

Abstract
The title compound, C17H26BrNO, exhibits a small twist between the amide residue and the benzene ring [C—N—C—C torsion angle = 29.4 (5)°]. In the crystal, the amido NH group is involved in N—H⋯O hydrogen bonding, which connects molecules into chains parallel to the c axis.
Related literature
For the related structure of a derivative with an alkyl-N-aryl substituent, see: Palakshamurthy et al. (2014 ▶), with an alkyl-N-phenylsulfonyl substituent, see: Shakuntala et al. (2011 ▶) and with a chloro-N-phenyl substituent, see: Betz et al. (2011 ▶). For details of the synthesis, see: Bentiss & Lagrenée (1999 ▶); Hill et al. (2007 ▶).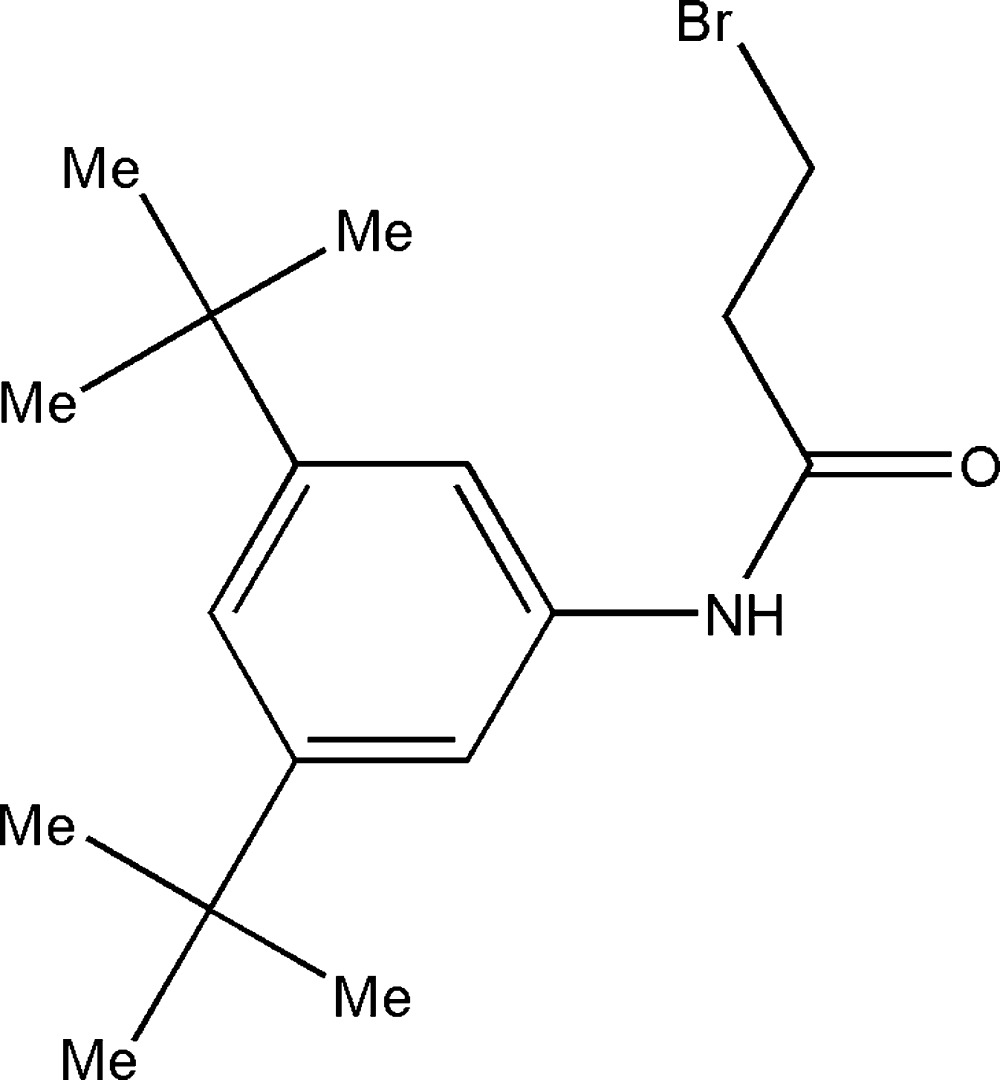
Experimental
Crystal data
C17H26BrNO
M r = 340.30
Monoclinic,

a = 15.666 (2) Å
b = 11.4885 (16) Å
c = 9.7829 (14) Å
β = 97.436 (4)°
V = 1745.9 (4) Å3
Z = 4
Mo Kα radiation
μ = 2.35 mm−1
T = 150 K
0.40 × 0.20 × 0.11 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2013 ▶) T min = 0.455, T max = 0.789
21557 measured reflections
4005 independent reflections
2738 reflections with I > 2σ(I)
R int = 0.055
Refinement
R[F 2 > 2σ(F 2)] = 0.050
wR(F 2) = 0.145
S = 1.04
4005 reflections
187 parameters
H-atom parameters constrained
Δρmax = 1.08 e Å−3
Δρmin = −0.65 e Å−3
Data collection: APEX2 (Bruker, 2013 ▶); cell refinement: SAINT (Bruker, 2013 ▶); data reduction: SAINT; program(s) used to solve structure: SIR92 (Altomare et al., 1994 ▶); program(s) used to refine structure: SHELXL2013 (Sheldrick, 2008 ▶); molecular graphics: CrystalMaker (CrystalMaker, 2014 ▶); software used to prepare material for publication: local programs.
Supplementary Material
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S1600536814012094/nk2223sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814012094/nk2223Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814012094/nk2223Isup3.cml
CCDC reference: 1005148
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1C⋯O1i | 0.88 | 2.01 | 2.889 (3) | 174 |
Symmetry code: (i)  .
.
Acknowledgments
The authors would like to thank Dr Aneta Borecki and Dr Paul Boyle (University of Western Ontario) for their help in data collection and refinement.
supplementary crystallographic information
S1. Comment
The title compound C17H25BrNO, is a brominated derivative of a secondary amide bearing a di-tert-butylbenzene ring. It exhibits a small twist between the amide residue and benzene ring [the C3—N1—C4—C5 torsion angle = 29.5 (4)°]. The N—H and C=O bonds are anti to each other (Fig. 1), as observed in many other derivatives (Shakuntala et al., 2011). In the structure, bond distances and angles are within normal range (Table 1) and comparable to reported values in amide derivatives (Palakshamurthy et al., 2014, Betz et al., 2011). The torsion angle of C3—N1—C4—C9 and C3—N1—C4—C5 are -153.2 (3)° and 29.5 (4)°, respectively. The amido NH group is involved in N—H···O [2.01 Å] hydrogen bonding, which connects molecules into chains parallel to c axis (Fig. 2).
S2. Experimental
Synthesis of (2,5-Bis(2-pyridyl)-1,3,4-oxadiazole)dimethylplatinum(II), [PtMe2(ox)]
A mixture of [Pt2Me4(µ-SMe2)2] (50 mg, 0.087 mmol)) (Hill et al., 2007) and ox (ox = 2,5-bis(2-pyridyl)-1,3,4-oxadiazole) (38 mg, 0.170Dr mmol) (Bentiss and Lagrenée, 1999) in dry ether (10 ml) was stirred for 1 h. A red precipitate resulted. The precipitate was isolated and washed with acetone (3 × 5 ml). The product was recrystallized from CH2Cl2. A yellow solid was produced and dried in vacuo.
The title compound was crystallized unintentionally from the reaction mixture of the complex (2,5-Bis(2-pyridyl)-1,3,4-oxadiazole)dimethylplatinum(II), [PtMe2(ox)] (0.05 g, 0.112 mmol) and, the commercially available N-(3,5-di-tert-butylphenyl)-3-bromopropanamide (0.052 g, 0.129 mmol) in acetone (15 ml) was stirred for 5 h at room temperature. The reaction color changed to yellow suspension. The solvent was evaporated under vacuum and the resulting solid was washed with water (2 × 10 ml) and pentane (3 × 10 ml). The isolated yellow solid is highly soluble in CH2Cl2 solvent, which was dried under high vacuum. Yield 87%. A suitable crystal for X-ray diffraction analysis was selected for data collection.
S3. Refinement
The hydrogen atoms were introduced at idealized positions and were allowed to ride on the parent atom, with C—H = 0.95–0.99 Å and N—H = 0.88 Å and Uiso(H) = 1.2–1.5Ueq(C,N).
Figures
Fig. 1.

The molecular structure of the title compound, with atom labels and anisotropic displacement ellipsoids (drawn at 50% probability level).
Fig. 2.

Partial molecular packing, showing the chains, parallel to c axis, formed via N—H···O hydrogen bonding (multi-rendered cylinders).
Crystal data
| C17H26BrNO | F(000) = 712 |
| Mr = 340.30 | Dx = 1.295 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 15.666 (2) Å | Cell parameters from 5619 reflections |
| b = 11.4885 (16) Å | θ = 2.2–26.0° |
| c = 9.7829 (14) Å | µ = 2.35 mm−1 |
| β = 97.436 (4)° | T = 150 K |
| V = 1745.9 (4) Å3 | Needle, colourless |
| Z = 4 | 0.40 × 0.20 × 0.11 mm |
Data collection
| Bruker APEXII CCD diffractometer | 2738 reflections with I > 2σ(I) |
| φ and ω scans | Rint = 0.055 |
| Absorption correction: multi-scan (SADABS; Bruker, 2013) | θmax = 27.6°, θmin = 2.2° |
| Tmin = 0.455, Tmax = 0.789 | h = −20→19 |
| 21557 measured reflections | k = −14→14 |
| 4005 independent reflections | l = −8→12 |
Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.050 | H-atom parameters constrained |
| wR(F2) = 0.145 | w = 1/[σ2(Fo2) + (0.0743P)2 + 1.5001P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.04 | (Δ/σ)max < 0.001 |
| 4005 reflections | Δρmax = 1.08 e Å−3 |
| 187 parameters | Δρmin = −0.65 e Å−3 |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.58935 (3) | 0.54272 (3) | 0.85526 (5) | 0.04516 (17) | |
| N1 | 0.61402 (17) | 0.2102 (2) | 0.8672 (2) | 0.0205 (6) | |
| H1C | 0.5990 | 0.2131 | 0.9508 | 0.025* | |
| O1 | 0.57853 (15) | 0.2805 (2) | 0.6495 (2) | 0.0292 (6) | |
| C1 | 0.4854 (2) | 0.4604 (3) | 0.7774 (4) | 0.0303 (8) | |
| H1A | 0.4344 | 0.4974 | 0.8092 | 0.036* | |
| H1B | 0.4790 | 0.4653 | 0.6756 | 0.036* | |
| C2 | 0.4901 (2) | 0.3353 (3) | 0.8208 (3) | 0.0234 (7) | |
| H2A | 0.4358 | 0.2957 | 0.7840 | 0.028* | |
| H2B | 0.4961 | 0.3308 | 0.9227 | 0.028* | |
| C3 | 0.56518 (19) | 0.2730 (3) | 0.7699 (3) | 0.0201 (6) | |
| C4 | 0.68642 (19) | 0.1401 (3) | 0.8510 (3) | 0.0201 (6) | |
| C5 | 0.74057 (19) | 0.1639 (3) | 0.7525 (3) | 0.0219 (7) | |
| H5 | 0.7282 | 0.2270 | 0.6904 | 0.026* | |
| C6 | 0.81286 (19) | 0.0950 (3) | 0.7452 (3) | 0.0226 (7) | |
| C7 | 0.8718 (2) | 0.1217 (3) | 0.6355 (3) | 0.0267 (7) | |
| C8 | 0.8922 (3) | 0.2519 (4) | 0.6334 (5) | 0.0476 (11) | |
| H8A | 0.9299 | 0.2674 | 0.5631 | 0.071* | |
| H8B | 0.9211 | 0.2757 | 0.7240 | 0.071* | |
| H8C | 0.8386 | 0.2959 | 0.6118 | 0.071* | |
| C9 | 0.8285 (2) | 0.0018 (3) | 0.8365 (3) | 0.0240 (7) | |
| H9 | 0.8774 | −0.0460 | 0.8308 | 0.029* | |
| C10 | 0.7749 (2) | −0.0236 (3) | 0.9359 (3) | 0.0222 (7) | |
| C11 | 0.7922 (2) | −0.1243 (3) | 1.0380 (3) | 0.0263 (7) | |
| C12 | 0.7155 (3) | −0.2089 (4) | 1.0182 (5) | 0.0467 (10) | |
| H12A | 0.6632 | −0.1682 | 1.0367 | 0.070* | |
| H12B | 0.7267 | −0.2745 | 1.0820 | 0.070* | |
| H12C | 0.7077 | −0.2378 | 0.9232 | 0.070* | |
| C13 | 0.8033 (3) | −0.0751 (3) | 1.1850 (4) | 0.0364 (9) | |
| H13A | 0.8532 | −0.0231 | 1.1975 | 0.055* | |
| H13B | 0.8122 | −0.1393 | 1.2514 | 0.055* | |
| H13C | 0.7515 | −0.0316 | 1.2001 | 0.055* | |
| C14 | 0.8740 (2) | −0.1920 (3) | 1.0187 (4) | 0.0355 (9) | |
| H14A | 0.8691 | −0.2226 | 0.9245 | 0.053* | |
| H14B | 0.8813 | −0.2567 | 1.0844 | 0.053* | |
| H14C | 0.9238 | −0.1400 | 1.0349 | 0.053* | |
| C15 | 0.7037 (2) | 0.0485 (3) | 0.9422 (3) | 0.0218 (6) | |
| H15 | 0.6666 | 0.0344 | 1.0100 | 0.026* | |
| C16 | 0.8254 (2) | 0.0852 (4) | 0.4948 (4) | 0.0385 (9) | |
| H16A | 0.7720 | 0.1299 | 0.4744 | 0.058* | |
| H16B | 0.8119 | 0.0020 | 0.4961 | 0.058* | |
| H16C | 0.8626 | 0.1004 | 0.4236 | 0.058* | |
| C17 | 0.9571 (2) | 0.0552 (4) | 0.6613 (4) | 0.0412 (10) | |
| H17A | 0.9459 | −0.0285 | 0.6509 | 0.062* | |
| H17B | 0.9849 | 0.0712 | 0.7549 | 0.062* | |
| H17C | 0.9949 | 0.0802 | 0.5945 | 0.062* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.0416 (3) | 0.0352 (2) | 0.0631 (3) | −0.00389 (17) | 0.0234 (2) | −0.00846 (19) |
| N1 | 0.0228 (13) | 0.0298 (15) | 0.0107 (12) | 0.0051 (11) | 0.0087 (10) | −0.0011 (10) |
| O1 | 0.0302 (13) | 0.0479 (15) | 0.0109 (10) | 0.0089 (11) | 0.0083 (9) | 0.0029 (10) |
| C1 | 0.0244 (17) | 0.038 (2) | 0.0295 (18) | 0.0105 (15) | 0.0071 (14) | 0.0062 (15) |
| C2 | 0.0170 (15) | 0.0324 (18) | 0.0221 (16) | 0.0020 (13) | 0.0082 (12) | −0.0002 (13) |
| C3 | 0.0179 (15) | 0.0277 (17) | 0.0157 (15) | −0.0025 (12) | 0.0057 (12) | −0.0035 (12) |
| C4 | 0.0169 (15) | 0.0278 (17) | 0.0164 (14) | −0.0003 (12) | 0.0055 (12) | −0.0042 (13) |
| C5 | 0.0202 (16) | 0.0322 (17) | 0.0142 (14) | 0.0000 (13) | 0.0059 (12) | 0.0012 (13) |
| C6 | 0.0182 (15) | 0.0329 (18) | 0.0175 (15) | −0.0009 (13) | 0.0053 (12) | −0.0063 (13) |
| C7 | 0.0209 (16) | 0.039 (2) | 0.0216 (16) | 0.0031 (14) | 0.0092 (13) | −0.0050 (14) |
| C8 | 0.044 (3) | 0.047 (3) | 0.059 (3) | −0.0080 (19) | 0.034 (2) | −0.004 (2) |
| C9 | 0.0204 (16) | 0.0315 (16) | 0.0205 (16) | 0.0034 (13) | 0.0040 (13) | −0.0070 (14) |
| C10 | 0.0228 (16) | 0.0280 (18) | 0.0158 (15) | −0.0019 (13) | 0.0027 (12) | −0.0049 (13) |
| C11 | 0.0258 (17) | 0.0294 (18) | 0.0236 (16) | 0.0040 (14) | 0.0021 (13) | 0.0014 (14) |
| C12 | 0.044 (2) | 0.037 (2) | 0.058 (3) | −0.0040 (18) | 0.001 (2) | 0.012 (2) |
| C13 | 0.045 (2) | 0.044 (2) | 0.0216 (17) | 0.0137 (17) | 0.0070 (16) | 0.0071 (16) |
| C14 | 0.042 (2) | 0.036 (2) | 0.0290 (19) | 0.0125 (17) | 0.0032 (16) | 0.0002 (16) |
| C15 | 0.0222 (16) | 0.0289 (17) | 0.0158 (14) | −0.0024 (13) | 0.0084 (12) | −0.0026 (13) |
| C16 | 0.036 (2) | 0.061 (3) | 0.0204 (17) | −0.0049 (18) | 0.0109 (15) | −0.0013 (17) |
| C17 | 0.028 (2) | 0.064 (3) | 0.035 (2) | 0.0075 (18) | 0.0159 (16) | −0.0010 (19) |
Geometric parameters (Å, º)
| Br1—C1 | 1.951 (4) | C9—C10 | 1.396 (5) |
| N1—C3 | 1.350 (4) | C9—H9 | 0.9500 |
| N1—C4 | 1.416 (4) | C10—C15 | 1.397 (4) |
| N1—H1C | 0.8800 | C10—C11 | 1.529 (5) |
| O1—C3 | 1.226 (4) | C11—C14 | 1.531 (5) |
| C1—C2 | 1.498 (5) | C11—C13 | 1.534 (5) |
| C1—H1A | 0.9900 | C11—C12 | 1.538 (5) |
| C1—H1B | 0.9900 | C12—H12A | 0.9800 |
| C2—C3 | 1.515 (4) | C12—H12B | 0.9800 |
| C2—H2A | 0.9900 | C12—H12C | 0.9800 |
| C2—H2B | 0.9900 | C13—H13A | 0.9800 |
| C4—C15 | 1.384 (4) | C13—H13B | 0.9800 |
| C4—C5 | 1.391 (4) | C13—H13C | 0.9800 |
| C5—C6 | 1.391 (4) | C14—H14A | 0.9800 |
| C5—H5 | 0.9500 | C14—H14B | 0.9800 |
| C6—C9 | 1.396 (5) | C14—H14C | 0.9800 |
| C6—C7 | 1.534 (4) | C15—H15 | 0.9500 |
| C7—C8 | 1.530 (6) | C16—H16A | 0.9800 |
| C7—C16 | 1.530 (5) | C16—H16B | 0.9800 |
| C7—C17 | 1.531 (5) | C16—H16C | 0.9800 |
| C8—H8A | 0.9800 | C17—H17A | 0.9800 |
| C8—H8B | 0.9800 | C17—H17B | 0.9800 |
| C8—H8C | 0.9800 | C17—H17C | 0.9800 |
| C3—N1—C4 | 127.9 (2) | C9—C10—C15 | 117.6 (3) |
| C3—N1—H1C | 116.1 | C9—C10—C11 | 122.7 (3) |
| C4—N1—H1C | 116.1 | C15—C10—C11 | 119.7 (3) |
| C2—C1—Br1 | 110.3 (2) | C10—C11—C14 | 112.6 (3) |
| C2—C1—H1A | 109.6 | C10—C11—C13 | 108.8 (3) |
| Br1—C1—H1A | 109.6 | C14—C11—C13 | 107.9 (3) |
| C2—C1—H1B | 109.6 | C10—C11—C12 | 109.0 (3) |
| Br1—C1—H1B | 109.6 | C14—C11—C12 | 108.4 (3) |
| H1A—C1—H1B | 108.1 | C13—C11—C12 | 110.0 (3) |
| C1—C2—C3 | 111.8 (3) | C11—C12—H12A | 109.5 |
| C1—C2—H2A | 109.2 | C11—C12—H12B | 109.5 |
| C3—C2—H2A | 109.2 | H12A—C12—H12B | 109.5 |
| C1—C2—H2B | 109.2 | C11—C12—H12C | 109.5 |
| C3—C2—H2B | 109.2 | H12A—C12—H12C | 109.5 |
| H2A—C2—H2B | 107.9 | H12B—C12—H12C | 109.5 |
| O1—C3—N1 | 124.3 (3) | C11—C13—H13A | 109.5 |
| O1—C3—C2 | 121.2 (3) | C11—C13—H13B | 109.5 |
| N1—C3—C2 | 114.5 (3) | H13A—C13—H13B | 109.5 |
| C15—C4—C5 | 120.7 (3) | C11—C13—H13C | 109.5 |
| C15—C4—N1 | 116.9 (3) | H13A—C13—H13C | 109.5 |
| C5—C4—N1 | 122.3 (3) | H13B—C13—H13C | 109.5 |
| C4—C5—C6 | 119.9 (3) | C11—C14—H14A | 109.5 |
| C4—C5—H5 | 120.1 | C11—C14—H14B | 109.5 |
| C6—C5—H5 | 120.1 | H14A—C14—H14B | 109.5 |
| C5—C6—C9 | 118.7 (3) | C11—C14—H14C | 109.5 |
| C5—C6—C7 | 119.4 (3) | H14A—C14—H14C | 109.5 |
| C9—C6—C7 | 121.9 (3) | H14B—C14—H14C | 109.5 |
| C8—C7—C16 | 109.3 (3) | C4—C15—C10 | 120.9 (3) |
| C8—C7—C17 | 108.2 (3) | C4—C15—H15 | 119.6 |
| C16—C7—C17 | 108.3 (3) | C10—C15—H15 | 119.6 |
| C8—C7—C6 | 110.5 (3) | C7—C16—H16A | 109.5 |
| C16—C7—C6 | 108.4 (3) | C7—C16—H16B | 109.5 |
| C17—C7—C6 | 112.1 (3) | H16A—C16—H16B | 109.5 |
| C7—C8—H8A | 109.5 | C7—C16—H16C | 109.5 |
| C7—C8—H8B | 109.5 | H16A—C16—H16C | 109.5 |
| H8A—C8—H8B | 109.5 | H16B—C16—H16C | 109.5 |
| C7—C8—H8C | 109.5 | C7—C17—H17A | 109.5 |
| H8A—C8—H8C | 109.5 | C7—C17—H17B | 109.5 |
| H8B—C8—H8C | 109.5 | H17A—C17—H17B | 109.5 |
| C6—C9—C10 | 122.3 (3) | C7—C17—H17C | 109.5 |
| C6—C9—H9 | 118.9 | H17A—C17—H17C | 109.5 |
| C10—C9—H9 | 118.9 | H17B—C17—H17C | 109.5 |
| Br1—C1—C2—C3 | −61.5 (3) | C9—C6—C7—C17 | −13.4 (4) |
| C4—N1—C3—O1 | −1.8 (5) | C5—C6—C9—C10 | −0.9 (5) |
| C4—N1—C3—C2 | 178.0 (3) | C7—C6—C9—C10 | −179.6 (3) |
| C1—C2—C3—O1 | −48.6 (4) | C6—C9—C10—C15 | −0.2 (5) |
| C1—C2—C3—N1 | 131.6 (3) | C6—C9—C10—C11 | −179.0 (3) |
| C3—N1—C4—C15 | −153.2 (3) | C9—C10—C11—C14 | 0.3 (4) |
| C3—N1—C4—C5 | 29.4 (5) | C15—C10—C11—C14 | −178.5 (3) |
| C15—C4—C5—C6 | 0.0 (5) | C9—C10—C11—C13 | 119.9 (3) |
| N1—C4—C5—C6 | 177.3 (3) | C15—C10—C11—C13 | −58.9 (4) |
| C4—C5—C6—C9 | 1.0 (5) | C9—C10—C11—C12 | −120.1 (4) |
| C4—C5—C6—C7 | 179.7 (3) | C15—C10—C11—C12 | 61.1 (4) |
| C5—C6—C7—C8 | 47.2 (4) | C5—C4—C15—C10 | −1.2 (5) |
| C9—C6—C7—C8 | −134.1 (3) | N1—C4—C15—C10 | −178.6 (3) |
| C5—C6—C7—C16 | −72.6 (4) | C9—C10—C15—C4 | 1.3 (5) |
| C9—C6—C7—C16 | 106.1 (4) | C11—C10—C15—C4 | −179.9 (3) |
| C5—C6—C7—C17 | 167.9 (3) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1C···O1i | 0.88 | 2.01 | 2.889 (3) | 174 |
| C5—H5···O1 | 0.95 | 2.41 | 2.931 (4) | 115 |
Symmetry code: (i) x, −y+1/2, z+1/2.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: NK2223).
References
- Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994). J. Appl. Cryst. 27, 435.
- Bentiss, F. & Lagrenée, M. (1999). J. Heterocycl. Chem. 36, 1029–1032.
- Betz, R., Gerber, T., Hosten, E., Siddegowda, M. S. & Yathirajan, H. S. (2011). Acta Cryst. E67, o2868. [DOI] [PMC free article] [PubMed]
- Bruker (2013). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- CrystalMaker Software (2014). CrystalMaker CrystalMaker Software, Bicester, Oxfordshire, England. www.crystalmaker.com
- Hill, G. S., Irwin, M. J., Levy, C. J., Rendina, L. M., Puddephatt, R. J., Andersen, R. A. & Mclean, L. (2007). Platinum(II) Complexes of Dimethyl Sulfide, in Inorganic Syntheses. New York: John Wiley & Sons Inc.
- Palakshamurthy, B. S., Suchetan, P. A., Sreenivasa, S., Lokanath, N. K. & Madhu Chakrapani Rao, T. (2014). Acta Cryst. E70, o223. [DOI] [PMC free article] [PubMed]
- Shakuntala, K., Foro, S. & Gowda, B. T. (2011). Acta Cryst. E67, o536. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S1600536814012094/nk2223sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814012094/nk2223Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814012094/nk2223Isup3.cml
CCDC reference: 1005148
Additional supporting information: crystallographic information; 3D view; checkCIF report