsely.
The construction of a dictionary can be considered as finding a suitable over-complete basis (frame), in which the signals of interest would be represented with far fewer non-zero coefficients, than in an arbitrary fixed basis such as a Fourier basis. The newly constructed basis is also called a dictionary. This dictionary can be used to assess how well the new signal fits the model represented by the dictionary, and therefore, it can be used as a new classification method.
In our experiment, we assume there are 2 groups of signals: one for drug resistant mutants, while the other group is non-drug resistant mutants. We construct two dictionaries for each group, respectively. After that, given a new signal (mutant, in our case), we use both dictionaries to represent this signal. By calculating and comparing the reconstruction error, the dictionary with the smaller error indicates that the signal belongs to this category. Based on the theory of the dictionary, it can be observed that the group number is not limited to 2, and such procedure could be used as a multi-group classification method. The 2 dictionaries for each set of drug resistance data were constructed and the classification performed as described in [36].
List of Abbreviations
HAART, Highly Active Antiretroviral Therapy; PR, HIV protease; PI, protease inhibitor; RT, HIV reverse transcriptase; NRTI, nucleoside RT inhibitor; NNRTI, non-nucleoside RT inhibitor; ANN, artificial neural networks; SVM, Support Vector Machine; APV, amprenavir; ATV, atazanavir; IDV, indinavir; LPV, lopinavir; NFV, nelfinavir; RTV, ritonavir; SQV, saquinavir; TPV, tipranvir; 3TC, lamivudine; ABC, abacavir; AZT, zidovudine; D4T, stavudine; DDI, didanosine; DLV, delaviridine; EFV, efavirenz; NVP, nevirapine; TDF, tenofovir.
Competing interests
The authors declare that they have no competing interests.
Table 13.
Accuracy (%) compared to other methods for HIV-1 PR inhibitors.
| ATV | NFV | IDV | LPV | SQV | TPV | |
|---|---|---|---|---|---|---|
| HIV-grade | 84.7 | 81.2 | 85.1 | 80.5 | 80.2 | 72.8 |
| ANRS | N/A | 78.1 | 85.1 | 87.0 | N/A | 59.7 |
| HIVdb | N/A | 83.4 | N/A | 83.9 | N/A | 76.8 |
| Rega | 84.4 | 82.2 | 85.6 | 84.0 | 69.3 | N/A |
| SVM | 95.5 | 96.0 | 94.6 | 96.2 | 94.6 | 96.1 |
| ANN | 95.8 | 94.4 | 93.4 | 96.3 | 95.7 | 95.1 |
| Sparse dictionary | 97.3 | 94.6 | 96.9 | 97.4 | 97.0 | 99.0 |
Table 14.
Accuracy (%) compared to other methods for HIV-1 RT NRTIs.
| 3TC | ABC | AZT | D4T | DDI | TDF | |
|---|---|---|---|---|---|---|
| HIV-grade | 91.5 | 89.7 | 94.6 | 88.1 | 89.7 | 80.7 |
| ANRS | 92.0 | 83.9 | 94.4 | 87.7 | 73.3 | 72.7 |
| HIVdb | 94.3 | 95.0 | 94.5 | 86.2 | 87.6 | 79.7 |
| Rega | 95.9 | 86.0 | 94.0 | 92.2 | 88.3 | 73.8 |
| SVM | 98.7 | 98.1 | 98.4 | 99.2 | 96.5 | 97.5 |
| ANN | 98.2 | 98.4 | 98.7 | 98.3 | 96.5 | 97.0 |
| Sparse dictionary | 91.8 | 91.5 | 93.2 | 87.9 | 81.6 | 85.2 |
Table 15.
Accuracy (%) compared to other methods for NNRTIs.
| NPV | DLV | EFV | |
|---|---|---|---|
| HIV-grade | 98.7 | N/A | 98.1 |
| ANRS | 94.8 | N/A | 97.9 |
| HIVdb | 98.4 | N/A | 98.7 |
| Rega | 98.6 | 96.8 | 98.7 |
| SVM | 98.2 | 98.3 | 99.1 |
| ANN | 98.3 | 98.6 | 98.6 |
| Sparse dictionary | 82.6 | 84.4 | 81.1 |
Authors' contributions
All authors designed the experiments. XY and RWH designed the algorithms. XY implemented the algorithms and ran the predictions. All authors interpreted the results and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Xiaxia Yu, Email: xyu3@student.gsu.edu.
Irene T Weber, Email: iweber@gsu.edu.
Robert W Harrison, Email: rwh@gsu.edu.
Acknowledgements
This research was supported, in part, by the National Institutes of Health grant GM062920 (ITW, RWH), and by a fellowship from the Georgia State University Molecular Basis of Disease Program (XY).
Declarations
The publication costs for this article were partly funded by the NIH-NIGMS grant U01GM062920.
This article has been published as part of BMC Genomics Volume 15 Supplement 5, 2014: Selected articles from the Third IEEE International Conference on Computational Advances in Bio and Medical Sciences (ICCABS 2013): Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/15/S5.
References
- UNAIDS Report on the Global AIDS Epidemic 2012. http://issuu.com/unaids/docs/20121120_unaids_global_report_2012
- Mehellou Y, De Clercq E. Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go. J Med Chem. 2010;15:521–538. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- Menéndez-Arias L. Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antiviral research. 2013;15:93–120. doi: 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- Ji J, Loeb LA. Fidelity of HIV-1 reverse transcriptase copying RNA in vitro. Biochemistry. 1992;15:954–958. doi: 10.1021/bi00119a002. [DOI] [PubMed] [Google Scholar]
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;15:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Agniswamy J, Shen C-H, Wang Y-F, Ghosh AK, Rao KV, Xu C-X. et al. Extreme multidrug resistant HIV-1 protease with 20 mutations is resistant to novel protease inhibitors with P1'-pyrrolidinone or P2-tris-tetrahydrofuran. J Med Chem. 2013;15:4017–4027. doi: 10.1021/jm400231v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber IT, Agniswamy J. HIV-1 protease:structural perspectives on drug resistance. Viruses. 2009;15:1110–1136. doi: 10.3390/v1031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccherini-Silberstein F, Svicher V, Sing T, Artese A, Santoro MM, Forbici F. et al. Characterization and structural analysis of novel mutations in human immunodeficiency virus type 1 reverse transcriptase involved in the regulation of resistance to nonnucleoside inhibitors. J Virol. 2007;15:11507–11519. doi: 10.1128/JVI.00303-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambuyzer L, Azijn H, Rimsky LT, Vingerhoets J, Lecocq P, Kraus G. et al. Short communication Compilation and prevalence of mutations associated with resistance to non-nucleoside reverse transcriptase inhibitors. Antiviral Therapy. 2009;15:103–109. [PubMed] [Google Scholar]
- Menéndez-Arias L. Molecular basis of human immunodeficiency virus drug resistance: an update. Antiviral Res. 2010;15:210–231. doi: 10.1016/j.antiviral.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Shafer R, Rhee S-Y, Pillay D, Miller V, Sandstrom P, Schapiro J. et al. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS. 2007;15:215–223. doi: 10.1097/QAD.0b013e328011e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch MS, Günthard HF, Schapiro JM, Vézinet FB, Clotet B, Hammer SM. et al. Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an International AIDS Society-USA panel. Clinical Infectious Diseases. 2008;15:266–285. doi: 10.1086/589297. [DOI] [PubMed] [Google Scholar]
- Vandamme A-M, Camacho RJ, Ceccherini-Silberstein F, De Luca A, Palmisano L, Paraskevis D. et al. European recommendations for the clinical use of HIV drug resistance testing: 2011 update. AIDS Rev. 2011;15:77–108. [PubMed] [Google Scholar]
- Vercauteren J, Vandamme A-M. Algorithms for the interpretation of HIV-1 genotypic drug resistance information. Antiviral Res. 2006;15:335–342. doi: 10.1016/j.antiviral.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Talbot A, Grant P, Taylor J, Baril J-G, Liu TF, Charest H. et al. Predicting tipranavir and darunavir resistance using genotypic, phenotypic, and virtual phenotypic resistance patterns: an independent cohort analysis of clinical isolates highly resistant to all other protease inhibitors. Antimicrob Agents Chemother. 2010;15:2473–2479. doi: 10.1128/AAC.00096-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier M, Pironti A, Berg T, Braun P, Däumer M, Eberle J. et al. HIV-GRADE: a publicly available, rules-based drug resistance interpretation algorithm integrating bioinformatic knowledge. Intervirology. 2012;15:102–107. doi: 10.1159/000331999. [DOI] [PubMed] [Google Scholar]
- Wang D, Larder B. Enhanced prediction of lopinavir resistance from genotype by use of artificial neural networks. J Infect Dis. 2003;15:653–660. doi: 10.1086/377453. [DOI] [PubMed] [Google Scholar]
- Drăghici S, Potter RB. Predicting HIV drug resistance with neural networks. Bioinformatics. 2003;15:98–107. doi: 10.1093/bioinformatics/19.1.98. [DOI] [PubMed] [Google Scholar]
- Rhee S-Y, Taylor J, Wadhera G, Ben-Hur A, Brutlag DL, Shafer RW. Genotypic predictors of human immunodeficiency virus type 1 drug resistance. Proc Natl Acad Sci. 2006;15:17355–17360. doi: 10.1073/pnas.0607274103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaer J, Høj L, Fox Z, Lundgren J. Prediction of phenotypic susceptibility to antiretroviral drugs using physiochemical properties of the primary enzymatic structure combined with artificial neural networks. HIV Med. 2008;15:642–652. doi: 10.1111/j.1468-1293.2008.00612.x. [DOI] [PubMed] [Google Scholar]
- Wang D, Larder B, Revell A, Montaner J, Harrigan R, De Wolf F. et al. A Comparison of three computational modelling methods for the prediction of virological response to combination HIV therapy. Artif Intell Med. 2009;15:63–74. doi: 10.1016/j.artmed.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N, Schmidt B, Walter H, Kaiser R, Lengauer T, Hoffmann D. et al. Diversity and complexity of HIV-1 drug resistance: a bioinformatics approach to predicting phenotype from genotype. Proc Natl Acad Sci. 2002;15:8271–8276. doi: 10.1073/pnas.112177799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerenwinkel N, Däumer M, Oette M, Korn K, Hoffmann D, Kaiser R. et al. Geno2pheno: estimating phenotypic drug resistance from HIV-1 genotypes. Nucleic Acids Res. 2003;15:3850–3855. doi: 10.1093/nar/gkg575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevin AD, DeGruttola V, Nijhuis M, Schapiro JM, Foulkes AS, Para MF. et al. Methods for investigation of the relationship between drug-susceptibility phenotype and human immunodeficiency virus type 1 genotype with applications to AIDS clinical trials group 333. J Infect Dis. 2000;15:59–67. doi: 10.1086/315673. [DOI] [PubMed] [Google Scholar]
- DiRienzo G, DeGruttola V, Larder B, Hertogs K. Nonparametric methods to predict HIV drug susceptibility phenotype from genotype. Stat Med. 2003;15:2785–2798. doi: 10.1002/sim.1516. [DOI] [PubMed] [Google Scholar]
- DiRienzo G, DeGruttola V. Collaborative HIV resistance-response database initiatives: sample size for detection of relationships between HIV-1 genotype and HIV-1 RNA response using a non-parametric approach. Antivir Ther. 2002;15(S71) [Google Scholar]
- Ravich VL, Masso M, Vaisman II. A combined sequence-structure approach for predicting resistance to the non-nucleoside HIV-1 reverse transcriptase inhibitor Nevirapine. Biophys Chem. 2011;15:168–172. doi: 10.1016/j.bpc.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Donoho DL, Elad M. Optimally sparse representation in general (nonorthogonal) dictionaries via ℓ1 minimization. Proc Natl Acad Sci. 2003;15:2197–2202. doi: 10.1073/pnas.0437847100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoho DL. Compressed sensing. IEEE Transactions on Information Theory. 2006;15:1289–1306. [Google Scholar]
- Mairal J, Elad M, Sapiro G. Sparse representation for color image restoration. IEEE Transactions on Image Processing. 2008;15:53–69. doi: 10.1109/tip.2007.911828. [DOI] [PubMed] [Google Scholar]
- Gao Y, Bouix S, Shenton M, Tannenbaum A. Sparse texture active contour. IEEE Transactions on Image Processing. 2013;15:3866–3878. doi: 10.1109/TIP.2013.2263147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elad M, Aharon M. Image denoising via sparse and redundant representations over learned dictionaries. IEEE Transactions on Image Processing. 2006;15:3736–3745. doi: 10.1109/tip.2006.881969. [DOI] [PubMed] [Google Scholar]
- Lou Y, Bertozzi AL, Soatto S. Direct sparse deblurring. J Math Imaging Vision. 2011;15:1–12. doi: 10.1007/s10851-010-0220-8. [DOI] [Google Scholar]
- Sprechmann P, Sapiro G. Dictionary learning and sparse coding for unsupervised clustering. IEEE International Conference on Acoustics Speech and Signal Processing. 2010. pp. 2042–2045.
- Wright J, Yang AY, Ganesh A, Sastry SS, Ma Y. Robust face recognition via sparse representation. IEEE Transactions on Pattern Analysis and Machine Intelligence. 2009;15:210–227. doi: 10.1109/TPAMI.2008.79. [DOI] [PubMed] [Google Scholar]
- Yu X, Weber I, Harrison R. SIAM International Conference on Data mining. Austin, TX, USA; 2013. Sparse Representation for HIV-1 Protease Drug Resistance Prediction; pp. 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;15:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P, Yu X, Harrison RW. Encoding protein structure with functions on graphs. IEEE International Conference on Bioinformatics and Biomedicine Workshops. 2011. pp. 338–344.
- Corbau R, Mori J, Phillips C, Fishburn L, Martin A, Mowbray C. et al. Lersivirine, a nonnucleoside reverse transcriptase inhibitor with activity against drug-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2010;15:4451–4463. doi: 10.1128/AAC.01455-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vapnik VN. The nature of statistical learning theory. Springer-Verlag New York Inc; 2000. [Google Scholar]
- Canu S, Grandvalet Y, Guigue V, Rakotomamonjy A. SVM and kernel methods MATLAB toolbox. Perception Systemes et Information, INSA de Rouen, Rouen, France. 2005;15 [Google Scholar]
- The MathWorks Inc. http://www.mathworks.com
- Hornik K, Stinchcombe M, White H. Multilayer feedforward networks are universal approximators. Neural Networks. 1989;15:359–366. doi: 10.1016/0893-6080(89)90020-8. [DOI] [Google Scholar]
- Howard D, Beale M. Neural Network Toolbox, for Use with MATLAB, User's Guide, Version 4. The MathWorks Inc. 2000. pp. 133–205.
- Larder B. Quantitative prediction of HIV-1 phenotypic drug resistance from genotypes: the virtual phenotype (VirtualPhenotype) Antiviral Ther. 2000;15:63–63. [Google Scholar]
- PuchhammerߚStöckl E, Steininger C, Geringer E, Heinz F. Comparison of virtual phenotype and HIV‐SEQ program (Stanford) interpretation for predicting drug resistance of HIV strains. HIV Med. 2002;15:200–206. doi: 10.1046/j.1468-1293.2002.00116.x. [DOI] [PubMed] [Google Scholar]
- Schmidt B, Walter H, Moschik B, Paatz C, Van Vaerenbergh K, Vandamme A-M. et al. Simple algorithm derived from a geno-/phenotypic database to predict HIV-1 protease inhibitor resistance. AIDS. 2000;15:1731–1738. doi: 10.1097/00002030-200008180-00007. [DOI] [PubMed] [Google Scholar]
- Zazzi M, Romano L, Venturi G, Shafer RW, Reid C, Dal Bello F. et al. Comparative evaluation of three computerized algorithms for prediction of antiretroviral susceptibility from HIV type 1 genotype. J Antimicrob Chemother. 2004;15:356–360. doi: 10.1093/jac/dkh021. [DOI] [PubMed] [Google Scholar]
- Agniswamy J, Shen C-H, Aniana A, Sayer JM, Louis JM, Weber IT. HIV-1 protease with 20 mutations exhibits extreme resistance to clinical inhibitors through coordinated structural rearrangements. Biochem. 2012;15:2819–2828. doi: 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Y, Boross PI, Wang Y-F, Gaddis L, Hussain AK, Leshchenko S. et al. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J Mol Biol. 2004;15:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- Ren J, Nichols C, Bird L, Chamberlain P, Weaver K, Short S. et al. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J Mol Biol. 2001;15:795–805. doi: 10.1006/jmbi.2001.4988. [DOI] [PubMed] [Google Scholar]
- Tu X, Das K, Han Q, Bauman JD, Clark AD Jr, Hou X. et al. Structural basis of HIV-1 resistance to AZT by excision. Nat Struct Mol Biol. 2010;15:1202–1209. doi: 10.1038/nsmb.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]