

ABSTRACT
The bacterium Streptococcus pneumoniae is one of the leading causes of fatal infections affecting humans. Intriguingly, phylogenetic analysis shows that the species constitutes one evolutionary lineage in a cluster of the otherwise commensal Streptococcus mitis strains, with which humans live in harmony. In a comparative analysis of 35 genomes, including phylogenetic analyses of all predicted genes, we have shown that the pathogenic pneumococcus has evolved into a master of genomic flexibility while lineages that evolved into the nonpathogenic S. mitis secured harmonious coexistence with their host by stabilizing an approximately 15%-reduced genome devoid of many virulence genes. Our data further provide evidence that interspecies gene transfer between S. pneumoniae and S. mitis occurs in a unidirectional manner, i.e., from S. mitis to S. pneumoniae. Import of genes from S. mitis and other mitis, anginosus, and salivarius group streptococci ensured allelic replacements and antigenic diversification and has been driving the evolution of the remarkable structural diversity of capsular polysaccharides of S. pneumoniae. Our study explains how the unique structural diversity of the pneumococcal capsule emerged and conceivably will continue to increase and reveals a striking example of the fragile border between the commensal and pathogenic lifestyles. While genomic plasticity enabling quick adaptation to environmental stress is a necessity for the pathogenic streptococci, the commensal lifestyle benefits from stability.
IMPORTANCE
One of the leading causes of fatal infections affecting humans, Streptococcus pneumoniae, and the commensal Streptococcus mitis are closely related obligate symbionts associated with hominids. Faced with a shortage of accessible hosts, the two opposing lifestyles evolved in parallel. We have shown that the nonpathogenic S. mitis secured harmonious coexistence with its host by stabilizing a reduced genome devoid of many virulence genes. Meanwhile, the pathogenic pneumococcus evolved into a master of genomic flexibility and imports genes from S. mitis and other related streptococci. This process ensured antigenic diversification and has been driving the evolution of the remarkable structural diversity of capsular polysaccharides of S. pneumoniae, which conceivably will continue to increase and present a challenge to disease prevention.
INTRODUCTION
Streptococcus pneumoniae is a leading cause of pneumonia, meningitis, septicemia, and middle ear infections (1). According to data from the World Health Organization, S. pneumoniae is the fourth most frequent cause of fatal infections worldwide (2). Intriguingly, the species is not related to other overt streptococcal pathogens but clusters within the mitis group of streptococci, which otherwise are important members of the commensal microbiota of the oral cavity and pharynx (3, 4). The unique pathogenic potential of S. pneumoniae among the species of the mitis group streptococci is explained by an array of virulence factors that provide escape of host immunity, such as the polysaccharide capsule and the IgA1 protease, in addition to surface-exposed proteins that enable adhesion to and destruction of host tissues (5, 6). In spite of relative conservation of its genome, some pneumococcal virulence factors show extensive structural diversity that ensures survival of the species after immunity has developed in response to infection or vaccination (5). One example is the capsular polysaccharide, which occurs in more than 90 distinct structures, encoded by serotype-specific capsular biosynthesis operons (cps), which, combined, add up to the same size as the complete pneumococcal genome (~2.1 Mb) (7). The 13 capsular polysaccharides most frequently associated with disease form the basis of a childhood vaccine currently implemented in most industrialized countries (8). However, frequent switching of capsular serotype (9–11) and the potential emergence of novel structures present a significant challenge to the continued successful prevention of pneumococcal infections.
Regulated natural competence for genetic transformation of pneumococci combined with induced lysis of noncompetent members of the same species enables frequent transfer of pathogenicity islands, exchange of complete virulence genes or fragments of them, and dissemination of antibiotic resistance within the species (12–17). In addition, recombination between S. pneumoniae, Streptococcus mitis, and Streptococcus oralis has been reported to be instrumental in the development and dissemination of resistance to beta-lactam antibiotics (18–20).
We previously proposed an evolutionary model suggesting that the species S. pneumoniae, S. mitis, and the more recently described Streptococcus pseudopneumoniae arose from a pneumococcus-like organism pathogenic to the immediate ancestor of hominids (3). Being almost exclusively adapted to humans and other hominids, their success conceivably is closely associated with the population size of susceptible hosts. Here we present evidence supporting this evolutionary model and demonstrate the genetic basis of how a dichotomy of distinct but successful bacterial lifestyles evolved in parallel within their host. The pathogenic lifestyle of the pneumococcus, dependent on continued import of genes from neighboring species, results in antigenic diversity that will continue to challenge the prevention of pneumococcal infections.
RESULTS
Phylogenetic relationships based on core genome sequences.
To shed light on the genetic processes that shaped the genomes of S. pneumoniae and its close commensal relatives, we explored new genomic information. Alignment of 35 genomes of S. pneumoniae, S. mitis, S. pseudopneumoniae, S. oralis, and Streptococcus infantis (see Table S1 in the supplemental material) identified a core of 822,537 nucleotides (nt). The number of polymorphic sites within this concatenated sequence was 292,227 (35.5%), of which 240,553 sites were parsimoniously informative (i.e., present in more than one strain). Phylogenetic reconstruction based on these core genome sequences confirmed our previous observation, based on selected housekeeping genes (3, 4), that S. pneumoniae is a single lineage in a cluster otherwise composed of S. mitis, that S. pseudopneumoniae takes up an intermediary position, and that all three species are well separated from S. oralis and S. infantis (Fig. 1). The average genetic distance of members of the S. mitis/S. pneumoniae/S. pseudopneumoniae cluster to the designated type strain of S. oralis, ATCC 35037, used as a common root, is slightly but significantly (P < 0.0001) greater for S. pneumoniae (0.001309 ± 0.0002) than for S. mitis (0.001278 ± 0.0008). This supports our hypothesis (3) that the S. pneumoniae lineage is the phylogenetically most ancient and only recently has been undergoing a population burst facilitated by the exponentially expanding human species, its primary host. Spreading vertically (21), success of the commensal species is not dependent on the host population size.
FIG 1 .
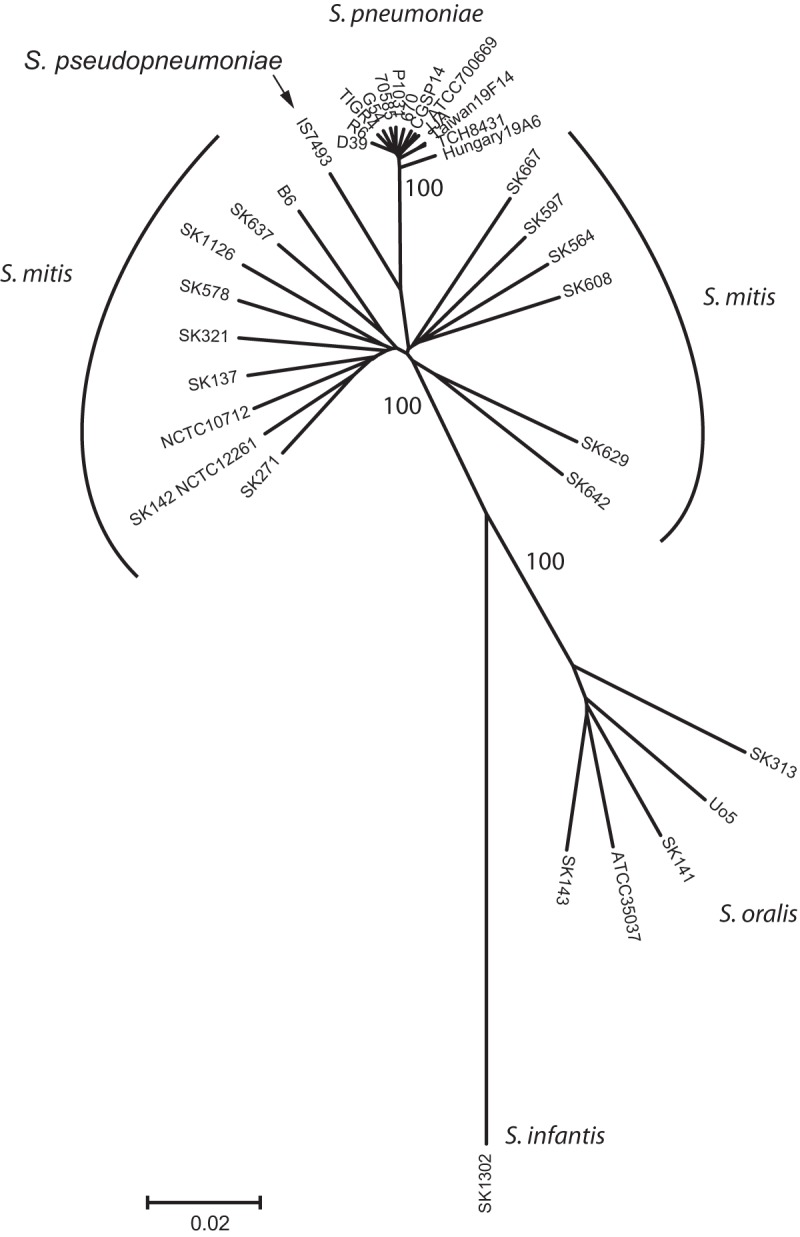
Phylogenetic tree of Streptococcus strains included in the study. The tree, generated by the minimum-evolution algorithm in MEGA version 5.2, was based on 822,537-nt sequences shared by all 35 genomes listed in Table S1 in the supplemental material. It illustrates that S. pneumoniae is a single lineage in a cluster otherwise composed of S. mitis and that S. pseudopneumoniae occupies an intermediary position. The bar represents the genetic distance.
Reductive evolution of the S. mitis genome.
The previously demonstrated sporadic occurrence of recognized S. pneumoniae virulence factors in S. mitis strains (3, 22, 23) was confirmed by detailed comparison of the gene contents of the 35 genomes. Most strikingly, 12 out of 15 S. mitis strains had a complete cps locus in the same genomic region as in S. pneumoniae. Likewise, assumed virulence factors like IgA1 protease and zinc metalloprotease C, neuraminidases A and B, autolysin, pneumolysin, several choline-binding proteins, and PavA were present in some strains of S. mitis and absent in others (Fig. 2).
FIG 2 .
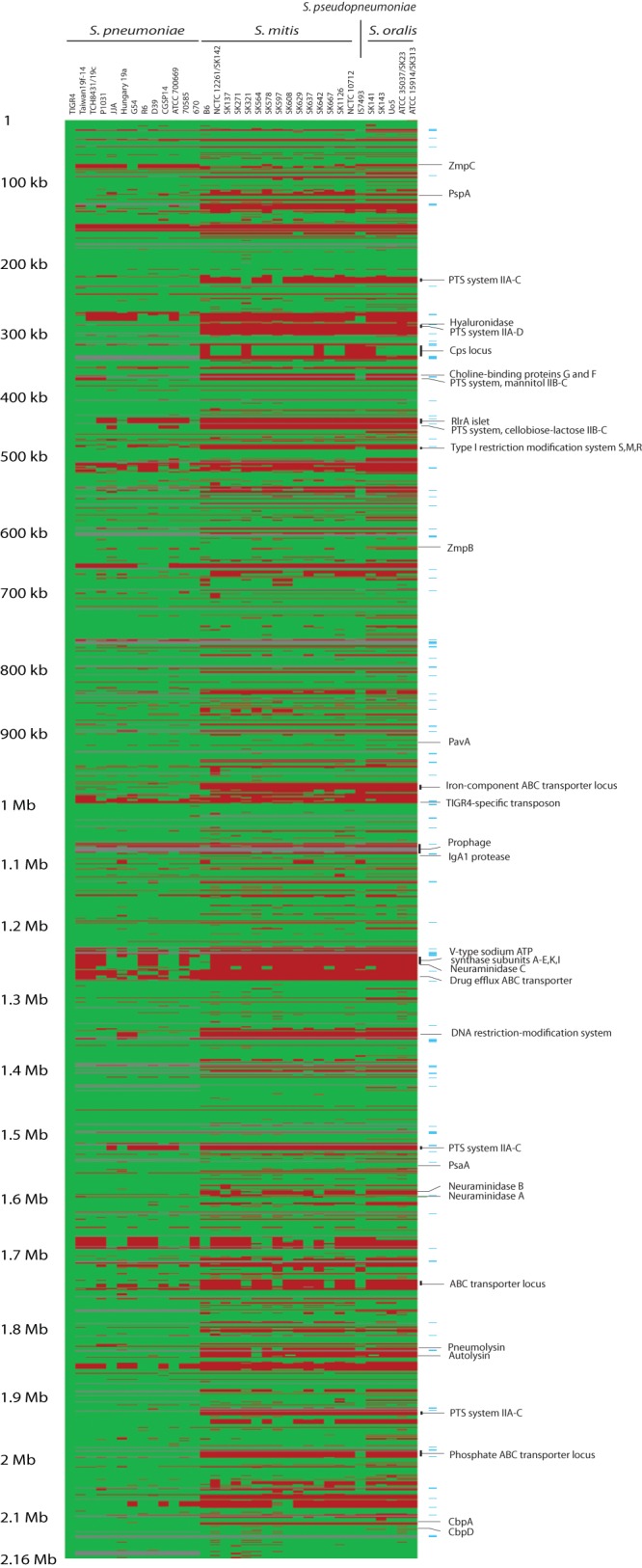
Comparative analysis of the gene contents of 35 genomes of S. pneumoniae, S. mitis, S. pseudopneumoniae, and S. oralis. The genome of S. pneumoniae TIGR4 served as a reference. Green indicates the presence and red the absence of genes. Transposase genes are indicated by light-blue horizontal lines on the right. The figure illustrates the strain-specific reductive evolution of S. mitis genomes, resulting in gene loss to various extents, including genes encoding virulence properties in S. pneumoniae.
To determine if such shared virulence genes represent pneumococcal genes transferred to S. mitis or genes ancestral to both, we generated phylogenetic trees of all predicted genes in S. pneumoniae TIGR4 and orthologs identified in all 35 genomes. In trees of virulence genes (one example is shown in Fig. S1A in the supplemental material), S. pneumoniae formed a tight cluster, whereas S. mitis strains formed more diverse lineages in patterns congruent with the core genome-based tree (Fig. 1). This proves that they are ancestral genes that have been diversifying in parallel with other parts of the genome and subsequently were lost by some S. mitis strains in a reductive evolutionary process. The loss is reflected in the S. mitis genomes being up to 15% smaller than those of S. pneumoniae (see Table S1). However, a surprising proportion (23.6%) of the 1,620 trees generated on the basis of nucleotide sequences of all genes (excluding transposases and genes unique to S. pneumoniae strains) showed clustering of S. pneumoniae genes among S. mitis genes. We interpret this as evidence of acquisition by S. pneumoniae strains of homologous gene sequences from strains of S. mitis. Although occasional trees identified the source of the gene sequence, the majority of transfers had as donors putative S. mitis clones not represented in our sample of the undoubtedly large global population of S. mitis (see Fig. S1B). The transfers from S. mitis to S. pneumoniae often affected several adjacent genes, amounting to sequences spanning from 116 bp to 10,600 bp, in full agreement with the sizes observed in an in vitro recombination experiment involving one strain each of S. mitis and S. pneumoniae (18). As shown in Table 1 and reflected in the phylogenetic tree in Fig. 1, Hungary19A was the strain of S. pneumoniae that acquired the largest proportion of genes (8.2% of genes, corresponding to 141 kb) from S. mitis. S. pseudopneumoniae showed extensive recombination between the S. pneumoniae lineage and S. mitis lineages, reflected in its intermediary position in the phylogenetic tree and its admixture of phenotypic traits of the two species (24). While 86% of the genes clustered with S. mitis 14% clustered with S. pneumoniae. No clear evidence of acquisition by S. mitis strains of gene sequences from S. pneumoniae was detected. However, as previously reported (18–20), genes encoding transpeptidases (“penicillin-binding proteins”), gyrase, and adjacent genes (e.g., orthologs of SP_0335, SP_0370, SP_0371, SP_1218, and SP_1662-1669) (25) revealed mosaic sequence structures (see Fig. S1C and D). This reflects multiple homologous recombination events between S. pneumoniae and S. mitis but often without clear traces of the direction of transfers.
TABLE 1 .
Numbers of gene replacements in S. pneumoniae strains imported from S. mitis
| Strain | Serotype | No. (%) of genes imported from S. mitisa |
|---|---|---|
| Hungary19A | 19A | 133 (8.2) |
| Taiwan19F | 19F | 88 (5.4) |
| CGSP14 | 14 | 85 (5.3) |
| ATCC 700669 | 23F | 72 (4.4) |
| P1031 | 1 | 71 (4.4) |
| TIGR4 | 4 | 61 (3.8) |
| TCH8431 | 19A | 60 (3.7) |
| 670 | 6B | 56 (3.5) |
| JJA | 14 | 56 (3.5) |
| G54 | 19F | 45 (2.8) |
| 70585 | 5 | 38 (2.4) |
| D39 | 2 | 28 (1.7) |
| R6 | Rough 2 | 27 (1.7) |
Based on analysis of 1,620 annotated genes shared by S. pneumoniae TIGR4 and other isolates.
Evolution of capsular polysaccharide diversity in S. pneumoniae.
Next, we tested the hypothesis that import of genes explains the extreme structural diversity of capsular polysaccharides in S. pneumoniae (n = 95), which has remained an enigma. The pneumococcal cps operons consist of 12 to 22 genes directly involved in synthesis and transport of the polysaccharides (7). Among these, the glycosyl transferases, glycosyl phosphotransferases, dehydrogenases, mutases, and epimerases are often unique to one or more serotypes and determine the distinct polysaccharide structure (26). We aligned each protein (n = 1575) encoded by the cps locus of the S. pneumoniae serotypes (7) to the NCBI nonredundant protein database. This provided evidence of extensive import of cps operon genes from numerous Streptococcus species, including other members of the mitis group (S. mitis, “Streptococcus mitis biovar 2,” S. oralis, S. infantis, Streptococcus sanguinis, Streptococcus parasanguinis, and Streptococcus peroris) and members of the more distant anginosus and salivarius groups. The number of genes imported from a single or several different donor species ranged from one gene to the entire cps locus (see Table S2 in the supplemental material). Imported genes included genes that were part of a cps operon in the donor, as well as genes with other glycosylation functions outside the cps locus.
The nucleotide identity between the putative donor and recipient cps genes ranged from 84 to 99%, presumably reflecting the time elapsed since the genetic transfer and/or the existence of donors not represented among the genome-sequenced streptococci. For instance, the membrane-associated flippase, responsible for transferring the oligosaccharide chains to the exterior of the pneumococcal membrane, is common to all pneumococcal cps operons except serotype 3 (7, 26). The genetic diversity among pneumococcal flippase genes is ~16 times larger than the overall diversity of the pneumococcal core genome (0.280% ± 0.056% versus 0.017% ± 0.003%), in support of a diverse origin of the gene among pneumococci (see Fig. S2 in the supplemental material). Alignments revealed >98% amino acid identities of flippases of several pneumococcal serotypes to those of strains of a range of Streptococcus taxa that are otherwise genetically more distant. The assumed direction of transfer was further supported by two gene-based observations. First, comparison of the genetic distance between genes from clonally independent strains of the respective serotypes of S. pneumoniae showed more conservation than among strains of donor species (Fig. 3). Second, several genes intact in the putative donor were pseudogenes in pneumococci. Comparison of serotypes belonging to the same serogroup (e.g., serogroups 7, 18, and 19) revealed that mutations resulting in pseudogenes, in some cases combined with import of additional genes from other donors or complete deletion of genes, have been driving the structural diversification within serogroups (see Fig. S3). As an example, an evolutionary model for the origin and diversification of the S. pneumoniae serogroup 19 is presented in Fig. 4.
FIG 3 .

Example of comparisons and genetic distances of cps locus genes among S. pneumoniae and S. mitis strains. The nucleotide sequence identity (%) of orthologous genes in S. pneumoniae serotype 2 and “S. mitis biovar 2” strains are shown for each flanking pair. Clonally independent strains of the respective serotypes of S. pneumoniae showed more conservation than strains of donor species, supporting the proposed transfer from S. mitis to S. pneumoniae.
FIG 4 .
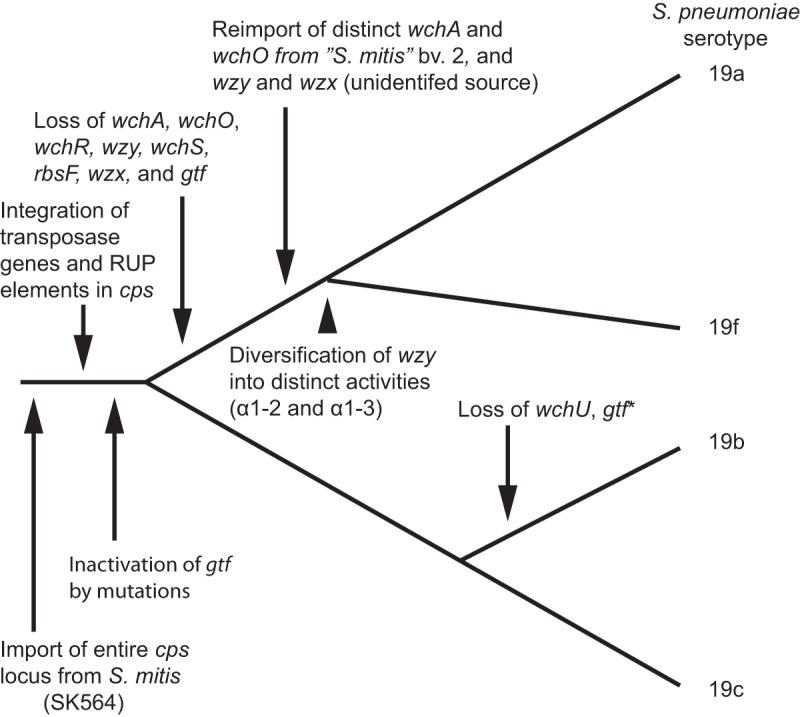
Phylogenetic model for acquisition and diversification of the four serogroup 19 S. pneumoniae serotypes. Acquisition of the entire capsular biosynthesis operon from S. mitis (SK564) introduced the serotype 19c capsule in S. pneumoniae. Subsequent incorporation of transposase and RUP sequences in the operon facilitated transfer to other strains of S. pneumoniae, in which allelic replacement with selected genes acquired from “S. mitis biovar 2” (this taxon is erroneously classified as a biovar of S. mitis), loss of genes, and gene mutations resulted in the structurally distinct capsular polysaccharides of serotypes 19b, 19a, and 19f. A detailed comparison of the cps operons of the four serogroup 19 serotypes is shown in Fig. S4 in the supplemental material.
Parallel evolution of genome plasticity and genome stability.
These findings indicate that interspecies gene transfer between S. pneumoniae and neighboring species is unidirectional, i.e., from other species to S. pneumoniae. This is supported by further observations. Competence for genetic transformation in pneumococci depends on 22 genes (27). Screening of the 35 genomes identified all 22 genes in all genomes of S. pneumoniae and S. pseudopneumoniae, whereas only 10 out of 15 S. mitis strains and none of the S. oralis strains possessed all. Up to 3 of the 22 essential genes were missing or significantly truncated in some strains (see Table S3 in the supplemental material), suggesting reduced or lack of transformation competence.
Several other genes facilitate the incorporation of foreign genetic elements in the pneumococcal genome. S. pneumoniae strains possess one of two complementary Dpn restriction-modification systems, DpnI or DpnII (28), that are part of the competence (com) regulon. It was recently demonstrated that induction of this system is necessary for optimal pathogenicity island transfer (29). While present in all S. pneumoniae strains, the majority of S. mitis strains lacked intact dpn loci (see Fig. S4 and Table S1 in the supplemental material). The genes were either missing in this location and in other parts of the genome or replaced by other genes, such as a transposase and an integrase in strain SK667. When present, alignments of the S. mitis Dpn locus genes with those of S. pneumoniae showed that they are ancestral genes diversified in parallel with other parts of the respective genomes. Interestingly, a third version of the locus (here termed DpnIII) was demonstrated in S. pneumoniae ATCC 700669, S. mitis SK578, and S. oralis ATCC 35037. In these strains, two genes on opposite strands and encoding a restriction enzyme resembling MutH of Escherichia coli and a DNA (cytosine-5-)-methyltransferase family protein constituted the locus. Other strains of S. oralis lacked Dpn locus genes. These observations suggest that the Dpn-associated function is under deterioration in S. mitis and S. oralis.
Transposases are widely used in bacteria to facilitate intra- and interstrain mobility of genes or islands of genes (30). The 13 S. pneumoniae strains possessed from 19 to 111 (median, 77) such elements distributed over the entire genome (Fig. 2), although some are degenerate, in agreement with their constant adaptation to the transforming genome. Notably, transposases are associated with cps operons of all pneumococcal serotypes, in most cases flanking the entire operon (7). Although most S. mitis and all S. oralis strains examined had complete cps operons, none included transposases. In general, S. mitis and S. oralis genomes harbored significantly fewer transposase genes (median number, 8) (see Table S1 in the supplemental material). One exception was S. mitis strain B6 (31), which in several ways, including the genome size, is exceptional among S. mitis strains.
Like transposases, repeat elements, including RUP (repeat units of pneumococcus) are assumed to facilitate genomic plasticity in addition to phase variation of genes (32, 33). In addition to facilitation of traditional homologous recombination, a recent report demonstrated that pneumococci can also generate diversity by transformation with fully homologous “self” DNA by generating a variety of merodiploids within a population facilitated by alternative pairing of repeat regions present in different parts of the genome (34). Analysis of the 35 genomes showed that pneumococcal genomes had 53 to 63 RUP elements, including one or two within the cps locus of all serotypes (except serotypes 5, 11a, and 23b), while S. mitis strains had either none or no more than three elements in the entire genome (see Table S1 in the supplemental material).
Bacterial defense systems against attack by foreign DNA include the clustered, regularly interspaced short palindromic repeat (CRISPR) loci. In agreement with a recent report (29), none of the S. pneumoniae possessed CRISPR sequences. This corroborates the finding that CRISPR loci artificially inserted into a pneumococcal genome were spontaneously ejected when under environmental stress (35). Likewise, the S. pseudopneumoniae strains did not possess CRISPR/Cas systems. However, 5 of the 15 S. mitis strains and 4 of the 5 S. oralis strains possessed CRISPR sequences (see Table S1 in the supplemental material). A few of the spacers showed sequence similarity to bacteriophage/prophage sequences, most of which are Streptococcus specific and in some cases are integrated in S. pneumoniae and S. pseudopneumoniae genomes (not shown).
DISCUSSION
One factor in the coevolution of obligate symbionts of humans that has so far received little attention is the impact of the susceptible host population size. This factor is of particular importance in pathogenic (i.e., parasitic) species that induce immunity or sometimes death, leaving the host nonaccessible for repeated colonization. Thus, successful survival of the pathogen requires a sizeable host population of sufficient density to allow spread between susceptible hosts and/or a capacity of the pathogen for constant antigenic change. In contrast, commensals that achieve a mutualistic lifestyle induce a tolerogenic response in the host’s immune system, allowing continued colonization and intimate and potentially lifelong association (36). Many species of the genus Streptococcus are almost exclusively adapted to humans and other hominids. S. pneumoniae is one of the most important pathogens affecting humans (2). Although it is a widespread colonizer particularly of children in day care centers, both colonization and infection result in rapid elimination (median duration, 19 days) by antibodies directed to the capsular polysaccharide and presumably other surface-exposed antigens (37). In contrast, the closely related S. mitis is a lifelong companion of all humans in the upper respiratory tract and is often present as mixed populations of multiple clones (38, 39). We have previously demonstrated that the two species share an immediate ancestor and have argued that the ancestor was a pneumococcus-like species presumably pathogenic to the immediate ancestor of hominids (3). The genome-based data obtained in this study support this model. Our results, furthermore, illustrate how a significant selection pressure resulting from a shortage of potential hosts (40) was handled by the S. pneumococcus-S. mitis-S. pseudopneumoniae ancestor in two opposing ways occurring in parallel. S. pneumoniae maintained its pathogenic potential, which facilitates horizontal spread, and optimized its genome plasticity (17). In contrast, harmonious coexistence by the majority of lineages becoming S. mitis was achieved by elimination of properties that challenge the host combined with increased genome stability (i.e., partial loss of competence genes, transposases, repeat elements, and the Dpn restriction-modification system, combined with acquisition of CRISPR/Cas sequences). Interestingly, these S. mitis lineages are now highly diverse and, according to traditional taxonomic standards, would represent separate species (3). An important factor in this diversification process has been the ecological and genetic isolation of clones colonizing distinct lineages of human hosts combined with a vertical spreading pattern. Our demonstration of various levels of loss of virulence-associated factors and properties contributing to genome plasticity among the examined strains of S. mitis (Fig. 2; see also Table S1 in the supplemental material) indicates that this is an ongoing process brought to different degrees of completion by individual S. mitis lineages. Future studies may reveal if this is reflected in the occasional ability of S. mitis strains to cause bacteremia or endocarditis in groups of predisposed patients (41, 42). Another result of the need of S. pneumoniae to expand its ecological niche may be the adaptation of certain clones to an equine host, which also included loss of virulence-associated genes (43).
Availability of a critical population of potential hosts (40) became an evolutionary bottleneck to the pathogen, reflected in the significant homogeneity of the core genome of today’s pneumococcus (Fig. 1). In addition to the expression of crucial virulence properties, life as a pathogen of the S. pneumoniae lineage required optimal genome plasticity, enabling antigenic diversity of surface structures. For example, the relative sequence diversification of the paralogous zinc metalloproteases IgA1 protease, ZmpB, and ZmpD is striking evidence of significantly enhanced selection for diversification of surface-exposed proteins in the pathogen S. pneumoniae compared to the closely related commensal streptococci (16). In addition to homologous recombination within the population of pneumococci, our results show that the need for diversification was remarkably solved by its continued exploitation of the gene pool of neighboring species. In some S. pneumoniae strains, up to 9% of the alleles of genes were imported from S. mitis (Table 1). This is an ongoing process facilitated by its colonization of an ecological niche, albeit briefly, where it frequently meets multiple members of related commensal species that serve as a genetic toolbox. Most remarkable is our finding that the previously enigmatic diversity of capsular polysaccharide structures expressed by S. pneumoniae is a direct result of gene import from several species of commensal streptococci, including S. mitis, the “S. mitis biovar 2” (mislabeled since it is more closely related to S. oralis [4]), S. oralis, S. infantis, S. sanguinis, S. parasanguinis, S. peroris, and members of the more distant anginosus and salivarius groups (see Table S3 in the supplemental material). In several serotypes, complete cps loci had been imported from a single donor, in some cases in several independent steps. In others, a mosaic of genes imported from distinct donors was evident. Contributing to the diversification that constitutes distinct serotypes belonging to the same serogroup (e.g., serogroups 7, 18, and 19) were mutations resulting in pseudogenes, import of additional genes from other donors, or complete deletion of genes (Fig. 3; see also Fig. S4). This process conceivably will continue to result in additional antigenic diversity that may challenge the currently successful prevention of pneumococcal infections by vaccination.
This is the first demonstration of how selective pressures resulting from a shortage of potential hosts was solved by bacteria in two opposing ways occurring in parallel. Harmonious coexistence by lineages becoming S. mitis was achieved by elimination of properties that challenge the host combined with increased genome stability. Life as a pathogen of the S. pneumoniae lineage required optimal genome plasticity combined with antigenic diversity of surface structures, including capsular polysaccharides, a challenge remarkably solved by its continued exploitation of the gene pool of neighboring species. More recently, success of the S. pneumoniae lineage reflected in the lineage-specific boost of the pneumococcus population has been ensured by the dramatic expansion of the susceptible host population.
MATERIALS AND METHODS
Bacterial genomes.
The 35 streptococcal genomes examined in the study are listed in Table S1 in the supplemental material together with NCBI accession numbers. A total of 11 genomes sequenced as a part of this study were generated using the 454 platform (GS20, FLX, and/or Titanium) and assembled with the Newbler assembler. Details on the libraries constructed, sequencing coverage, and assembler version used are available in the GenBank entries.
Alignment of genomes.
A multiple whole-genome nucleotide alignment of contigs or complete chromosomes from the 35 whole genomes was generated using the software program Mugsy (44), and clusters of syntenic orthologs across the genomes were obtained with Mugsy-Annotator (45).
Phylogenetic analyses.
A phylogenetic tree based on the concatenated core genome sequences from the Mugsy alignment was generated using the minimum-evolution algorithm according to the maximum composite likelihood model in the software program MEGA 5.2 (46) and validated by bootstrap analysis based on 500 replications. Recombination in selected genes was visualized using the program SplitsTree 4 (47).
Bioinformatics tools and analyses.
Annotated genome sequences from the 35 genomes (see Table S1 in the supplemental material) and Mugsy-Annotator clusters of syntenic orthologs were loaded into the Sybil comparative genomics software package (48) for comparative analyses.
To determine the extent of recombination between S. pneumoniae and the related commensal species, we aligned nucleotide sequences within Mugsy-Annotator clusters and generated minimum-evolution phylogenetic trees in MEGA5.2. A total of 1,620 trees (excluding transposases and genes unique to S. pneumoniae strains) were generated and manually examined.
The presence or absence of annotated genes based on Mugsy-Annotator clusters was detected in Sybil and confirmed by blastn analysis (49). Figure 2 was generated by loading profiles of gene presence and absence into the MeV interface (50). RUP (repeated unit of pneumococcus) elements were identified by searching TIGR4 RUP sequences (32) with blastn against the 35 genomes.
Genetic distances, i.e., the number of base substitutions per site from averaging over all sequence pairs, were determined in MEGA5.2 using the maximum composite likelihood model (51) based on aligned single genes or concatamers of six multilocus sequence type (MLST) genes of S. pneumoniae (52).
CRISPR regions were identified using the CRISPR finder tool (http://crispr.u-psud.fr).
Nucleotide sequence accession numbers.
The Whole Genome Shotgun projects have been deposited at DDBJ/EMBL/GenBank under the following accession numbers: Streptococcus mitis SK137, JPFS00000000; Streptococcus mitis SK271, JPGW00000000; Streptococcus mitis SK1126, JPFT00000000; Streptococcus mitis SK629, JPFU00000000; Streptococcus mitis SK667, JPFV00000000; Streptococcus mitis SK642, JPFW00000000; Streptococcus mitis SK637, JPFX00000000; Streptococcus mitis SK578, JPFY00000000; Streptococcus mitis SK608, JPFZ00000000; Streptococcus oralis SK141, JPGA00000000; Streptococcus oralis SK143, JPGB00000000. The versions described in this paper are versions XXXX01000000.
SUPPLEMENTAL MATERIAL
Phylogenetic trees based on aligned nucleotide sequences of selected genes in S. pneumoniae and related species. The tree in panel A was generated in MEGA 5.2 using the minimum-evolution algorithm, and the numbers on branches represent bootstrap values. Trees in panels B, C, and D were generated with the SplitsTree 4 software program. (A) The position of the pneumolysin gene (ply) from two strains of S. mitis and one strain of S. pseudopneumoniae distant from the cluster of S. pneumoniae gene sequences shows that the ply genes are ancestral genes that have been diversifying in parallel with other parts of the respective genomes. (B) Clustering of several S. pneumoniae gene sequences among S. mitis genes (indicated by arrows) is evidence of transfer from S. mitis to S. pneumoniae strains. The intermediary position of strain P1031 illustrates transfer of part of the gene. (C and D) Tree generated in SplitsTree, illustrating extensive intra- and interspecies recombination between genes encoding the penicillin-binding protein 1A (orthologs of SP_0369) (C) and the neighboring gene encoding recombination protein U (orthologs of SP_0370) (D). Download
Phylogenetic tree based on aligned amino acid sequences of the flippase protein involved in capsular polysaccharide biosynthesis. The tree was generated in MEGA 5.2 using the minimum-evolution algorithm. The tree illustrates significant sequence diversity and clustering of S. pneumoniae sequences with sequences of flippases from distantly related Streptococcus species, indicated by arrows. The bar indicates the genetic distance. Download
Comparison of cps operon structures in S. mitis SK564 and in S. pneumoniae operons encoding serotypes of serogroup 19. Gray connecting boxes indicate genes that were part of the same cluster of syntenic orthologs. The S. mitis SK564 and S. pneumoniae serotype 19c operons are identical apart from the transposon gene (tnp) and the mutated glf gene in the latter. Genes in serotype 19f and 19a operons presented in hatched color represent allelic replacements relative to SK564 and serotype 19c and 19b acquired from different donors, including “S. mitis biovar 2” (this taxon is erroneously classified as a biovar of S. mitis). Download
Organization of the Dpn locus in representative S. pneumoniae, S. mitis, and S. pseudopneumoniae strains. S. pneumoniae strains possess one of two complementary Dpn restriction-modification systems, DpnI or DpnII. The DpnI organization consists of dpnI, encoding an atypical restriction enzyme cleaving methylated double-stranded DNA, and dpnD, of unknown function. The DpnI locus is shown for strain TIGR4 and is representative of the S. pneumoniae strains Taiwan19F, JJA, R6, D39, CGSP14, and TCH8431/19A. The DpnII organization, consisting of the methylase DpnIIA, the DpnIIB restrictase, and the DpmM double-stranded DNA methylase, shown for S. pneumoniae P1031, is also found in S. pneumoniae strains 670, 70585, and Hungary19A. Only two S. mitis strains (SK321 and SK629) possessed the DpnI locus. However, in both strains the dpnD gene was disrupted by a frameshift, and in addition, SK321 harbored an in-frame stop codon. Three S. mitis strains (B6, SK1126, and SK597) had a DpnII-like locus encoding DpmM and DpnIIA and a type II restriction enzyme (MjaIII) distinct from the DpnIIB restrictase of pneumococcal strains. In strains B6 and SK1126, a gene encoding a 270-amino-acid (aa) protein showing 89% amino acid identity to a hypothetical protein in Streptococcus sp. HPH0090 interspersed the gene encoding this restrictase and DpnA, while a 622-bp sequence with no open reading frame or homology to any sequence in the NCBI database took this place in SK597. In the majority of S. mitis strains, the dpn genes were either missing in this location and in other parts of the genome or replaced by other genes, such as a transposase and an integrase in strain SK667. S. mitis SK578 and S. pneumoniae ATCC 700669 had neither of the two described Dpn operons either in the usual region or in any other parts of the genome. Instead, both had two genes in opposite directions encoding a restriction enzyme resembling MutH of Escherichia coli and a DNA (cytosine-5-)-methyltransferase family protein presumably constituting a third version of the Dpn locus (DpnIII). The same locus structure was identified in S. oralis strain ATCC 35037, whereas other strains of S. oralis lacked these genes (not shown). In S. pseudopneumoniae strain IS7493, the locus included two transposase genes flanking a gene encoding a lipid A core-O-antigen-ligase-like enzyme. However, homologues of DpnIII genes were found elsewhere in the genome. The S. infantis genome showed no evidence of dpn genes. Alignments of the Dpn locus genes in S. pneumoniae and S. mitis strains showed that they are ancestral genes diversified in parallel with other parts of the respective genomes. Download
Streptococcus genomes examined in this study with selected characteristics
S. pneumoniae capsular polysaccharide biosynthesis (cps) operon genes imported from other bacterial species and their respective donors; due to their widespread occurrence and conservation, the regulatory genes wzg, wzh, wzd, and wze and the four rhamnose pathway genes (rmlA to rmlD) are not included; the figures represent the sequence identity of the gene to that of the donor relative to the sequence identity of the core genomes of the recipient and S. pneumoniae TIGR4
Presence in S. mitis, S. oralis, and S. pseudopneumoniae of genes shown to be essential in S. pneumoniae for transformation competence
ACKNOWLEDGMENTS
This work was supported by grant 10-083748 from the Danish Research Council for Health and Disease and internal funds from the University of Maryland School of Medicine. We acknowledge the use of the pneumococcal MLST database, which is located at Imperial College London and is funded by the Wellcome Trust.
We thank members of the Genomics Resource Center and the Informatics Resource Center at the Institute for Genome Sciences for their support.
M.K. and H.T. designed the research. D.R.R. established the Sybil comparative genomics database. M.K., A.J., H.B., and H.T. performed the analyses. M.K. and H.T. wrote the first draft of the manuscript, which was edited and approved by all authors.
Footnotes
Citation Kilian M, Riley DR, Jensen A, Brüggemann H, Tettelin H. 2014. Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. mBio 5(4):e01490-14. doi:10.1128/mBio.01490-14.
REFERENCES
- 1. Mitchell TJ. 2003. The pathogenesis of streptococcal infections: from tooth decay to meningitis. Nat. Rev. Microbiol. 1:219–230. 10.1038/nrmicro771 [DOI] [PubMed] [Google Scholar]
- 2. Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5:685–694. 10.1016/S1473-3099(05)70267-X [DOI] [PubMed] [Google Scholar]
- 3. Kilian M, Poulsen K, Blomqvist T, Håvarstein LS, Bek-Thomsen M, Tettelin H, Sørensen UB. 2008. Evolution of Streptococcus pneumoniae and its close commensal relatives. PLoS One 3:e2683. 10.1371/journal.pone.0002683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bishop CJ, Aanensen DM, Jordan GE, Kilian M, Hanage WP, Spratt BG. 2009. Assigning strains to bacterial species via the internet. BMC Biol. 7:3. 10.1186/1741-7007-7-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitchell AM, Mitchell TJ. 2010. Streptococcus pneumoniae: virulence factors and variation. Clin. Microbiol. Infect. 16:411–418. 10.1111/j.1469-0691.2010.03183.x [DOI] [PubMed] [Google Scholar]
- 6. Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6:288–301. 10.1038/nrmicro1871 [DOI] [PubMed] [Google Scholar]
- 7. Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, Donohoe K, Harris D, Murphy L, Quail MA, Samuel G, Skovsted IC, Kaltoft MS, Barrell B, Reeves PR, Parkhill J, Spratt BG. 2006. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2:E31. 10.1371/journal.pgen.0020031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Plosker GL. 2013. 13-Valent pneumococcal conjugate vaccine: a review of its use in infants, children, and adolescents. Paediatr. Drugs 15:403–423. 10.1007/s40272-013-0047-z [DOI] [PubMed] [Google Scholar]
- 9. Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434. 10.1126/science.1198545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Croucher NJ, Finkelstein JA, Pelton SI, Mitchell PK, Lee GM, Parkhill J, Bentley SD, Hanage WP, Lipsitch M. 2013. Population genomics of post-vaccine changes in pneumococcal epidemiology. Nat. Genet. 45:656–663. 10.1038/ng.2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wyres KL, Lambertsen LM, Croucher NJ, McGee L, von Gottberg A, Liñares J, Jacobs MR, Kristinsson KG, Beall BW, Klugman KP, Parkhill J, Hakenbeck R, Bentley SD, Brueggemann AB. 2013. Pneumococcal capsular switching: a historical perspective. J. Infect. Dis. 207:439–449. 10.1093/infdis/jis703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Claverys JP, Prudhomme M, Mortier-Barrière I, Martin B. 2000. Adaptation to the environment: Streptococcus pneumoniae, a paradigm for recombination-mediated genetic plasticity? Mol. Microbiol. 35:251–259. 10.1046/j.1365-2958.2000.01718.x [DOI] [PubMed] [Google Scholar]
- 13. Morrison DA, Lee MS. 2000. Regulation of competence for genetic transformation in Streptococcus pneumoniae: a link between quorum sensing and DNA processing genes. Res. Microbiol. 151:445–451. 10.1016/S0923-2508(00)00171-6 [DOI] [PubMed] [Google Scholar]
- 14. Johnsborg O, Håvarstein LS. 2009. Regulation of natural genetic transformation and acquisition of transforming DNA in Streptococcus pneumoniae. FEMS Microbiol. Rev. 33:627–642. 10.1111/j.1574-6976.2009.00167.x [DOI] [PubMed] [Google Scholar]
- 15. Claverys JP, Håvarstein LS. 2007. Cannibalism and fratricide: mechanisms and raisons d’être. Nat. Rev. Microbiol. 5:219–229. 10.1038/nrmicro1613 [DOI] [PubMed] [Google Scholar]
- 16. Bek-Thomsen M, Poulsen K, Kilian M. 2012. Occurrence and evolution of the paralogous zinc metalloproteases IgA1 protease, ZmpB, ZmpC, and ZmpD in Streptococcus pneumoniae and related commensal species. mBio 3(5):e00303-12. 10.1128/mBio.00303-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnston C, Campo N, Bergé MJ, Polard P, Claverys JP. 2014. Streptococcus pneumoniae, le transformiste. Trends Microbiol. 22:113–119. 10.1016/j.tim.2014.01.002 [DOI] [PubMed] [Google Scholar]
- 18. Sauerbier J, Maurer P, Rieger M, Hakenbeck R. 2012. Streptococcus pneumoniae R6 interspecies transformation: genetic analysis of penicillin resistance determinants and genome-wide recombination events. Mol. Microbiol. 86:692–706. 10.1111/mmi.12009 [DOI] [PubMed] [Google Scholar]
- 19. Coffey TJ, Dowson CG, Daniels M, Spratt BG. 1993. Horizontal spread of an altered penicillin-binding protein 2B gene between Streptococcus pneumoniae and Streptococcus oralis. FEMS Microbiol. Lett. 110:335–339. 10.1111/j.1574-6968.1993.tb06345.x [DOI] [PubMed] [Google Scholar]
- 20. Dowson CG, Coffey TJ, Kell C, Whiley RA. 1993. Evolution of penicillin resistance in Streptococcus pneumoniae; the role of Streptococcus mitis in the formation of a low affinity PBP2B in S. pneumoniae. Mol. Microbiol. 9:635–643. 10.1111/j.1365-2958.1993.tb01723.x [DOI] [PubMed] [Google Scholar]
- 21. Fitzsimmons S, Evans M, Pearce C, Sheridan MJ, Wientzen R, Bowden G, Cole MF. 1996. Clonal diversity of Streptococcus mitis biovar 1 isolates from the oral cavity of human neonates. Clin. Diagn. Lab. Immunol. 3:517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnston C, Hinds J, Smith A, van der Linden M, Van Eldere J, Mitchell TJ. 2010. Detection of large numbers of pneumococcal virulence genes in streptococci of the mitis group. J. Clin. Microbiol. 48:2762–2769. 10.1128/JCM.01746-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Madhour A, Maurer P, Hakenbeck R. 2011. Cell surface proteins in S. pneumoniae, S. mitis and S. oralis. Iran. J. Microbiol. 3:58–67 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3279804/ [PMC free article] [PubMed] [Google Scholar]
- 24. Rolo D, Simões AS, Domenech A, Fenoll A, Liñares J, de Lencastre H, Ardanuy C, Sá-Leão R. 2013. Disease isolates of Streptococcus pseudopneumoniae and non-typeable S. pneumoniae presumptively identified as atypical S. pneumoniae in Spain. PLoS One 8:e57047. 10.1371/journal.pone.0057047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506. 10.1126/science.1061217 [DOI] [PubMed] [Google Scholar]
- 26. Aanensen DM, Mavroidi A, Bentley SD, Reeves PR, Spratt BG. 2007. Predicted functions and linkage specificities of the products of the Streptococcus pneumoniae capsular biosynthetic loci. J. Bacteriol. 189:7856–7876. 10.1128/JB.00837-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peterson SN, Sung CK, Cline R, Desai BV, Snesrud EC, Luo P, Walling J, Li H, Mintz M, Tsegaye G, Burr PC, Do Y, Ahn S, Gilbert J, Fleischmann RD, Morrison DA. 2004. Identification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol. Microbiol. 51:1051–1070. 10.1046/j.1365-2958.2003.03907.x [DOI] [PubMed] [Google Scholar]
- 28. Lacks SA, Mannarelli BM, Springhorn SS, Greenberg B. 1986. Genetic basis of the complementary DpnI and DpnII restriction systems of S. pneumoniae: an intercellular cassette mechanism. Cell 46:993–1000. 10.1016/0092-8674(86)90698-7 [DOI] [PubMed] [Google Scholar]
- 29. Johnston C, Martin B, Granadel C, Polard P, Claverys JP. 2013. Programmed protection of foreign DNA from restriction allows pathogenicity island exchange during pneumococcal transformation. PLoS Pathog. 9:e1003178. 10.1371/journal.ppat.1003178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dobrindt U, Hochhut B, Hentschel U, Hacker J. 2004. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2:414–424. 10.1038/nrmicro884 [DOI] [PubMed] [Google Scholar]
- 31. Denapaite D, Brückner R, Nuhn M, Reichmann P, Henrich B, Maurer P, Schähle Y, Selbmann P, Zimmermann W, Wambutt R, Hakenbeck R. 2010. The genome of Streptococcus mitis B6—what is a commensal? PLoS One 5:E9426. 10.1371/journal.pone.0009426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oggioni MR, Claverys JP. 1999. Repeated extragenic sequences in prokaryotic genomes: a proposal for the origin and dynamics of the RUP element in Streptococcus pneumoniae. Microbiology 145:2647–2653 [DOI] [PubMed] [Google Scholar]
- 33. Saunders NJ, Jeffries AC, Peden JF, Hood DW, Tettelin H, Rappuoli R, Moxon ER. 2000. Repeat-associated phase variable genes in the complete genome sequence of Neisseria meningitidis strain MC58. Mol. Microbiol. 37:207–215. 10.1046/j.1365-2958.2000.02000.x [DOI] [PubMed] [Google Scholar]
- 34. Johnston C, Caymaris S, Zomer A, Bootsma HJ, Prudhomme M, Granadel C, Hermans PW, Polard P, Martin B, Claverys JP. 2013. Natural genetic transformation generates a population of merodiploids in Streptococcus pneumoniae. PLoS Genet. 9:e1003819. 10.1371/journal.pgen.1003819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bikard D, Hatoum-Aslan A, Mucida D, Marraffini LA. 2012. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 12:177–186. 10.1016/j.chom.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 36. Sansonetti PJ. 2011. To be or not to be a pathogen: that is the mucosally relevant question. Mucosal Immunol. 4:8–14. 10.1038/mi.2010.77 [DOI] [PubMed] [Google Scholar]
- 37. Ekdahl K, Ahlinder I, Hansson HB, Melander E, Mölstad S, Söderström M, Persson K. 1997. Duration of nasopharyngeal carriage of penicillin-resistant Streptococcus pneumoniae: experiences from the South Swedish Pneumococcal Intervention Project. Clin. Infect. Dis. 25:1113–1117. 10.1086/516103 [DOI] [PubMed] [Google Scholar]
- 38. Hohwy J, Reinholdt J, Kilian M. 2001. Population dynamics of Streptococcus mitis in its natural habitat. Infect. Immun. 69:6055–6063. 10.1128/IAI.69.10.6055-6063.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bek-Thomsen M, Tettelin H, Hance I, Nelson KE, Kilian M. 2008. Population diversity and dynamics of Streptococcus mitis, Streptococcus oralis, and Streptococcus infantis in the upper respiratory tract of adults, determined by a nonculture strategy. Infect. Immun. 76:1889–1896. 10.1128/IAI.01511-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cavalli-Sforza LL, Feldman MW. 2003. The application of molecular genetic approaches to the study of human evolution. Nat. Genet. 33:266–275. 10.1038/ng1113 [DOI] [PubMed] [Google Scholar]
- 41. Bochud PY, Eggiman P, Calandra T, Van Melle G, Saghafi L, Francioli P. 1994. Bacteremia due to viridans streptococcus in neutropenic patients with cancer: clinical spectrum and risk factors. Clin. Infect. Dis. 18:25–31. 10.1093/clinids/18.1.25 [DOI] [PubMed] [Google Scholar]
- 42. Hoshino T, Fujiwara T, Kilian M. 2005. Use of phylogenetic and phenotypic analyses to identify non-hemolytic streptococci from bacteremic patients. J. Clin. Microbiol. 43:6073–6085. 10.1128/JCM.43.12.6073-6085.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Whatmore AM, King SJ, Doherty NC, Sturgeon D, Chanter N, Dowson CG. 1999. Molecular characterization of equine isolates of Streptococcus pneumoniae: natural disruption of genes encoding the virulence factors pneumolysin and autolysin. Infect. Immun. 67:2776–2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Angiuoli SV, Salzberg SL. 2011. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27:334–342. 10.1093/bioinformatics/btq665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Angiuoli SV, Dunning Hotopp JC, Salzberg SL, Tettelin H. 2011. Improving pan-genome annotation using whole genome multiple alignment. BMC Bioinformatics 12:272. 10.1186/1471-2105-12-272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- 48. Riley DR, Angiuoli SV, Crabtree J, Dunning Hotopp JC, Tettelin H. 2012. Using Sybil for interactive comparative genomics of microbes on the web. Bioinformatics 28:160–166. 10.1093/bioinformatics/btr652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378. 10.1016/S0076-6879(06)11009-5 [DOI] [PubMed] [Google Scholar]
- 51. Tamura K, Nei M, Kumar S. 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U. S. A. 101:11030–11035. 10.1073/pnas.0404206101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Enright M, Spratt BG. 1998. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology 144:3049–3060. 10.1099/00221287-144-11-3049 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic trees based on aligned nucleotide sequences of selected genes in S. pneumoniae and related species. The tree in panel A was generated in MEGA 5.2 using the minimum-evolution algorithm, and the numbers on branches represent bootstrap values. Trees in panels B, C, and D were generated with the SplitsTree 4 software program. (A) The position of the pneumolysin gene (ply) from two strains of S. mitis and one strain of S. pseudopneumoniae distant from the cluster of S. pneumoniae gene sequences shows that the ply genes are ancestral genes that have been diversifying in parallel with other parts of the respective genomes. (B) Clustering of several S. pneumoniae gene sequences among S. mitis genes (indicated by arrows) is evidence of transfer from S. mitis to S. pneumoniae strains. The intermediary position of strain P1031 illustrates transfer of part of the gene. (C and D) Tree generated in SplitsTree, illustrating extensive intra- and interspecies recombination between genes encoding the penicillin-binding protein 1A (orthologs of SP_0369) (C) and the neighboring gene encoding recombination protein U (orthologs of SP_0370) (D). Download
Phylogenetic tree based on aligned amino acid sequences of the flippase protein involved in capsular polysaccharide biosynthesis. The tree was generated in MEGA 5.2 using the minimum-evolution algorithm. The tree illustrates significant sequence diversity and clustering of S. pneumoniae sequences with sequences of flippases from distantly related Streptococcus species, indicated by arrows. The bar indicates the genetic distance. Download
Comparison of cps operon structures in S. mitis SK564 and in S. pneumoniae operons encoding serotypes of serogroup 19. Gray connecting boxes indicate genes that were part of the same cluster of syntenic orthologs. The S. mitis SK564 and S. pneumoniae serotype 19c operons are identical apart from the transposon gene (tnp) and the mutated glf gene in the latter. Genes in serotype 19f and 19a operons presented in hatched color represent allelic replacements relative to SK564 and serotype 19c and 19b acquired from different donors, including “S. mitis biovar 2” (this taxon is erroneously classified as a biovar of S. mitis). Download
Organization of the Dpn locus in representative S. pneumoniae, S. mitis, and S. pseudopneumoniae strains. S. pneumoniae strains possess one of two complementary Dpn restriction-modification systems, DpnI or DpnII. The DpnI organization consists of dpnI, encoding an atypical restriction enzyme cleaving methylated double-stranded DNA, and dpnD, of unknown function. The DpnI locus is shown for strain TIGR4 and is representative of the S. pneumoniae strains Taiwan19F, JJA, R6, D39, CGSP14, and TCH8431/19A. The DpnII organization, consisting of the methylase DpnIIA, the DpnIIB restrictase, and the DpmM double-stranded DNA methylase, shown for S. pneumoniae P1031, is also found in S. pneumoniae strains 670, 70585, and Hungary19A. Only two S. mitis strains (SK321 and SK629) possessed the DpnI locus. However, in both strains the dpnD gene was disrupted by a frameshift, and in addition, SK321 harbored an in-frame stop codon. Three S. mitis strains (B6, SK1126, and SK597) had a DpnII-like locus encoding DpmM and DpnIIA and a type II restriction enzyme (MjaIII) distinct from the DpnIIB restrictase of pneumococcal strains. In strains B6 and SK1126, a gene encoding a 270-amino-acid (aa) protein showing 89% amino acid identity to a hypothetical protein in Streptococcus sp. HPH0090 interspersed the gene encoding this restrictase and DpnA, while a 622-bp sequence with no open reading frame or homology to any sequence in the NCBI database took this place in SK597. In the majority of S. mitis strains, the dpn genes were either missing in this location and in other parts of the genome or replaced by other genes, such as a transposase and an integrase in strain SK667. S. mitis SK578 and S. pneumoniae ATCC 700669 had neither of the two described Dpn operons either in the usual region or in any other parts of the genome. Instead, both had two genes in opposite directions encoding a restriction enzyme resembling MutH of Escherichia coli and a DNA (cytosine-5-)-methyltransferase family protein presumably constituting a third version of the Dpn locus (DpnIII). The same locus structure was identified in S. oralis strain ATCC 35037, whereas other strains of S. oralis lacked these genes (not shown). In S. pseudopneumoniae strain IS7493, the locus included two transposase genes flanking a gene encoding a lipid A core-O-antigen-ligase-like enzyme. However, homologues of DpnIII genes were found elsewhere in the genome. The S. infantis genome showed no evidence of dpn genes. Alignments of the Dpn locus genes in S. pneumoniae and S. mitis strains showed that they are ancestral genes diversified in parallel with other parts of the respective genomes. Download
Streptococcus genomes examined in this study with selected characteristics
S. pneumoniae capsular polysaccharide biosynthesis (cps) operon genes imported from other bacterial species and their respective donors; due to their widespread occurrence and conservation, the regulatory genes wzg, wzh, wzd, and wze and the four rhamnose pathway genes (rmlA to rmlD) are not included; the figures represent the sequence identity of the gene to that of the donor relative to the sequence identity of the core genomes of the recipient and S. pneumoniae TIGR4
Presence in S. mitis, S. oralis, and S. pseudopneumoniae of genes shown to be essential in S. pneumoniae for transformation competence