

Abstract
Purpose.
In many cell types, the E3 ubiquitin ligase, c-Cbl, induces ligand-dependent ubiquitylation of the epidermal growth factor receptor (EGFR) and targets the receptor for lysosomal degradation. The goal of this study was to determine whether c-Cbl is a negative regulator of EGFR in the corneal epithelium and if it can be inhibited to promote corneal epithelial homeostasis.
Methods.
Expression and activity of c-Cbl were blocked in immortalized human corneal epithelial cells (hTCEpi) using RNAi and pharmacological agents ([4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo-d-3,4-pyrimidine] or PP1). Following c-Cbl inhibition, cells were assessed for ligand-dependent receptor ubiquitylation, receptor phosphorylation, and in vitro wound healing. Subsequent experiments used PP1 in hTCEpi cells and monitored in vivo murine corneal epithelial wound healing.
Results.
Knockdown and inhibition of c-Cbl decreased ligand-dependent ubiquitylation of the EGFR and prolonged receptor activity as measured by tyrosine phosphorylation. Further, these treatments also increased the extent of ligand-dependent corneal epithelial wound healing in vitro and in vivo.
Conclusion.
Manipulating the duration of EGFR activity can enhance the rate of restoration of the corneal epithelial layer. Based on our findings, c-Cbl is a new therapeutic target to enhance EGFR-mediated corneal epithelial homeostasis that bypasses the limitations of previous approaches.
Keywords: EGFR, endocytic trafficking, corneal epithelium, corneal wound healing
The epidermal growth factor receptor (EGFR) is a critical component of corneal epithelial homeostasis; c-Cbl is a negative regulator of EGFR activity and inhibition of c-Cbl promotes EGFR-dependent corneal epithelial homeostasis.
Introduction
One of the keys to healthy vision is maintaining an intact corneal epithelium. It must be an impermeable barrier to exogenous agents (small particles, bacteria, viruses, and so forth) and fully differentiated such that it properly refracts light. There are a number of factors that can disrupt the integrity of the corneal epithelium, including injury, side effects from medications, and an indirect consequence of disease.1–4 The goal of this research was to identify the molecular mechanisms that maintain the homeostasis of the corneal epithelium with the long-term goal of developing novel therapeutic strategies to restore damaged tissue.
The epidermal growth factor receptor (EGFR) is a well-established mediator of corneal epithelial homeostasis.5,6 In experimental models, ligand-mediated activation of this receptor accelerates corneal epithelial wound healing.1,5,6 Patients taking inhibitors of the EGFR (i.e., erlotinib and cetuximab) as anticancer therapy have an increased incidence of corneal erosions.1,7 However, therapeutic administration of recombinant EGF has had mixed results in patients with compromised corneal epithelium.8–11 There are numerous potential explanations to explain the discrepancies between experimental and clinical data. One possibility is that the ligand-receptor complex undergoes downregulation after administration of exogenous growth factor. That is, on ligand binding, the ligand-receptor internalizes and is targeted for degradation, thereby limiting the magnitude and duration of EGFR signaling.12 Alternatively, the addition of exogenous ligand may not be effective because of the relatively high levels of endogenous EGF.13 With these potential explanations in mind, we have redirected our efforts to identify new ways of sustaining receptor activity once ligand is bound.
In this regard, we have targeted the endocytic pathway, a dynamic, cellular process that controls the level of cell surface receptor expression, the duration of receptor activity, and interaction with downstream signaling molecules.14 Endocytosis is a constitutive process that occurs both in the presence and absence of ligand binding; in the unliganded, inactive state, EGFR endocytosis occurs slowly and quickly recycles back to the plasma membrane. In the presence of growth factor, the ligand-receptor complex internalizes much more rapidly, and for many ligands, gets targeted to the lysosome for degradation, which effectively terminates signaling. For additional EGFR signaling to occur, the receptor must be newly synthesized and trafficked to the plasma membrane to make it accessible to extracellular growth factor. The generation of new receptors is a lengthy and energy-intensive process.
The route of EGFR endocytosis has been fairly well delineated.14 Receptors enter the cell via clathrin-enriched membrane domains that invaginate into the cell (called clathrin-coated pits) and subsequently pinch off to form intracellular clathrin-coated vesicles. The clathrin is shed from the vesicle and is recycled for further use; the resulting intermediate vesicle then fuses with an early endosome where it can be sorted for its cellular destination: recycling to the plasma membrane,15 the nucleus,16 endoplasmic reticulum,17 or lysosome for degradation.18 During this process, the EGFR gets ubiquitylated, allowing it to interact with the Endosomal Sorting Complexes Required for Transport (ESCRT) machinery.19 Specifically, the ubiquitylated EGFR binds to tumor suppressor gene 101 (TSG101, part of the ESCRT-1 complex), which guides the receptor from the limiting membrane of the late endosome/multivesicular body (LE/MVB) into interluminal vesicles (ILVs).20 A fusion reaction allows transfer of the contents of the LE/MVB (i.e., ILVs) to the lysosome for degradation.21
In cancer cells, EGFR ubiquitylation is mediated by the E3 ubiquitin ligase, cellular Casitas B-lineage Lymphoma gene (c-Cbl).22 Certain acute myeloid leukemias, as well as other cancers, are characterized by mutated c-Cbl, which prevents the downregulation of signaling proteins, like EGFR.23 The result is cells that are more aggressive in migration and proliferation. Here, we examine the role of c-Cbl as a regulator of EGFR degradation in human corneal epithelial cells and test the hypothesis that antagonizing c-Cbl activity will prolong EGFR activity and sustain its signaling.
In corneal epithelial cells, c-Cbl mediates ubiquitylation of the EGFR in a ligand-dependent manner. Further, inhibition of c-Cbl expression by RNA interference (RNAi) will attenuate ligand-dependent EGFR ubiquitylation, alter trafficking of the EGF-EGFR complex, and promote EGFR-mediated cell migration. Pharmacological antagonism of c-Cbl also decreases ligand-stimulated EGFR ubiquitylation and enhances EGF-stimulated corneal epithelial wound healing in vitro and basal corneal epithelial wound healing in vivo. Thus, c-Cbl is a promising new target to enhance corneal epithelial homeostasis.
Materials and Methods
Cell Culture
Human telomerase-immortalized corneal epithelial cells (hTCEpi) were obtained from Geron Corp. (Menlo Park, CA, USA), as described previously.24 Cells were maintained in growth media (Defined Keratinocyte Media with growth supplement; Invitrogen, Grand Island, NY, USA) containing 100 U/mL penicillin and 100 U/mL streptomycin at 37°C in 5% CO2.
Primary human corneal epithelial cells were derived from corneas that were unusable for transplantation (Oklahoma Lion's Eye Bank, Oklahoma City, OK, USA).15 Use of human tissue adhered to the tenets of the Declaration of Helsinki.
Generation of Lentivirus and Production of hTCEpi (-c-Cbl) Cells
A sequence targeting c-Cbl (5′-CCCGTACTATCTTGTCAAGATA-3′) was obtained from RNAi Codex.25 Sense and antisense oligos (IDT) were annealed and ligated into BfuA1 cut pEN_TTRmiRc2 (AddGene, Cambridge, MA, USA). Next, pEN_TTRmiRc2 and pSLIK Neo (AddGene) were in vitro recombined using Gateway LR Clonase II (Invitrogen) to create pSLIK shRNA c-Cbl Neo.26 Constructs were confirmed by DNA sequencing. Lentiviruses were prepared by calcium phosphate transfection of HEK293 cells plated on poly-l-lysine (Sigma, St. Louis, MO, USA)-coated plates with 20 μg transfer vector, 6 μg pMD2.G, 10 μg pMDL/RRE g/p, and 5 μg pRSV-REV per 10-cm plate. Cell supernatant was collected 48 hours and 72 hours posttransfection, filtered with a 0.2-μm filter, and precipitated using polyethylene glycol. Virus was resuspended in Dulbecco's modified Eagle's medium (DMEM) and added to cells in the presence of 8 μg/mL of polybrene (Sigma) overnight. Infected cells were grown and selected with hTCEpi cell growth media supplemented with 800 ng/mL G418 (Gibco/Invitrogen, Grand Island, NY, USA) for 2 weeks.
Cell Lysate Preparation and Immunoblotting
Cell lysates were generated and immunoblots were performed as described previously.27 Proteins were detected using antibodies against EGFR (SC-03; Santa Cruz Biotechnology, Santa Cruz, CA, USA), phosphotyrosine (pY99) (SC-7020; Santa Cruz Biotechnology), c-Cbl (Cell Signaling, Danvers, MA, USA), ubiquitin (Ub) (SC-8017; Santa Cruz Biotechnology), α-tubulin (T6199; Sigma), horseradish peroxidase (HRP)-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Pierce, Rockford, IL, USA). Immunoblots were visualized by enhanced chemiluminescence using a Fotodyne imaging system (Fotodyne, Inc., Hartland, WI, USA).
In Vitro Wound Healing Assays
In vitro wound healing was measured as previously described.13 The area was photographed at each time point using an Olympus IX70 microscope with a ×2 objective. Images were captured using QCapture Pro 6.0 software (QImaging, Surrey, British Columbia, Canada). The uncovered area at each time point was quantified using ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA). Scratch Assay and Boyden Chamber Assays (Corning Life Sciences, Tweksbury, MA, USA) were performed and quantified as previously described.15
Immunoprecipitation of Ubiquitylated EGFRs
Epidermal growth factor receptor ubiquitylation was monitored using a modification of a protocol by Visser Smit et al.28 Corneal epithelial cells (primary, hTCEpi, or hTCEpi [-c-Cbl]) were serum starved for 2 hours, treated with 100 ng/mL EGF in K-SFM for 10 minutes, and harvested in a chilled EGFR-UB lysis buffer (0.5% Triton x-100/50 mM Tris pH 7.5/150 mM NaCl/1 mM EDTA/1 mM sodium orthovanadate/10 mM sodium fluoride) supplemented with 2 mM phenylmethyl sulfonyl fluoride (PMSF) (Calbiochem, Billerica, MA, USA)/16 μM G5 Ubiquitin isopeptidase inhibitor I (Santa Cruz Biotechnology). Cell lysates were prepared and immunoprecipitated with 1 μg EGFR antibody, Ab-1, incubated at 4°C overnight followed by another 2-hour incubation at 4°C with protein A/G + Agarose (Santa Cruz Biotechnology). Immunoprecipitates were washed thrice in chilled EGFR-UB lysis buffer. Proteins were eluted with 6× SDS sample buffer and separated by electrophoresis on a 7.5% SDS-PAGE and immunoblotted for EGFR, Ub, pY99, or c-Cbl as indicated. Immunoblots were quantified using ImageJ software, taking care to make sure the exposures were in the linear range.
Radioligand Secretion
Cells were incubated for 7 minutes with 1 ng/mL 125I-EGF (Perkin Elmer, Waltham, MA, USA) at 37°C in binding buffer (DMEM/20 mM HEPES/0.1% bovine serum albumin [BSA], pH 7.3). Cells were washed twice with PBS and twice with binding buffer to remove unbound radioligand. Cells were incubated with prewarmed, 37°C binding buffer and returned to 37°C. At each time point, the media was collected (extracellular fraction) and the remaining cells were solubilized in 1% NP-40/20 mM Tris pH 7.4 (intracellular fraction). The amount of intact 125I-EGF was determined by precipitation with 10% trichloroacetic acid for 60 minutes at 4°C. Intact and soluble fractions were separated by centrifugation; associated radioactivity was determined using a Beckman Gamma Counter (Beckman Coulter, Inc., Brea, CA, USA). In Figure 3A, data are plotted as the percentage intact secreted radioligand (extracellular intact protein/[extracellular intact and extracellular degraded]). In Supplemental Figure S1, data are plotted as the amount of intracellular radioactivity/total cell associated radioactivity (extracellular + intracellular).
Figure 3.

The inducible knockdown of c-Cbl promotes EGF-EGFR recycling and enhances corneal epithelial cell migration. Parental hTCEpi (top) and hTCEpi (-c-Cbl) (bottom) cells were incubated without and with 1 μg/mL doxycycline for 72 hours. (A) Recycling of the 125I-EGF-EGFR complex was monitored by measuring the amount of secreted intact radioligand. Data are plotted as the average ± SEM percentage of intact, extracellular radioligand at each time point (n = 3). (B) Brightfield images of hTCEpi and hTCEpi (-c-Cbl) in an in vitro healing assay taken at 16 hours after treatment with or without EGF (1.6 nM) and doxycycline (1 μg/mL). (C) Cell migrations were analyzed as the difference from the plug's removal (0 hour) compared with the growth after 16 hours of treatment. Data are plotted as the average ± SEM percentage of area covered (wound healed) (n = 3). Analysis by a two-way ANOVA with Bonferroni post hoc test. ***P < 0.001, n ≥ 3 with three to five replicate values. (D) Representative images 8 hours post wounding from a Scratch assay using confluent dishes of parental hTCEpi cells or hTCEpi(-Cbl) cells (bottom). Doxycycline and EGF treatments are indicated. Quantification of Scratch assays at (E) 8 hours and (F) 16 hours after wounding. Data were analyzed with a two-way ANOVA with Bonferroni post hoc test. *P < 0.05; ****P < 0.001, n ≥ 3 with three to five replicate values.
FACS Analysis
DsRed-positive cells after 48 hours of induction with doxycycline (2 μg/mL) were enriched by an Influx Cell Sorter (BD Biosciences, Franklin Lakes, NJ, USA) at the University of Oklahoma Health Sciences Center Flow Cytometry and Imaging Facility. Cells were maintained with or without doxycycline for an additional 48 hours.
Cell Treatment With PP1
The Src inhibitor PP1 (4-amino-5-[4-methylphenyl]-7-[t-butyl]pyrazolo-d-3,4-pyrimidine) (EMD Millipore, Billerica, MA, USA) was solubilized in DMSO at a concentration of 50 mM. For tissue culture experiments, cells were incubated for 2 hours at 37°C with the indicated concentrations of PP1 (0.025 % DMSO was used as a control) in serum-free media.
ELISA for EGFR Phosphorylation Assay
High-binding ELISA plates (R&D Systems, Minneapolis, MN, USA) were coated with 100 μL 8 μg/mL EGFR antibody (EMD Millipore, Winooski, VT, USA) and incubated overnight at 4°C. The antibody coating was washed thrice with 1 × PBS-0.05%TWEEN and plates were incubated for 2 hours at room temperature with 300 μL 5% BSA/PBS blocking buffer. Lysates were treated with 10 ng/mL and EGF harvested in radioimmunoprecipitation assay buffer supplemented with 2 mM PMSF. Known concentrations of lysate were added to the plate, volumed to 100 μL with buffer, and incubated at 4°C overnight. The plate was washed three times and 100 μL antiphosphotyrosine antibody 4G10 (EMD Millipore) (250 ng/mL) was added. The plate was incubated 1 hour at room temperature, washed three times, and 100 μL HRP-conjugated goat anti-mouse (1:2500) added for 1 hour at room temperature. The plate was washed in buffer (three times), developed with ABTS HRP substrate (KPL, Gaithersburg, MD, USA), and stopped with 1% SDS. Absorbance was measured using Biotek plate reader at 405 nm.
In Vivo Murine Corneal Epithelial Wounding
Adult female C57BL6/J mice (Jackson Laboratory, Bar Harbor, ME, USA) between the ages of 8 and 10 weeks were anesthetized with an intraperitoneal injection of ketamine (50 mg/kg) and xylazine (5 mg/kg) (Butler Schein, Dublin, OH, USA). The central epithelium was demarcated with a 1.5-mm-diameter biopsy punch and removed with a 0.5-mm burr using the AlgerbrushII (Alger Company, Inc., Lago Vista, TX, USA) taking care not to disrupt the basement membrane.29 Wounded eyes were pretreated with eye drops containing PBS with DMSO (0.0001%) or with PP1 (10 nM) for 15 minutes; AG1478 (1 μg/mL) was part of the pretreatment solution where indicated. Following pretreatment, new eye drops composed of PBS with or without EGF (100 ng/mL) were applied to the wound. At each time point (0, 16, 24, 40 hours) the corneal wounds were visualized using sterile fluorescein sodium ophthalmic strips USP (Fluorets; Chauvin Laboratory, Aubenas, France) damped with sterile PBS. Wounds were examined and photographed at ×3 magnification with a stereoscopic zoom microscope (SMZ1000; Nikon, Tokyo, Japan) equipped with a digital sight DS-Fi2 camera (Nikon). At each time point, eyes were subjected to the same treatment as at time zero. The wound areas were measured using Image J software. All treatment of animals was in accordance with the ARVO statement for the use of animals in ophthalmic and vision research and approved by the University of Louisville Institutional Animal Care and Use Committee (IACUC#12046).
Results
To determine if the EGFR was ubiquitylated in response to EGF in corneal epithelial cells, both primary (HCEC) and immortalized (hTCEpi) cells were treated with and without EGF, solubilized, and the EGFR was immunoprecipitated (Fig. 1A). Immunoprecipitates were immunoblotted with antibodies against ubiquitin, phosphotyrosine, and total EGFR. Both primary and immortalized cells exhibited an EGF-dependent induction of EGFR ubiquitylation (Fig. 1B, top) and phosphorylation (Fig. 1B, bottom).
Figure 1.
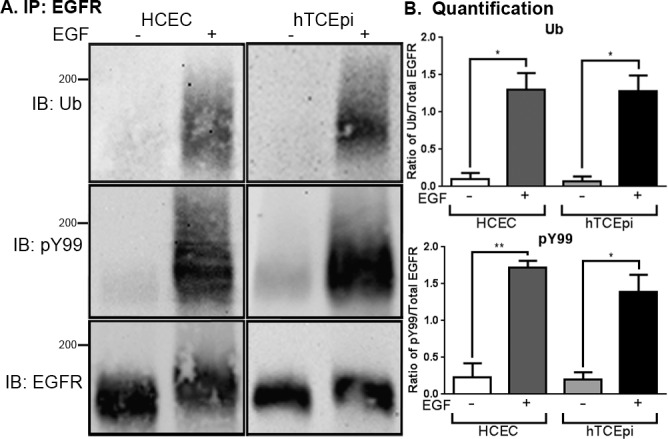
Epidermal growth factor–dependent ubiquitylation of the EGFR in primary and immortalized corneal epithelial cells. (A) Primary human corneal epithelial cells and immortalized corneal epithelial cells (hTCEpi) were treated without (−) or with (+) 100 ng/mL EGF for 10 minutes. Cell lysates were prepared and immunoprecipitated for the EGFR. The resulting immunoprecipitate was solved by 7.5% SDS-PAGE, and immunoblotted for ubiquitin (Ub), phosphotyrosine (pY99), or the EGFR; 200-kDa molecular weight standard is indicated on the left of the immunoblot. Shown in a representative experiment that was repeated three times. (B) Densitometric analysis of multiple experiments as performed in (A). Data are plotted as the ratio of ubiquitylated EGFR to total EGFR (top) and of phosphorylated EGFR to total EGFR (bottom) (average ± SEM; n = 3). Data were analyzed with a paired Student's t-test. *P < 0.05; **P < 0.01.
To determine if c-Cbl was mediating EGFR ubiquitylation, we generated stable cell lines with an inducible knockdown of c-Cbl and DsRed expression; therefore, induction of the shRNA can be monitored by DsRed expression. Clonal isolates of these cells (termed hTCEpi[-c-Cbl]) were generated after G418 treatment. Doxycycline-inducible expression of the DsRed can be seen in most of the cells after 72 hours (Fig. 2A). Although there is only a 50% decrease in c-Cbl expression (Figs. 2B, 2D, bottom), that knockdown was sufficient to decrease EGF-dependent receptor ubiquitylation approximately 60% (Figs. 2C, 2D, top). Importantly, there was no effect on ligand-stimulated EGFR phosphorylation (Figs. 2C, 2D, middle).
Figure 2.

Knockdown of c-Cbl reduces EGF-dependent ubiquitylation of the EGFR in corneal epithelial cells. Stable hTCEpi cells lines encoding for tetracycline-regulatable c-Cbl-specific shRNA and DsRed (hTCEpi[-c-Cbl]) were generated as described in Materials and Methods. Human telomerase-immortalized corneal epithelial cells were treated without (−Dox) or with (+Dox) 2 μg/mL of doxycycline in growth media for 72 hours. (A) Cell images were collected using a Nikon Ti-E microscope under Brightfield and CY5 settings. Identical settings were used for both treated and untreated cells. (B) Varying amounts cell lysates (20 μg, 10 μg, and 5 μg decreasing concentrations are represented by the thickness of the black triangle) from hTCEpi (-c-Cbl) cells treated without (−Dox) or with (+Dox) 1 μg/mL of doxycycline for 72 hours were immunoblotted for either c-Cbl or α-tubulin (α-Tub) as indicated. The 200-kDa and 45-kDa molecular weight standards are indicated on the left of the immunoblots. (C) Human telomerase-immortalized corneal epithelial cells were treated without (−) or with (+) doxycycline and/or 100 ng/mL EGF as indicated. Cell lysates were prepared and immunoprecipitated for the EGFR. The immunoprecipitate was resolved for 7.5% SDS-PAGE and immunoblotted for Ub, pY99, or the EGFR. A portion of the total cell lysate was resolved on a gel and immunoblotted for c-Cbl. Molecular weight standards are indicated on the left of the blot. Shown is a representative experiment repeated at least three times. (D) Densitometric analysis of three immunoblots from the experiment performed in (C). Data are plotted as the ratio of ubiquitylated EGFR to total EGFR (top), phosphorylated EGFR to total EGFR (middle), or c-Cbl (average ± SEM; n = 3). Data were analyzed with a paired Student's t-test. *P < 0.05; **P < 0.01.
Knockdown of c-Cbl altered the endocytic trafficking of the EGF-EGFR complex, by promoting recycling of the EGF-EGFR complex. Using 125I-EGF to monitor trafficking of the complex, we observed that there is an increase in the amount of intact 125I-EGF secreted from the cells when c-Cbl is knocked down as compared with parental hTCEpi cells and hTCEpi (-C-Cbl) cells without c-Cbl knockdown (Fig. 3A). This is consistent with the notion that the ligand-receptor complex that has bypassed lysosomal degradation and was recycled. Importantly, knockdown of c-Cbl did not change the rate of 125I-EGF-EGFR internalization (Supplemental Fig. S1). Together, these data indicate this level of c-Cbl knockdown and accompanying decrease in receptor ubiquitylation were sufficient to alter the endocytic trafficking of the EGF-EGFR complex.
Next, we wanted to know whether these changes in trafficking were sufficient to affect EGF-dependent cell migration. Using the parental and c-Cbl knockdown cells, we monitored cell migration in vitro using three different assays: an in vitro wound healing assay (Figs. 3B, 3C), a Scratch assay (Figs. 3D–F, Supplemental Fig. S2), and a Boyden chamber migration assay (Supplemental Fig. S3). Cell migration into a 2-mm “wound” was measured as the percentage of the initial acellular area that was covered with cells after 16 hours (Fig. 3C). In control cells, EGF-stimulated cell migration increased from approximately 38% in control cells to 64% in the cells with attenuated c-Cbl expression. Reduced c-Cbl also results in an increase of basal rate of cell migration. This is likely due to the level of constitutive EGFR activity stimulated by release of endogenous EGFR ligands (i.e., HB-EGF), as previously reported.30,31 Similar results were observed when migration was tested with a Scratch assay. The increase in cell migration in hTCEpi (-c-Cbl) cells was so much faster than the parental cells, data were collected at both 8 and 16 hours after wounding (Figs. 3D–F, Supplemental Fig. S2). At 8 hours after wounding, the loss of c-Cbl resulted in more ligand-dependent and independent cell migration than control cells (Fig. 3E); 16 hours after wounding, all control cells exhibited ligand-dependent cell migration, but the differences compared with the c-Cbl knockdown cells were less pronounced, presumably due to closing of the wound. Knockdown of c-Cbl also enhanced cell migration when directly measured using a Boyden chamber assay (Corning Life Sciences; Supplemental Fig. S3), although in this assay the addition of doxycycline inhibited cell migration.
Together, these studies provide in vitro evidence for c-Cbl negatively regulating EGFR-mediated corneal epithelial cell migration. However, RNAi is not a practical approach for in vivo studies, primarily because the kinetics of protein knockdown (typically 48–72 hours) would not offer a significant advantage to the physiological rate of tissue restoration.
The initial plan was to use a pharmacological agent that would directly antagonize c-Cbl activity. In the absence of such a commercial compound, we decided to indirectly inhibit c-Cbl, by using an Src inhibitor. The PP1 is a cell-permeable Src family kinase inhibitor.32 Previous work by Kassenbrock et al.33 demonstrated that at 50 μM, there is a complete inhibition of ligand-mediated EGFR ubiquitylation in MCF12A cells. We observed a similar inhibition of EGFR ubiquitylation in hTCEpi cells with 12 μM PP1 (Fig. 4A, left). However, subsequent experiments reveal this concentration inhibited all cell migration (data not shown). At much lower concentrations of PP1 (0.01 μM), we were still able to achieve approximately 60% inhibition of EGFR ubiquitylation (Figs. 4A, right, 4B), with no apparent changes to cell morphology. In the presence of 0.01 μM PP1, there was sustained EGF-dependent, EGFR phosphorylation for 2 hours (Fig. 4E). Based on the average area under of the curve (AUC), this is an approximately 20% increase in total EGFR phosphorylation (AUC = 111.0 ± 8.4 without PP1 versus AUC = 133.0 ± 4.2 with PP1). To determine if lower concentrations of PP1 could potentiate in vitro wound healing, we administered PP1 for 30 minutes before EGF treatment and monitored wound healing while both PP1 and EGF were present (Figs. 4C, 4D). Low doses of PP1 were sufficient to enhance EGF-mediated corneal epithelial wound healing in vitro.
Figure 4.

The Src inhibitor PP1 enhances migration and attenuates ligand-dependent EGFR ubiquitylation. (A) Human telomerase-immortalized corneal epithelial cells were pretreated without or with 12 μM (left) or 0.01 μM (right) as indicated. Cells were treated without (−) or with (+) 100 ng/mL EGF for 10 minutes. Cell lysates were prepared and immunoprecipitated with EGFR. The immunoprecipitation was divided into thirds, resolved by 7.5% SDS-PAGE, transferred to nitrocellulose, and immunoblotted with antibodies against Ub, pY99, and total EGFR. Shown are representative experiments repeated at least three times. (B) Quantification of EGFR after treatment with 0.01 μM PP1. Data are plotted as the ratio of ubiquitylated EGFR to total EGFR (Ub/EGFR) and phosphorylated EGFR to total EGFR (pY99/EGFR) (average ± SEM; n = 3). Data were analyzed with a paired Student's t-test. *P < 0.05. (C) Brightfield images of hTCEpi cells in vitro healing assay taken at 16 hours with and without EGF (10 ng/mL) after pretreatment of PP1 at various concentrations. (D) Cell migrations were analyzed as the difference in area from the plug removal (outer line) compared with 16 hours of EGF treatment (inner line). Data are plotted as the average ± SEM of area covered (wound healed). Analysis by a paired Student's t-test. **P < 0.05; *P < 0.1, n ≥ 3 with three to five replicate values. (E) The kinetics of EGFR phosphorylation were monitored with an ELISA assay. Plotted are the levels of phosphotyrosine activity associated with the isolated EGFR (average ± SEM, n = 4).
Finally, we wanted to determine if inhibition of PP1 would increase corneal epithelial wound healing in vivo (Fig. 5). A priori, we predicted that PP1 alone would enhance wound healing due to the high basal levels of EGF found in tears.13 Wounded eyes were treated with and without PP1 and in the absence and presence of EGF (Fig. 5). The PP1 promoted corneal epithelial wound healing in the absence of EGF, but did not accelerate wound healing in the presence of EGF. Importantly, when the EGFR inhibitor AG1478 was added, there was no increase in in vivo corneal epithelial wound healing. These data indicate that PP1 is mediating its effect via the EGFR.
Figure 5.

The PP1 attenuates EGF-mediated EGFR ubiquitylation and enhances EGFR corneal epithelial wound healing in vitro. A 1.5-mm-diameter wound was generated on the corneal epithelium of a mouse as described in Materials and Methods. Eyes were treated with various combinations of PBS, PP1 (0.01 μM), and EGF (100 ng/mL), as indicated on the figure. Shown are representative photographs from experiments repeated four to five times per condition. (B) Percentage of initial wound that has healed at each time point. Data are plotted as the average ± SEM, n = 4–5 eyes/condition. (C–E) Data from (B) replotted to more clearly show the (C) 16-hour, (D) 24-hour, and (E) 40-hour time points. **P < 0.05; *P < 0.1. Data were analyzed using a two-way ANOVA with a Bonferroni post hoc. Data from AG1478 mice were compared with mice with the same treatment in the absence of AG1478.
Discussion
Increased EGFR activity directly correlates with an increased rate of corneal epithelial wound healing. This study has focused on identifying mechanisms to increase the duration of EGFR activity rather than the magnitude. We have explored a mechanism to divert the EGF-EGFR complex from lysosomal degradation and sustain its signaling to ultimately enhance the cellular events associated with corneal epithelial wound healing. We find that attenuating c-Cbl activity, either through RNAi or pharmacological inhibitors, causes a decrease in receptor ubiquitylation, altered trafficking of the ligand-receptor complex, and an increase in EGFR phosphorylation. Ultimately, these biochemical changes culminate in enhanced corneal epithelial wound healing as measured by in vitro and in vivo assays. The increased in vivo wound healing is independent of exogenous ligand, likely due to the high levels of basal EGF found in tears.13 This interpretation is supported by the fact that the EGFR inhibitor, AG1478, inhibits the PP1-mediated increase in in vivo corneal epithelial wound healing.
The contribution of the endocytic pathway in EGFR-mediated corneal epithelial cell migration has been established previously. Transforming growth factor–α (TGF-α) is an endogenous EGFR ligand that more efficaciously promotes corneal epithelial cell migration as compared with EGF.15 In addition, due to the increased sensitivity of TGF-α to pH, the ligand more readily dissociates from the receptor in the more acidic early endosome.34 The unbound receptor can recycle back to the plasma membrane, rather than target to the lysosome for degradation. Interestingly, TGF-α–EGFR complexes also are ubiquitylated less than EGF-EGFR complexes.35 Our studies demonstrate that a pharmacological agent can inhibit EGFR ubiquitylation to cause sustained EGFR activity. This is particularly useful given that the addition of exogenous EGFR ligands do not increase EGFR-mediated wound healing in vivo,13 indicating the use of TGF-α may not be an effective strategy.
These experiments required balancing the PP1-mediated inhibition of Src to prevent EGFR ubiquitylation and inhibition of the other nonreceptor tyrosine kinases (i.e., Lck, Fyn, Hck) that are required for cell viability and migration. These “off-target” effects of PP1 likely limit its pharmacologic effect. Although PP1 has been previously reported to inhibit EGFR ubiquitylation, that report was largely focused on receptor biochemistry and the resulting cell biology was not explored.33 Others have examined the effect of PP1 and its analog (PP2) on corneal epithelial homeostasis. Gao et al.36 have shown that the addition of PP1 blocks corneal epithelial cells (A6[1] cells) in scratch-wound assay. However, these experiments were performed using 10 μM PP1, which is 225-fold over the half maximal inhibitory concentration (IC50) to inhibit Src (IC50 = 44 nM). Other studies, by Xu et al.,37 used PP2 (at 12.5 μM), and demonstrated that at primary corneal epithelial cells had reduced cell migration in the presence of PP2. It is important to note that our studies deliberately use submaximal concentrations of PP1 that are below the IC50 for inhibiting Src (170 nM) to minimize off-target effects. By design, we achieve only a partial decrease in Src activity and subsequently EGFR ubiquitylation. The PP1-mediated reduction in EGFR ubiquitylation is similar to what we observed in our hTCEpi(c-Cbl) knockdown cells and was able to increase the extent of EGFR-mediated cell migration.
It is striking that the modest decreases in ligand-mediated EGFR ubiquitylation have such profound effects on corneal epithelial cell biology in vitro and in vivo. Biologically, this may mean receptor ubiquitylation is such an important regulator of EGFR signaling, and even weak inhibition evokes biological changes. Another possibility is that the biochemical assays (Figs. 1, 2, 4) use supraphysiological levels of EGF that show only minor decreases in receptor ubiquitylation. However, under physiological ligand concentrations, the difference is more profound. Alternatively, the explanation may be a combination of the two reasons.
Although this study provides evidence that manipulation of c-Cbl is a bona fide therapeutic strategy, it also highlights the potential of more targeted inhibitors of EGFR–c-Cbl interactions. First, PP1 inhibits multiple nonreceptor tyrosine kinases. More-specific agents would minimize the deleterious effects of inhibiting nonreceptor tyrosine kinases on cell viability. In addition, in the absence of off-target effects, it is likely a c-Cbl activity could be inhibited to a greater extent and perhaps further accelerate the rate of wound healing. Finally, if an inhibitor targeted only EGFR–c-Cbl interactions, the other receptor tyrosine kinases that c-Cbl ubiquitylates (i.e., hepatocyte growth factor, platelet-derived growth factor, nerve growth factor receptors)38–40 would not have perturbed activity. Of course, inhibiting the ubiquitylation (and subsequent degradation) of other receptors is another strategy for improving corneal epithelial wound healing. However, our AG1478 experiments indicate that in hTCEpi cells, the major role of c-Cbl is through the EGFR. Although our focus has been on the inhibition of c-Cbl, one can not overlook the strategy of attenuating receptor ubiquitylation by enhancing the target activation of the deubiquitinating enzymes as an alternative strategy to promote receptor recycling.41
Acknowledgments
Supported by National Institutes of Health Grants 1R01GM092874-01A1 (BPC) and 1R01EY021497-01 (BPC).
Disclosure: J.S. Rush, None; M.A. Boeving, None; W.L. Berry, None; B.P. Ceresa, None.
References
- 1. Johnson KS, Levin F, Chu DS. Persistent corneal epithelial defect associated with erlotinib treatment. Cornea. 2009; 28: 706–707 [DOI] [PubMed] [Google Scholar]
- 2. Oliva MS, Schottman T, Gulati M. Turning the tide of corneal blindness. Indian J Ophthalmol. 2012; 60: 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosenberg ME, Tervo TM, Immonen IJ, Muller LJ, Gronhagen-Riska C, Vesaluoma MH. Corneal structure and sensitivity in type 1 diabetes mellitus. Invest Ophthalmol Vis Sci. 2000; 41: 2915–2921 [PubMed] [Google Scholar]
- 4. Whitcher JP, Srinivasan M, Upadhyay MP. Corneal blindness: a global perspective. Bull World Health Organ. 2001; 79: 214–221 [PMC free article] [PubMed] [Google Scholar]
- 5. Imanishi J, Kamiyama K, Iguchi I, Kita M, Sotozono C, Kinoshita S. Growth factors: importance in wound healing and maintenance of transparency of the cornea. Prog Retin Eye Res. 2000; 19: 113–129 [DOI] [PubMed] [Google Scholar]
- 6. Zieske JD, Takahashi H, Hutcheon AEK, Dalbone AC. Activation of epidermal growth factor receptor during corneal epithelial migration. Invest Opthalmol Vis Sci. 2000; 41: 1346–1355 [PubMed] [Google Scholar]
- 7. Foerster CG, Cursiefen C, Kruse FE. Persisting corneal erosion under cetuximab (Erbitux) treatment (epidermal growth factor receptor antibody). Cornea. 2008; 27: 612–614 [DOI] [PubMed] [Google Scholar]
- 8. Daniele S, Frati L, Fiore C, Santoni G. The effect of the epidermal growth factor (EGF) on the corneal epithelium in humans. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1979; 210: 159–165 [DOI] [PubMed] [Google Scholar]
- 9. Dellaert MM, Casey TA, Wiffen S, et al. Influence of topical human epidermal growth factor on postkeratoplasty re-epithelialisation. Br J Ophthalmol. 1997; 81: 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kandarakis AS, Page C, Kaufman HE. The effect of epidermal growth factor on epithelial healing after penetrating keratoplasty in human eyes. Am J Ophthalmol. 1984; 98: 411–415 [DOI] [PubMed] [Google Scholar]
- 11. Pastor JC, Calonge M. Epidermal growth factor and corneal wound healing. A multicenter study. Cornea. 1992; 11: 311–314 [DOI] [PubMed] [Google Scholar]
- 12. Ceresa BP. Spatial regulation of epidermal growth factor receptor signaling by endocytosis. Int J Mol Sci. 2012; 14: 72–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peterson JL, Phelps ED, Doll MA, Schaal S, Ceresa BP. Analysis of EGFR ligands found in human tears reveals differences in corneal epithelial wound healing and ligand induced EGFR signaling. Invest Ophthalmol Vis Sci. 2014; 55: 2870–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009; 315: 683–696 [DOI] [PubMed] [Google Scholar]
- 15. McClintock JL, Ceresa BP. Transforming growth factor-α (TGF-α) enhances corneal epithelial cell migration by promoting EGFR recycling. Invest Ophthalmol Vis Sci. 2010; 51: 3455–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang YN, Yamaguchi H, Hsu JM, Hung MC. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene. 2010; 29: 3997–4006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liao HJ, Carpenter G. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol Biol Cell. 2007; 18: 1064–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Opresko LK, Chang CP, Will BH, Burke PM, Gill GN, Wiley HS. Endocytosis and lysosomal targeting of epidermal growth factor receptors are mediated by distinct sequences independent of the tyrosine kinase domain. J Biol Chem. 1995; 270: 4325–4333 [DOI] [PubMed] [Google Scholar]
- 19. Babst M, Odorizzi G, Estepa EJ, Emr SD. Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic. 2000; 1: 248–258 [DOI] [PubMed] [Google Scholar]
- 20. Razi M, Futter CE. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol Biol Cell. 2006; 17: 3469–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem. 2009; 284: 12110–12124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joazeiro CAP, Wing SS, Huang H-K, Leverson JD, Hunter T, Liu Y-C. The tyrosine kinase negative regulator c-Cbl as a RING-Type, E2-dependent ubiquitin-protein ligase. Science. 1999; 286: 309–312 [DOI] [PubMed] [Google Scholar]
- 23. Caligiuri MA, Briesewitz R, Yu J, et al. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood. 2007; 110: 1022–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robertson DM, Li L, Fisher S, et al. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Invest Ophthalmol Vis Sci. 2005; 46: 470–478 [DOI] [PubMed] [Google Scholar]
- 25. Olson A, Sheth N, Lee JS, Hannon G, Sachidanandam R. RNAi codex: a portal/database for short-hairpin RNA (shRNA) gene-silencing constructs. Nucleic Acids Res. 2006; 34: D153–D157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shin KJ, Wall EA, Zavzavadjian JR, et al. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc Natl Acad Sci U S A. 2006; 103: 13759–13764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rush JS, Quinalty LM, Engelman L, Sherry DM, Ceresa BP. Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. J Biol Chem. 2012; 287: 712–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Visser Smit GD, Place TL, Cole SL, et al. Cbl controls EGFR fate by regulating early endosome fusion. Sci Signal. 2009; 2: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharma GD, He J, Bazan HE. p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between MAP kinase cascades. J Biol Chem. 2003; 278: 21989–21997 [DOI] [PubMed] [Google Scholar]
- 30. Xu K-P, Ding Y, Ling J, Dong Z, Yu F-S. Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004; 45: 813–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoshioka R, Shiraishi A, Kobayashi T, et al. Corneal epithelial wound healing impaired in keratinocyte-specific HB-EGF-deficient mice in vivo and in vitro. Invest Ophthalmol Vis Sci. 2010; 51: 5630–5639 [DOI] [PubMed] [Google Scholar]
- 32. Amoui M, Draber P, Draberova L. Src family-selective tyrosine kinase inhibitor, PP1, inhibits both Fc epsilonRI- and Thy-1-mediated activation of rat basophilic leukemia cells. Eur J Immunol. 1997; 27: 1881–1886 [DOI] [PubMed] [Google Scholar]
- 33. Kassenbrock CK, Hunter S, Garl P, Johnson GL, Anderson SM. Inhibition of Src family kinases blocks epidermal growth factor (EGF)-induced activation of Akt, phosphorylation of c-Cbl, and ubiquitination of the EGF receptor. J Biol Chem. 2002; 277: 24967–24975 [DOI] [PubMed] [Google Scholar]
- 34. Rutten MJ, Dempsey PJ, Luttropp CA, et al. dentification of an EGF/TGF-alpha receptor in primary cultures of guinea pig gastric mucous epithelial cells. Am J Physiol. 1996; 270: G604–G612 [DOI] [PubMed] [Google Scholar]
- 35. Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol. 2002; 156: 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao CY, Stepp MA, Fariss R, Zelenka P. Cdk5 regulates activation and localization of Src during corneal epithelial wound closure. J Cell Sci. 2004; 117: 4089–4098 [DOI] [PubMed] [Google Scholar]
- 37. Xu K-P, Yin J, Yu F-S. Src-family tyrosine kinases in wound- and ligand-induced epidermal growth factor receptor activation in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006; 47: 2832–2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garcia-Guzman M, Larsen E, Vuori K. The proto-oncogene c-Cbl is a positive regulator of Met-induced MAP kinase activation: a role for the adaptor protein Crk. Oncogene. 2000; 19: 4058–4065 [DOI] [PubMed] [Google Scholar]
- 39. Yokouchi M, Wakioka T, Sakamoto H, et al. APS, an adaptor protein containing PH and SH2 domains, is associated with the PDGF receptor and c-Cbl and inhibits PDGF-induced mitogenesis. Oncogene. 1999; 18: 759–767 [DOI] [PubMed] [Google Scholar]
- 40. Takahashi Y, Shimokawa N, Esmaeili-Mahani S, et al. Ligand-induced downregulation of TrkA is partly regulated through ubiquitination by Cbl. FEBS Lett. 2011; 585: 1741–1747 [DOI] [PubMed] [Google Scholar]
- 41. Kimura Y, Tanaka K. Regulatory mechanisms involved in the control of ubiquitin homeostasis. J Biochem. 2010; 147: 793–798 [DOI] [PubMed] [Google Scholar]