

Abstract
Background
Apolipoprotein E (ApoE) is the major apolipoprotein present in the high-density lipoprotein-like particles in the central nervous system (CNS). ApoE is involved in various protective functions in CNS including cholesterol transport, anti-inflammatory, and antioxidant effects. An ApoE peptide would be expected to exert protective effects on neuroinflammation.
Objective
To determine the effects of an ApoE mimetic peptide Ac-hE18A-NH2 on amyloid-β pathology.
Method
Using human APP/PS1ΔE9 transgenic mice and in vitro studies, we have evaluated the effect of an ApoE mimetic peptide, Ac-hE18A-NH2, on amyloid plaque deposition and inflammation.
Results
Administration of Ac-hE18A-NH2 to APP/PS1ΔE9 mice for 6 weeks (50 μg/mouse, 3 times a week) significantly improved cognition with a concomitant decrease in amyloid plaque deposition and reduced activated microglia and astrocytes, and increased brain ApoE levels. Oligomeric Aβ42 (oAβ42) and oxidized PAPC (ox-PAPC) inhibited secretion of ApoE in U251 cells, a human astrocyte cell line, and this effect was ameliorated in the presence of peptide Ac-hE18A-NH2. The peptide also increased Aβ42 uptake in a cell line of human macrophages.
Conclusions
Peptide Ac-hE18A-NH2 attenuates the effects of oxidative stress on ApoE secretion, inhibits amyloid plaque deposition, and thus could be beneficial in the treatment of Alzheimer’s disease.
Keywords: Alzheimer’s disease, amyloid-β, apolipoprotein E, blood-brain barrier, oxidized phospholipid, peptide mimetics
INTRODUCTION
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder characterized by the deposition of amyloid-β (A-β) plaques and neurofibrillary tangles (NFTs) in the brain [1]. Increased levels of A-β oligomers and synaptic impairment are thought to play a pivotal role in AD pathogenesis [2]. Microglia participate in the innate immunity in the brain, and they are also considered to be the major cell type responsible for inflammation-mediated neurotoxicity [3]. Early microglial activation appears to delay progression of AD-like pathology [4]. However, as AD progresses, microglia become dysfunctional and fail to phagocytose and/or degrade A-β, further contributing to disease progression [5]. Recent large scale genome wide association studies indicate that both inflammation and lipid metabolism are involved in the development of sporadic, late-onset AD [6].
Apolipoprotein E (ApoE) is a protein consisting of 299 amino acids with a molecular mass of 34 kDa. It has two domains, the N-terminal domain (residues 1–191) containing the receptor binding region and the C-terminal domain (residues 223–299) containing the lipid binding region [7]. As a component of lipoproteins, it is involved in cholesterol and lipid transport and metabolism [8, 9]. Recently it has also been shown to possess anti-inflammatory and antioxidant properties [10, 11]. The liver is the major site for synthesis and secretion of peripheral ApoE [12]. ApoE in the central nervous system (CNS) is synthesized and secreted by astrocytes and microglia, since it cannot cross the blood-brain barrier (BBB) [13]. In humans ApoE has three major protein isoforms (ApoE2, ApoE3, and ApoE4). The association of the ε4 allele of ApoE as a genetic risk factor for AD has been well established, accounting for between 50–60% of the genetic variation in the disease [14, 15]. Although the mechanisms by which ApoE isoforms affect risk of AD are not entirely understood, there is strong evidence that ApoE isoforms differentially modulate Aβ metabolism and accumulation. Since ApoE has a crucial role in Aβ-dependent and Aβ-independent AD pathogenic pathways, it is logical to consider strategies that regulate its expression and function. The genotype-dependent decrease in CNS ApoE levels mirrors the relative risk of developing AD, suggesting that low levels of total ApoE exhibited by ε4 carriers [16, 17] and by human ApoE4 knock-in mice [18–20], may directly contribute to the disease progression, perhaps due to reduced ApoE function in lipid metabolism, synaptic repair, and Aβ clearance. Thus, increasing the expression of ApoE irrespective of genotype might prevent or slow the progression of AD.
Based on the observation that ApoE possesses dual domains, Ac-hE18A-NH2, a 28 amino acid residue peptide, was designed in our laboratory. This ApoE mimetic peptide contains the cationic putative receptor-binding region of human ApoE (hE; residues 141–150, LRKLRKRLLR) covalently linked to the class A amphipathic helical model peptide 18A (DWLKAFYDKVAEKLKEAF). Details of the design of this peptide have been previously described [21, 22]. Peptide Ac-hE18A-NH2 reduces plasma cholesterol levels in dyslipidemic animal models and inhibits atherosclerotic plaque formation [23]. Analogous to ApoE, this peptide has also been shown to possess anti-inflammatory and recycling properties and capacity to efflux cellular cholesterol [24].
As inflammation and cholesterol are linked to AD, we hypothesized that the dual-domain anti-inflammatory peptide would exert protective effects in a mouse model of AD. Here we show that Ac-hE18A-NH2 inhibits amyloid deposition when administered to APP/PS1ΔE9 transgenic mice with a concomitant decrease in the number of activated microglia and astrocytes. Using an in vitro system we have shown for the first time that soluble oligomeric Aβ42 as well as oxidative stress inhibits ApoE secretion by astrocytes, and that this is ameliorated in the presence of peptide Ac-hE18A-NH2.
MATERIALS AND METHODS
Materials
Peptide Ac-hE18A-NH2 with the sequence Ac-LRKLRKRLLRDWLKAFYDKVAEKLKEAF-NH2 was synthesized by the solid phase peptide synthesis method using fluorenylmethyloxycarbonyl (FMOC) amino acids and suitable protected amino acids as described previously [24]. The peptides were purified by preparative HPLC, and the purity and identity of the peptides were determined by analytical HPLC and mass spectrometry. For obtaining 14C-labeled peptides, during the last step of the synthesis 14C-labeled acetic acid (American Radiolabeled Chemicals, Inc. St. Louis, MO) was used to acetylate the N-terminus of the peptide instead of normal acetic acid. Oxidized lipids (ox-PAPC) were prepared by 72 h air oxidation of palmytoylarachadonyl phosphatidyl choline (PAPC) purchased from Avanti Polar Lipids (Birmingham, AL). Aβ42 was purchased from EZbiolab Inc, USA and soluble oligomeric Aβ42 was prepared as described [25].
Animals
Male APP/PS1ΔE9 mice were purchased from Jackson Laboratories, Bar Harbor, ME (Strain name B6C3-Tg [APPswe,PSEN1ΔE9]85Db0/J; stock number 004462) [26]. A breeding colony was established by breeding male APP/PS1ΔE9-mice with female B6C3F1/J (Jackson Laboratories, Bar Harbor, ME). The animals were genotyped for the presence of trans-gene by PCR amplification of genomic DNA extracted from 1 mm tail clippings. Two groups of 4 month old male APP/PS1ΔE9 were used for the study (n = 8 in each group). One group received peptide Ac-hE18A-NH2 by retro-orbital administration (50 μg/ mouse) 3 times a week for 6 weeks. The control group received an equal volume of saline. Mice were housed under standard conditions in conventional cages and were given standard rodent diet and water ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Water maze
Spatial memory was evaluated by the water maze test as described earlier [26]. Briefly, the mice were tested in a water maze consisting of a round pool (diameter, 112 cm) filled with water (22°C) to a height of 31 cm. An escape platform (8 cm diameter) was submerged 1/2 cm below the water surface in the Northeast (NE) quadrant. The acquisition of the spatial task consisted of placing the mouse in the water next to and facing the wall successively in south (S), north (N), east (E), and west (W) positions. In each trial, the mouse was allowed to swim until it found the hidden platform or until 60 s had elapsed, at which point the mouse was guided to the platform. The time required to find the hidden platform is referred to as escape latency. Shorter escape latency values represent improved cognitive function. The movement of the mice was tracked by the Ethovision system (Noldus, Leesburg) for four trials daily for 5 days. The day after completion of the acquisition phase, a “probe trial” was also conducted by removing the platform and placing the mouse facing the south side. The amount of time mice spent in the target quadrant, where the platform was present earlier, was measured in a single one minute trial.
Brain tissue preparation
Mice were anesthetized with ketamine/xylazine, and blood was collected by cardiac puncture. After whole-body perfusion with 0.9% saline, brains were harvested. The brains were cut sagittally into left and right hemispheres. The right hemisphere was fixed in buffered formaldehyde for histological analysis. The left hemisphere was snap frozen in liquid nitrogen and stored at −80°C until biochemical analysis.
Plasma cholesterol
Plasma total cholesterol levels were determined colorimetrically using commercial reagents (Infinity cholesterol reagent, Sigma, St. Louis, MO).
Enzyme-linked immunosorbent assay
The cortex and hippocampus was homogenized with 15 volumes of ice-cold tris-buffered saline (TBS; pH 7.4) with protease inhibitors (Sigmafast protease inhibitor tablets, Sigma). The sample was centrifuged at 20,000g for 20 min at 4°C. Supernatant (TBS extract) was transferred to a new tube and stored at − 80°C until analyzed. The pellet was washed with 50 μl of cold TBS. 400 μl of 5 M guanidine hydrochloride (GuHCl) containing complete protease inhibitor was added to the pellet. The sample was vortexed and incubated at room temperature for 4 h. Homogenate was spun at 20,000 rpm for 20 min at 4°C. The supernatant (guanidine extract) was transferred to new tubes and stored at −80°C until analyzed. Total protein levels in soluble and insoluble fractions were assayed using the BCA (bicinchoninic acid) protein assay reagent method (Pierce). Interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) in the TBS soluble fractions were measured using commercially available ELISA kits (R&D systems, Minneapolis, MN) according to manufacturer’s protocol. ApoE levels in the soluble fractions of brain homogenates were determined by commercially available mouse ApoE ELISA kit (TSZ ELISA, Framingham, MA) according to manufacturer’s protocol. No co-reaction with the peptide was detected by adding peptide in increasing levels to the ELISA plate in the absence of brain materials (data not shown). All values were expressed as amount per total protein.
Immunohistochemistry
Free floating sections were cut using a sliding, freezing microtome at 30 μm and the sections were used for immunohistochemical analysis. The primary antibody used for assessing amyloid load was 6E10 (a mono-clonal antibody raised against peptides 1–16 of Aβ, Covance, Dedham, MA) and visualized using Vectastain ABC kit (Vector laboratories, Burlingame, CA). Activated microglia were detected with rabbit polyclonal antibody to ionized calcium binding adaptor molecule-1 (Iba-1; Wako, 1:250). Activated astrocytes were detected using rabbit polyclonal antibody to glial fibrillary acidic protein (GFAP; Sigma, 1:100). Microglia and astrocytes were visualized using Vectastain rabbit IgG kit (Vector laboratories, Burlingame, CA) and Extra avidin Peroxidase kit (Sigma, St.Louis, MO) respectively.
Microscopic image analysis
Image analysis was performed on four coronal sections per brain. The sections were digitalized using a Nikon Eclipse E600 microscope with camera, and the images were converted to grayscale using the Paint Shop Pro 7 program. To analyze amyloid depositions and activated microglial and astrocyte burden, the area covered by Aβ (i.e., amyloid burden), Iba-1 positive area (i.e., activated microglia), and GFAP positive area (i.e., reactive astrocytes) were measured through the cortex and hippocampus using the Scion Image (NIH) program. Burden was defined as the percentage of stained surface of total area.
Tissue bioavailability
APP/PS1ΔE9 mice (n = 3) were injected with 14C-labeled Ac-hE18A-NH2. The injected volume was 200 μl at a concentration of 2.2 mg/ml with a specific activity of 148227 cpm/mg via tail vein. 6 h post administration, mice were perfused with 30 ml saline, brain harvested, homogenized and 14C radioactivity was counted.
Cell culture
The U251-MG astroglioma cell line (kind gift from Dr. Etty Benveniste) was grown in DMEM/Ham’s F-12 medium supplemented with 10 mM HEPES, 2 mM l-glutamine, 100 U/ml penicillin, 10 μg/ml streptomycin, and 10% FBS as described previously [27, 28]. Cells were treated with Ac-hE18A-NH2 at different concentrations for 24 h. The cell viability was assessed by commercially available TOX-1 kit (MTT assay, Sigma). THP-1 monocytes obtained from American Tissue Culture Collection (ATCC) were maintained in RPMI-1640 medium (ATCC, USA) containing 10% FBS, 2 mM HEPES, 1.5 g/L sodium bicarbonate, 50 U/ml penicillin, 50 μg/ml streptomycin, and 50 μM β-mercaptoethanol at 37°C in 5% CO2. The monocytes were differentiated into macrophages by the addition of phorbol myristate acetate.
Aβ42 uptake
THP-1 monocyte derived macrophages were washed twice with serum free media (SFM) and fresh SFM was added to the cells. Soluble Aβ42 was added to the media at concentration of 2 μg/ml along with peptide Ac-hE18A-NH2 at various concentrations. Cells treated with Aβ42 alone were considered as control. The cells were incubated at 37°C for 3 h. The medium was then removed and the cells were washed twice with PBS. To remove cell surface-bound Aβ, the cells were incubated with 0.05% trypsin/EDTA for 20 min. The cells were then pelleted by centrifugation and the pellet was washed twice with PBS. Following centrifugation, the cells were lysed in lysis buffer (containing Complete protease inhibitor, Roche) and incubated at 4°C for 30 min. The cell lysates were then cleared by centrifugation at 14,000× g. Protein content was measured using a BCA protein assay (Thermo Scientific). Aβ42 was quantitated using commercially available ELISA kit (Life Technologies).
Secretion of ApoE
To measure the ability of peptide to overcome the oxidized lipid-mediated inhibition of ApoE secretion, U251 astrocytes were incubated overnight with and without peptide (5 μg/ml) following pretreatment with or without different amounts of ox-PAPC or oAβ42 for 6 h. Amounts of ApoE secreted into the media were determined by human ApoE ELISA (Mabtech Inc., Cincinnati, OH). To determine whether the ELISA assay detects ApoE mimetic peptide, a control experiment was carried out using various concentrations of Ac-hE18A-NH2, and performing ELISA. The ApoE ELISA assay was specific for hApoE as it did not detect peptide (data not shown).
Statistical analysis
Data were expressed as mean ± SEM. Comparison of treatment groups were performed by student’s t-test. SigmaStat software (Systat Software Inc.) was used for all statistical analysis. p < 0.05 was considered statistically significant.
RESULTS
Administration of peptide Ac-hE18A-NH2 did not decrease plasma cholesterol levels
We chose the dose, frequency, and duration of peptide treatment (50 μg/mouse, 3 times per week for 6 weeks), based on the observation that in earlier experiments with the same parameters the peptide was able to inhibit atherosclerotic plaque formation in hyperlipidemic mouse models [23, 29]. Peptide Ac-hE18A-NH2 has been shown to lower plasma cholesterol dramatically in hyperlipidemic mouse models but not in C57BL/6J mice on normal diet [30]. Thus we analyzed plasma cholesterol levels in saline and peptide administered mice at baseline and after 6 weeks of treatment. These mice are normolipidemic with high-density lipoprotein being the major component. In agreement with our earlier observations [30], we did not find differences between plasma cholesterol values in saline (from 141.3 ± 8.0 mg/dl at baseline to 156.2 ± 12.5 mg/dl at euthanasia) versus peptide administered mice (from 165.3 ± 10.3 mg/dl to 170.9 ± 13.8 mg/dl). Retro-orbital (RO) administration is one way of administering the peptide intravenously, and this mode of administration has a distinct advantage over tail vein administration that it allows multiple administrations. We compared the known effect of peptide Ac-hE18A-NH2 on plasma cholesterol by injecting ApoE null mice with same dose of the peptide either via tail vein or RO. The 5 min and 5 h change in plasma cholesterol levels were identical compared to baseline in both route of administration [tail vein: 52 ± 4% (5 min) and 14 ± 1% (5 h) and RO: 52 ± 3% (5 min) and 17.2 ± 2% (5 h)]. Thus we have administered peptide Ac-hE18A-NH2 via RO in studies using multiple injections.
Administration of peptide improves cognitive function
One week prior to the end of the experiment, the mice were assessed for their spatial memory and retention in the water maze. This test is widely used to evaluate spatial learning and memory ability in rats and mice [31]. As shown in Fig. 1A, mice treated for 6 weeks with the ApoE mimetic peptide Ac-hE18A-NH2 showed improvement in their spatial memory by day 5 of the test period by finding the hidden platform significantly faster than saline treated mice (p < 0.05). Retention of the memory for the location of the platform was tested in a probe trial in which on the 6th day after starting the training, the platform was removed and mice were allowed to swim in the pool for 60 s. The amount of time mice spent in the target quadrant was noted. The peptide treated mice spent more time than saline treated mice in the target quadrant, showing a trend for improved memory retention (Fig. 1B), suggesting that the ApoE mimetic peptide improved memory acquisition with a trend toward improved memory retention.
Fig 1.
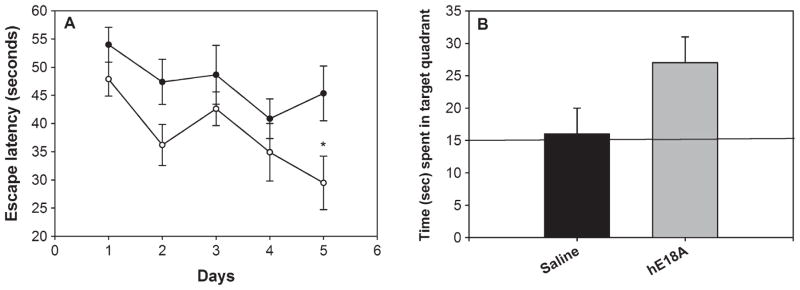
ApoE mimetic peptide improves behavioral deficits in APP/PS1ΔE9 mice. Mice were treated with either saline (n = 8) or Ac-hE18A-NH2 (n = 8) (50 μg/day), 3 times a week for 6 weeks via retro-orbital sinus (RO). A) Mice treated with peptide (open circle) found the hidden platform in the water maze test significantly faster than control mice (closed circle) (p < 0.05) by day 5. Each point represents the average of four daily trials. B) During the probe trial, where the mice were allowed to swim for 60 s without the hidden platform, peptide treated mice (grey bar) spent more time in the quadrant where the platform was previously hidden, compared to control mice (dark bar). The line indicates the chance level (25%) of performance. Values are mean ± SEM. *p < 0.05.
ApoE mimetic peptide inhibits amyloid plaque deposition
We next evaluated Aβ immunoreactivity in brain sections from control and peptide-treated mice using antibody 6E10 which recognizes amino acids 1–17 of human Aβ [32]. As shown in Fig. 2A and B, Ac-hE18A-NH2 administered mice have fewer plaques in the dorsal cortex and hippocampus compared to control mice treated with saline. Quantitative analysis of the amyloid burden, defined as the percentage of area occupied by immunoreactive stain in the measurement field (Fig. 2C), showed statistically significant differences between the control and peptide treated groups (p < 0.05) in both the cortex and hippocampus.
Fig 2.

Administration of peptide Ac-hE18A-NH2 inhibits deposition of amyloid plaque. Brain coronal sections were immunostained with 6E10 antibody for amyloid load. A, B: Representative photomicrographs of saline and peptide treated mice respectively. C: Quantification of percent amyloid load, showing significant improvement in the peptide treated group (n = 8) compared to saline treated group (n = 8). Values expressed as mean ± SEM. *p < 0.05.
Gliosis is inhibited by Ac-hE18A-NH2
Ac-hE18A-NH2 has been shown to possess anti-inflammatory properties [23, 24]. Thus, in addition to the analysis of Aβburden, we evaluated the treatment effect of peptide Ac-hE18A-NH2 on inflammatory responses, characterized by activated microglia and reactive astrocytes which has been shown to be associated with AD [33]. Brain sections were immunostained with Iba-1 (for activated microglia) and GFAP (for reactive astrocytes). As shown in Fig. 3A, transgenic mice treated with saline exhibited GFAP-positive astrocytes in the entire cortex and hippocampus. However, treatment with peptide for 6 week (Fig. 3B) was able to significantly inhibit reactive astrocytes by about 22% (p < 0.05). Similarly, compared with saline-treated mice, the number of Iba-1 immunopositive activated microglia in the peptide Ac-hE18A-NH2 treated mice were significantly reduced (Fig. 4, p < 0.05). Together, these results indicate that the peptide Ac-hE18A-NH2 can reduce the number of activated microglia and reactive astrocytes, corroborating the anti-inflammatory properties of this peptide as have been previously established [24].
Fig 3.

GFAP reactive astrocytes are reduced when mice are administered with apoE mimetic peptide. Reactive astrocytes were analyzed by staining with GFAP antibody. Photomicrographs of representative sections are shown (A: saline treatment; B: Ac-hE18A-NH2 treatment). Percentage GFAP staining demonstrated significant attenuation of activated gliosis by treatment with Ac-hE18A-NH2 (grey bar, n = 8) versus control (dark bar, n = 8) (C) (*p < 0.05). Error bars indicate SEM.
Fig 4.

Peptide Ac-hE18A-NH2 inhibits number of activated microglia. Iba-1 antibody was used to immunostain activated microglial cells. Representative photomicrographs (A: saline; B: peptide treatment) and quantitation of percent of activated microglial cells (Iba-1) (C) demonstrate a significant reduction in peptide treated mice (grey bar, n = 8) compared to control mice (dark bar, n = 8). Values are expressed as mean ± SEM. *p < 0.05.
Suppression of neuroinflammatory mediators by ApoE mimetic peptide
Neuroinflammation is a process resulting from an abnormally high or chronic activation of glia (microglia and astrocytes). “Overactive” states of glia result in increased secretion of pro-inflammatory and oxidative stress molecules such as interleukin 1β (IL-1β), IL-6, and tumor necrosis factor α (TNF-α), which can lead to neuronal damage and death. Neuronal damage or death can also induce glial activation, thereby resulting in a detrimental cycle of neuroinflammation. Thus we analyzed whether the protective effects exhibited by peptide administration on gliosis would translate to the inhibition of pro-inflammatory cytokines released by activated glial cells. For this we determined the levels of IL-6 and TNF-α in the TBS soluble fraction of the brain homogenates of control and peptide treated mice by ELISA. As shown in Fig. 5, both cytokines were decreased significantly in the peptide-treated group, indicating that the mechanism by which the peptide inhibits amyloid deposition may be due to its anti-inflammatory properties.
Fig 5.
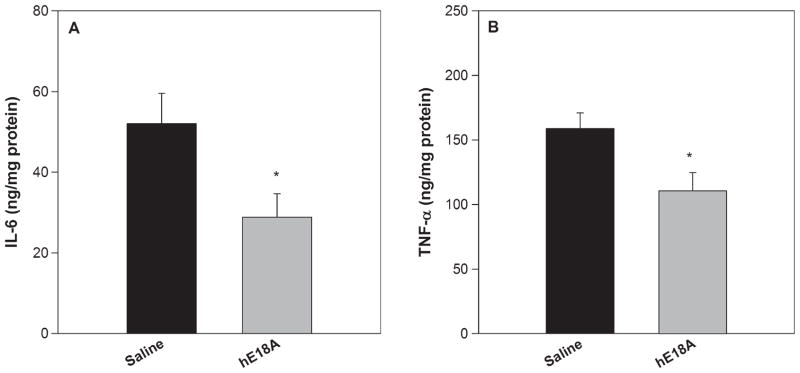
IL-6 and TNF-α levels are decreased in presence of the ApoE mimetic peptide. TBS soluble fractions of brain homogenates were analyzed for levels of IL-6 (A) and TNF-α (B) using ELISA. Treatment with Ac-hE18A-NH2 inhibited the secretion of both IL-6 and TNF-α significantly compared to control mice. Values are expressed as mean ± SEM. *p < 0.05.
Ac-hE18A-NH2 increases Aβ42 uptake in THP-1 monocyte derived macrophages
THP-1 human monocytes have been extensively used to investigate the Aβ-induced proinflammatory response and display a similar pattern of activation to that of microglial cells [34, 35]. It has been shown that aggregated Aβ42 induces TNF-α production from a human monocyte cell line via toll-like receptors [36]. As we have seen that peptide Ac-hE18A-NH2 inhibits TNF-α secretion, we wanted to determine the effect of peptide on Aβ42 uptake. For this we utilized THP-1 monocyte-derived macrophages. The cells were treated with Aβ42 at a concentration of 2 μg/ml in the presence and absence of various concentrations of peptide Ac-hE18A-NH2. As shown in Fig. 6, peptide Ac-hE18A-NH2 enhanced Aβ42 uptake significantly in a dose dependent manner compared to control. Even the lowest concentration of the peptide (1 μg/ml) enhanced Aβ42 uptake more than 2-fold compared to cells without Ac-hE18A-NH2.
Fig 6.
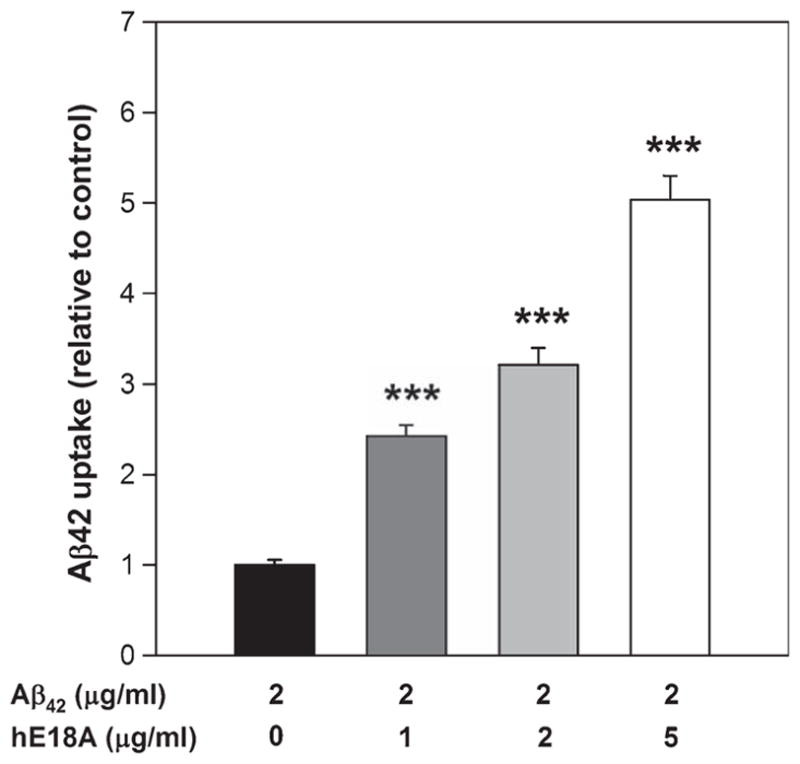
Peptide Ac-hE18A-NH2 enhances Aβ42 uptake by THP-1 monocyte derived macrophages. THP-1 monocyte derived macrophages were incubated with Aβ42 (2 μg/ml) in the presence and absence of various concentrations of peptide Ac-hE18A-NH2 for 3 h at 37°C. The cells were washed with PBS, incubated with trypsin to remove cell surface bound Aβ42, and lysed in lysis buffer. The cell-internalized Aβ42 was then assessed by ELISA. Values were calculated per mg protein of cell pellet and expressed relative to control cells. Mean ± SEM. N = 6 for each condition. ***p < 0.001.
ApoE mimetic peptide crosses BBB
Half-life of the peptide in plasma is 0.0266 h in rapid phase and 1.61 h in slow phase [30]. To analyze if the peptide can cross BBB and to quantitate the amount of peptide in brain, 14C- labeled Ac-hE18A-NH2 was used. 200 μl (2.2 mg/ml) of 14C- Ac-hE18A-NH2 with a specific activity of 148227 cpm/mg was injected into hAPP/PS1ΔE9 mice (n = 3) via tail vein, as we did not want to compromise the BBB. 6 h after administration, mice were anesthetized and perfused with saline and brain was harvested. Brain was immediately homogenized in a Dounce homogenizer with 60% acetonitrile/water, spun at 14,000 rpm for 30 min and the radioactivity in the supernatant was counted. A 6-h time point was used since we know from previous work that blood levels of peptide at this time point is negligible and would thus help avoid false positive results due to blood contamination [30]. We found 0.163 ± 0.011 μM peptide in the brain.
Peptide Ac-hE18A-NH2 is not toxic to glial cells
Administration of peptide for 6 weeks to APP/ PS1ΔE9 mice resulted in inhibition of amyloid plaque deposition with a concomitant decrease in number of activated glial cells and pro-inflammatory cytokines. Thus we wanted to further explore the anti-inflammatory properties of this peptide. To achieve this, we used U251 astrocytes, a human astrocytic cell line. We tested the toxicity of the peptide by treating U251 cells with different concentrations of peptide Ac-hE18A-NH2 (0 to 10 μg/ml) for 24 h and conducted an MTT assay to evaluate cell viability. The results indicated that there was no toxicity of the peptide compared to control at any of the tested concentrations (Fig. 7).
Fig 7.
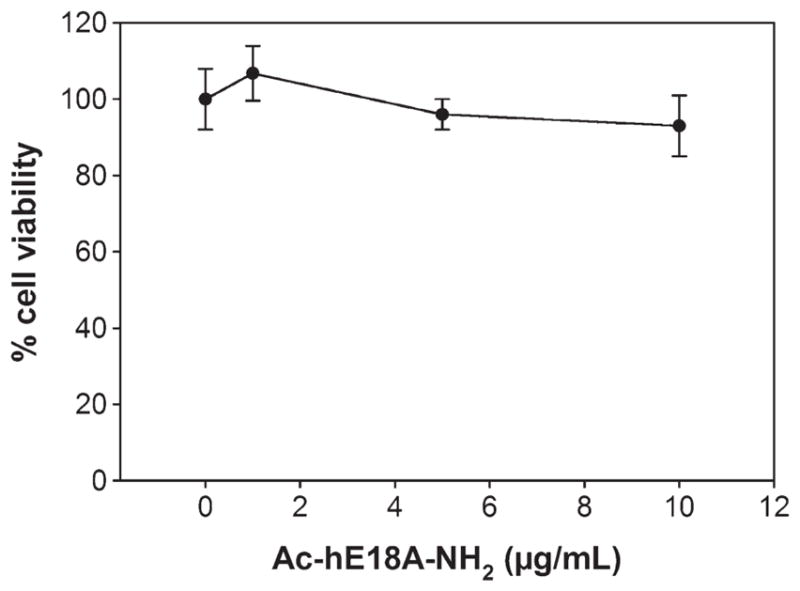
Ac-hE18A-NH2 is not toxic to astrocytes. Human astroglioma cell line U251 was treated with Ac-hE18A-NH2 at the indicated concentration for 24 h. The cell viability was assessed by MTT reduction assay (Sigma). The tested dose (up to 10 μg/ml) was not toxic to astrocytes. Values expressed as mean ± SD. n = 4 from at least 3 different experiments.
Peptide Ac-hE18A-NH2 increases ApoE levels
In the CNS, astrocytes are the major source of ApoE, which is lipidated by the transporter protein ABCA-1 and is involved in the transport of cholesterol, lipids, and Aβ peptide. As the peptide can cross the BBB, we wanted to analyze its effect on ApoE levels in CNS. ApoE levels in TBS soluble brain homogenates were determined by ELISA. As shown in Fig. 8, compared to mice that were treated with saline, ApoE levels were significantly increased in mice treated with peptide Ac-hE18A-NH2.
Fig 8.
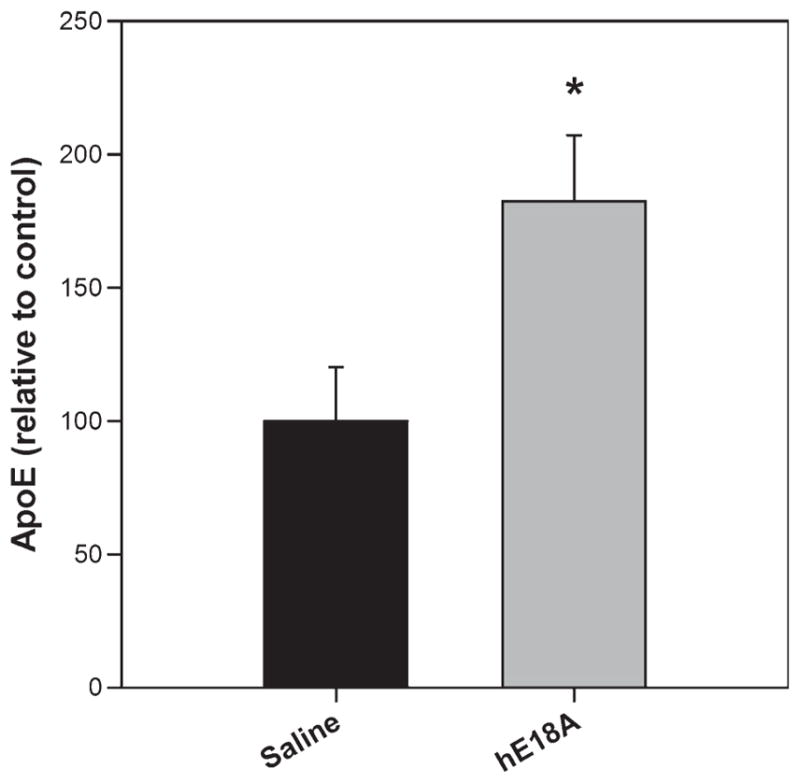
Ac-hE18A-NH2 increases ApoE levels. TBS soluble brain homogenate was analyzed for the amount of ApoE by ELISA. Mice treated with peptide Ac-hE18-NH2 had significantly higher levels of ApoE, compared to saline treated mice. Values are calculated per mg protein in brain homogenate and expressed as relative to saline treatment. n = 6 per condition. Values are expressed as mean ± SEM. *p < 0.05.
Oxidative stress inhibits ApoE secretion which is ameliorated by peptide Ac-hE18A-NH2. Recently we have shown that treating HepG2 cells or human THP-1 monocyte-derived macrophages with ox-PAPC resulted in inhibition of ApoE secretion, and that this was ameliorated by the addition of peptide Ac-hE18A-NH2 in a dose dependent manner [29]. As oxidative stress has been shown to be a component of AD, we speculated that the anti-inflammatory effects seen in mice treated with Ac-hE18A-NH2 could partly be due to enhanced ApoE secretion, which by itself has been shown to be anti-inflammatory [11]. Human U251 astrocytes were treated with different concentrations of either ox-PAPC (Fig. 9A) or oAβ42 (Fig. 9B) for 24 h and ApoE secreted into media was analyzed by ELISA. In agreement with our earlier observation in macrophages, secretion of ApoE was inhibited by ox-PAPC as well as oAβ42 treatments. However, the inhibitory effect on ApoE secretion was overcome when peptide Ac-hE18A-NH2 was added to the media for 18 h in a dose dependent manner after pre-incubation with ox- PAPC or oAβ42. Furthermore, treatment by Ac-hE18A-NH2 by itself was able to enhance ApoE secretion by about 25%.
Fig 9.
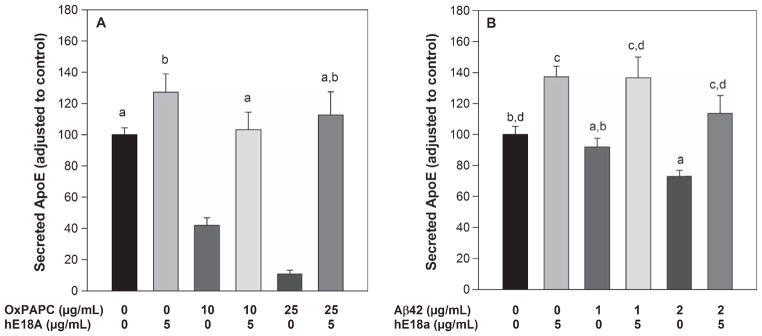
ApoE mimetic peptide Ac-hE18A-NH2 ameliorates the inhibitory effect of oAβ42 and Ox-PAPC on apoE secretion in astrocytes. U251 cells were treated with OxPAPC (A) or oligomeric Aβ42 (B) for 6 h at the indicated concentration. Cells were then treated with Ac-hE18A-NH2 at the indicated concentration for a further 18 h. At the end of 24 h, ApoE secreted into media was analyzed by ELISA. Cells without any treatment served as controls. Data were expressed as percentage of control. Values are mean ± SEM. n = 6 per treatment. Like letters (a, b, c, d) represents the groups that are NOT statistically significant from each other.
DISCUSSION
Previous work from our laboratory has shown that the ApoE mimetic peptide Ac-hE18A-NH2 possesses cholesterol-lowering and anti-inflammatory properties and is effective in inhibiting atherosclerotic plaque formation in dyslipidemic animal models [23, 24, 29]. In this study, we show that this peptide is also capable of inhibiting cognitive deficits observed in APP/PS1ΔE9 transgenic mice with a concomitant decrease in amyloid plaque deposition. Impaired acquisition and retention of spatial memory in APP/PS1ΔE9 transgenic mice is consistent with previous reports [37]. Administration of Ac-hE18A-NH2 reduced the escape latency significantly by day 5 of the water maze test (Fig. 1A) and showed a trend toward improved spatial memory retention (i.e., probe trial; Fig. 1B) compared to saline-treated APP/PS1ΔE9 transgenic mice, indicating improvement in spatial memory capability. Treatment with peptide also significantly reduced amyloid load, a neuropathological hallmark of AD (Fig. 2). This was accompanied by reduced pro-inflammatory cytokine levels, which are released by activated microglia (Fig. 5). Glial activation contributes to release of pro-inflammatory molecules which may at least in part cause neuronal toxicity observed in the disease [38]. We found that treatment with Ac-hE18A-NH2 resulted in a 13% reduction in the amount of activated microglia detected by staining for Iba-1 (Fig. 4) and a 22% decrease in reactive astrocytes detected by staining for GFAP (Fig. 3). Also, peptide Ac-hE18A-NH2 was able to significantly dampen the microglial release of pro-inflammatory factors IL-6 and TNF-α (Fig. 5) in accordance with earlier work by Datta et al. [24], which showed that peptide Ac-hE18A-NH2 inhibits LPS-induced VCAM-1, IL-6, and MCP-1 expression in human umbilical vein endothelial cells and THP-1 monocyte-derived macrophages. Thus, the protective effects we have observed could partly be due to the direct anti-inflammatory effects of the ApoE mimetic peptide. One drawback in our study is that we do not have a peptide control. However, earlier work in our lab has shown that a control peptide Ac-nhE18A-NH2 (with residues 151–160 of hApoE covalently linked to peptide 18A) had no effect on plasma cholesterol but enhanced aortic sinus lesion compared to Ac-hE18A-NH2 treated ApoE null mice [23].
There are several studies which suggest that reduced ApoE levels, observed in human ε4 carriers and in ApoE4 knock-in mouse models, might contribute to AD [16, 20]. Thus, the effect of peptide in enhancing neural levels of ApoE may be an additional mechanism by which the peptide may prevent or slow the progression of AD. To exert such an effect in the brain, it is important that some peptide crosses the BBB. We have shown that indeed the peptide Ac-hE18A-NH2 can cross BBB (0.13% of the injected dose). As this peptide has been shown to possess recycling properties, we speculate that the amount of peptide that enters the CNS is capable of exerting the beneficial effects. The observed increase in neural ApoE levels (Fig. 8) may reflect this.
Several studies have shown beneficial effects of LXR agonists [39, 40] and ApoE based therapies [41] in mouse models of AD, wherein treatment upregulates ApoE and ABCA-1 thereby inhibiting amyloid pathology. In these studies, total mouse ApoE levels were increased in the CNS and it is known that ApoE in mice and other species behaves functionally like human ApoE3, which is protective [42]. It is also possible that increased levels of ApoE4 may generate deleterious effects such as slowing Aβ clearance through the BBB [43], augmenting Aβ neurotoxicity [44]. Thus there is debate in the field as to whether it is better to increase [40, 41] or decrease [45, 46] human ApoE levels (regardless of isoform) to reduce Aβ level and improve AD pathology. However, in our studies peptide Ac-hE18A-NH2 has been shown to increase ApoE levels and improve amyloid pathology in a mouse model of AD.
Both Aβ and oxidative stress have been shown to play a significant role in the development and progression of AD. The appearance of oxidative stress markers are one of the earliest changes in AD brain [47]. Aβ induces oxidative stress in vivo and in vitro [48–50] and oxidative stress is itself is able to induce the increased production of Aβ [51–53]. Our studies show that peptide Ac-hE18A-NH2 enhances the uptake of Aβ 42 by THP-1 monocyte derived macrophages in a dose dependent manner (Fig. 6). In this study we have shown for the first time that ApoE secretion by astrocytes in cell culture is inhibited in the presence of oxidized lipids or in the presence of oligomeric Aβ 42 and treatment with the ApoE mimetic peptide overcomes this inhibitory effect (Fig. 9). Thus, the peptide-mediated enhanced secretion of ApoE may partly contribute to its anti-inflammatory property. Even though we have observed increased ApoE levels and concomitant improvement in amyloid pathology in mice treated with peptide Ac-hE18A-NH2, at present we do not know the direct contribution of peptide versus the beneficial effects of enhanced ApoE levels. Similarly, to better understand the role of ApoE levels per se in amyloid pathology, further studies are required wherein peptide is administered to AD model mice expressing different human ApoE isoforms and assess the resulting effect on amyloid pathology.
CONCLUSIONS
Administration of ApoE mimetic peptide Ac-hE18A-NH2 improves spatial memory and inhibits amyloid plaque deposition in a mouse model of AD. The ApoE mimetic peptide is also effective in reducing gliosis with a concomitant decrease in the levels of pro-inflammatory cytokines IL-6 and TNF-α and an increase in brain ApoE levels. These anti-inflammatory effects of peptide Ac-hE18A-NH2 may perhaps in part be due to its ability to enhance ApoE secretion which is known to be anti-inflammatory, or due to direct anti-inflammatory properties.
Acknowledgments
These studies were supported by Institute for the Study of Aging (S.P.H), grants R01 HL090803 and P01 HL34343 from the NIH, and by a gift from the Julius H. Caplan Charity Foundation, Inc. in memory of Miles D. Garber Jr., M.D. The behavioral studies were performed at the Behavioral Assessment core, Dept. of Neurobiology, UAB which is supported by P30 NS47466.
Footnotes
Authors’ disclosures available online (http://www.jalz.com/disclosures/view.php?id=1725).
References
- 1.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 4.Simrad AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 5.Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, Lossinsky AS, Graves MC, Gustavson A, Sayre J, Sofroni E, Suarez T, Chiappelli F, Bernard G. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis. 2005;7:221–232. doi: 10.3233/jad-2005-7304. [DOI] [PubMed] [Google Scholar]
- 6.Naj AC, Jun G, Beecham GW, Wang LS, Varadarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradley WA, Gilliam EB, Gotto AM, Jr, Gianturco SH. Apolipoprotein E degradation in human very low density lipoprotein by protease(s): Chemical and biological consequences. Biochem Biophys Res Commun. 1982;109:1360–1367. doi: 10.1016/0006-291x(82)91927-1. [DOI] [PubMed] [Google Scholar]
- 8.Mahley RW. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 9.Beisiegel U, Weber W, Inrke G, Herzx J, Stanley KK. The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature. 1989;341:162–164. doi: 10.1038/341162a0. [DOI] [PubMed] [Google Scholar]
- 10.Getz GS, Reardon CA. Apolipoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J Lipid Res. 2009;50:S156–S161. doi: 10.1194/jlr.R800058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jofrey-Monseny L, Minihane AM, Rimbach G. Impact of apoE genotype on oxidative stress, inflammation, and disease risk. Mol Nutr Food Res. 2008;52:131–145. doi: 10.1002/mnfr.200700322. [DOI] [PubMed] [Google Scholar]
- 12.Mahley RW, Rall SC., Jr Apolipoprotein E: Far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 13.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Rosed AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in the late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 15.Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004;25:641–650. doi: 10.1016/j.neurobiolaging.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 16.Ringman JM, Elashoff D, Geschwind DH, Welch BT, Gylys KH, Lee C, Cummings JL, Cole GM. Plasma signaling proteins in persons at genetic risk for Alzheimer disease: Influence of APOE genotype. Arch Neurol. 2012;69:757–764. doi: 10.1001/archneurol.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beffert U, Cohn JS, Petit-Turcotte C, Tremblay M, Aumont N, Ramassamy C, Davignon J, Poirier J. Apolipoprotein E and b-amyloid levels in the hippocampus and frontal cortex of Alzheimer’s disease subjects are disease-related and apolipoprotein E genotype dependent. Brain Res. 1999;843:87–94. doi: 10.1016/s0006-8993(99)01894-6. [DOI] [PubMed] [Google Scholar]
- 18.Bales KR, Liu F, Wu S, Lin S, Koger D, Delong C, Hansen JC, Sullivan PM, Paul SM. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck GW, Weeber EJ, Bu G, Yu C, LaDu MJ. ApoE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287:41774–41786. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anantharamaiah GM, Datta G, Garber DW. Toward the design of peptide mimetics of antiatherogenic apolipoproteins A-I and E. Curr Sci. 2001;81:53–65. [Google Scholar]
- 22.Sharifov OF, Nayyar G, Garber DW, Handattu SP, Mishra VK, Goldberg D, Anantharamaiah GM, Gupta H. Apolipoprotein E mimetics and cholesterol-lowering properties. Am J Cardiovasc Drugs. 2011;11:371–381. doi: 10.2165/11594190-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 23.Nayyar G, Handattu SP, Monroe CE, Chaddha M, Datta G, Mishra VK, Keenum TD, Palgunachari MN, Garber DW, Anantharamaiah GM. Two adjacent domains (141–150 and 151–160) of apo E covalently linked to a class A amphipathic helical peptide exhibit opposite atherogenic effects. Atherosclerosis. 2010;213:449–457. doi: 10.1016/j.atherosclerosis.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Datta G, White CR, Dashti N, Chaddha M, Palgunachari MN, Gupta H, Handattu SP, Garber DW, Anantharamaiah GM. Anti-inflammatory and recycling properties of an apolipoprotein mimetic peptide, Ac-hE18A-NH(2) Atherosclerosis. 2010;208:134–141. doi: 10.1016/j.atherosclerosis.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 26.Handattu SP, Garber DW, Monroe CE, van Groen T, Kadish I, Nayyar G, Cao D, Palgunachari MN, Li L, Anantharamaiah GM. Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2009;34:525–534. doi: 10.1016/j.nbd.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winkler MK, Benveniste EN. Transforming growth factor-β inhibition of cytokine-induced vascular cell adhesion molecule-1 expression in human astrocytes. GLIA. 1998;22:171–179. doi: 10.1002/(sici)1098-1136(199802)22:2<171::aid-glia8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 28.Lee Y-J, Benveniste EN. STAT-1α expression is involved in IFN-γ induction of the class II transactivator and class II MHC genes. J Immunol. 1996;157:1559–1568. [PubMed] [Google Scholar]
- 29.Handattu SP, Nayyar G, Garber DW, Palgunachari MN, Monroe CE, Keenum TD, Mishra VK, Datta G, Anantharamaiah GM. Two apolipoprotein E mimetic peptides with similar cholesterol reducing properties exhibit differential atheroprotective effects in LDL-R null mice. Atherosclerosis. 2012;227:58–64. doi: 10.1016/j.atherosclerosis.2012.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garber DW, Handattu S, Aslan I, Datta G, Chaddha M, Anantharamaiah GM. Effect of an arginine-rich amphipathic helical peptide on plasma cholesterol in dyslipidemic mice. Atherosclerosis. 2003;168:229–237. doi: 10.1016/s0021-9150(03)00101-1. [DOI] [PubMed] [Google Scholar]
- 31.Brandeis R, Brandys Y, Yehuda S. The use of the Morris Water Maze in the study of memory and learning. Int J Neurosci. 1989;48:29–69. doi: 10.3109/00207458909002151. [DOI] [PubMed] [Google Scholar]
- 32.Kim KS, Wen GY, Bancher C, Chen CMJ, Sapienza VJ, Hong H, Wisniewski HM. Detection and quantitation of amyloid beta-peptide with 2 monoclonal antibodies. Neurosci Res Commun. 1990;7:113–122. [Google Scholar]
- 33.Heneka MT, Sastre M, Dumitresscu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O’Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPAR g agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ 1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 34.Klegeris A, Walker DG, McGeer PL. Interaction of Alzheimer b-amyloid peptide with the human monocytic cell line THP-1 results in a protein kinase C-dependent secretion of tumor necrosis factor-a. Brain Res. 1997;747:114–121. doi: 10.1016/s0006-8993(96)01229-2. [DOI] [PubMed] [Google Scholar]
- 35.Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, Brunden KR. Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J Neurochem. 2000;74:1017–1025. doi: 10.1046/j.1471-4159.2000.0741017.x. [DOI] [PubMed] [Google Scholar]
- 36.Udan ML, Ajit D, Crouse NR, Nichols MR. Toll-like receptors 2 and 4 mediate Aβ (1–42) activation of the innate immune response in a human monocytic cell line. J Neurochem. 2008;104:524–533. doi: 10.1111/j.1471-4159.2007.05001.x. [DOI] [PubMed] [Google Scholar]
- 37.Malm TM, Livonen H, Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Kanninen K, Salminen A, Auriola S, Van Groen T, Tanila H, Koistinaho J. Pyrrolodine Dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting β-amyloid burden. J Neurosci. 2007;27:3712–3721. doi: 10.1523/JNEUROSCI.0059-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalaria RN. Microglia and Alzheimer’s disease. Curr Opin Hematol. 1999;6:15–24. doi: 10.1097/00062752-199901000-00004. [DOI] [PubMed] [Google Scholar]
- 39.Mandrekar-Colucchi S, Carlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated-receptor- g mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci. 2012;32:10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL. ATP-binding cassette transporter A1 mediates the beneficial effects of liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/ presenilin 1 mice. J Biol Chem. 2010;285:34144–34154. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Rstivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatters DM, Peters-Lebeu CA, Weisgraber KH. Apolipoprotein E structure: Insight into function. Trends Biochem Sci. 2006;31:445–454. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 43.Deane R, Sagare E, Hamm K, Parisi M, Lan S, Finn MB, Holtzman DM, Zlokovic BV. ApoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manelli AM, Bulfinch LC, Sullivan PM, LaDu MJ. Aβ 42 neurotoxicity in primary co-cultures: Effect of apoE isoform and Aβ conformation. Neurobiol Aging. 2007;28:1139–1147. doi: 10.1016/j.neurobiolaging.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bien-Ly N, Gillispie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. Neurobiol Dis. 2012;32:4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduce amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S, Smith MA. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol. 2000;59:1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- 48.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer’s disease. Proc Natl Acad Sci U S A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murakami K, Irie K, Ohigashi H, Hara H, Nagao M, Shimizu T, Shirasawa T. Formation stabilization model of the 42-mer Abeta radical: Implications for the long-lasting oxidative stress in Alzheimer’s disease. J Am Chem Soc. 2005;127:15168–15174. doi: 10.1021/ja054041c. [DOI] [PubMed] [Google Scholar]
- 50.Tabner BJ, El-Agnaf OM, Turnbull S, German MJ, Paleologou KE, Hayashi Y, Cooper LJ, Fullwood NJ, Allsop D. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem. 2005;280:35789–35792. doi: 10.1074/jbc.C500238200. [DOI] [PubMed] [Google Scholar]
- 51.Paola D, Domenicotti C, Nitti M, Vitali A, Borghi R, Cottalasso D, Zaccheo D, Odetti P, Strocchi P, Mainari UM, Tabaton M, Pronzato MA. Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and beta II PKCs in NT2 cells. Biochem Biophys Res Commun. 2000;268:642–646. doi: 10.1006/bbrc.2000.2164. [DOI] [PubMed] [Google Scholar]
- 52.Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 53.Tong Y, Zhou W, Fung V, Christensen MA, Qing H, Sun X, Song W. Oxidative stress potentiates BACE1 gene expression and Abeta generation. J Neural Transm. 2005;112:455–469. doi: 10.1007/s00702-004-0255-3. [DOI] [PubMed] [Google Scholar]