

The basic building units of the hydrous periodate Pb3(IO4(OH)2)2 are three Pb2+ cations and two IO4(OH)2 3− anions. The octahedral anions are arranged in a distorted hexagonal rod packing, with the cations (each with a coordination number of eight) located in between.
Keywords: crystal structure, lead, periodate, non-merohedral twinning
Abstract
The structure of the title compound, trilead(II) bis[dihydroxidotetraoxidoiodate(VII)], was determined from a crystal twinned by non-merohedry with two twin domains present [twin fraction 0.73 (1):0.27 (1)]. It contains three Pb2+ cations and two IO4(OH)2 3− anions in the asymmetric unit. Each of the Pb2+ cations is surrounded by eight O atoms (cut-off value = 3.1 Å) in the form of a distorted polyhedron. The octahedral IO4(OH)2 3− anions are arranged in rows extending parallel to [021], forming a distorted hexagonal rod packing. The cations and anions are linked into a framework structure. Although H-atom positions could not be located, O⋯O distances suggest medium-strength hydrogen-bonding interactions between the IO4(OH)2 octahedra, further consolidating the crystal packing.
Chemical context
Lead and mercury can both exist in different oxidation states and each of the two elements exhibits a peculiar crystal chemistry. In the case of Pb2+-containing compounds, the crystal chemistry is mainly dominated by the stereoactive 6s 2 lone-pair of lead (Holloway & Melnik, 1997 ▶), whereas Hg2+-containing compounds show a strong preference for a linear coordination of mercury (Breitinger, 2004 ▶). In this respect, it appears surprising that for some Pb2+- and Hg2+-containing compounds an isotypic relationship exists, e.g. for PbAs2O6 (Losilla et al., 1995 ▶) and HgAs2O6 (Mormann & Jeitschko, 2000b ▶; Weil, 2000 ▶), or for the mineral descloizite PbZn(VO4)OH (Hawthorne & Faggiani, 1979 ▶) and the synthetic phase HgZn(AsO4)OH (Weil, 2004 ▶). With this in mind, it seemed interesting to study the relation between phases in the systems HgII–IVII–O–H and PbII–IVII–O–H. Whereas in the system HgII–IVII–O–H two compounds have been structurally characterized, viz. Hg3(IO4(OH)2)2 (Mormann & Jeitschko, 2000a ▶) and Hg(IO3(OH)3) (Mormann & Jeitschko, 2001 ▶), a phase in the system PbII–IVII–O–H has not yet been structurally determined, although several lead(II) periodate phases have been reported to exist. Willard & Thompson (1934 ▶) claimed to have obtained only one phase with composition Pb3H4(IO6)2 in the system PbII–IVII–O–H. However, Drátovský & Matějčková (1965a ▶,b ▶) reported the existence of three phases with composition Pb3(IO5)2·H2O, Pb2I2O9·3H2O and Pb4I2O11·5H2O in this system. To shed some light on the conflicting composition of the Pb:I 3:2 phase [Pb3H4(IO6)2 versus Pb3(IO5)2·H2O with a lower water content], the synthetic procedure described by Willard & Thompson (1934 ▶) was repeated for crystal growth of this lead periodate. The current structure determination of the obtained crystals showed the composition Pb3H4(IO6)2 as reported by Willard & Thompson (1934 ▶) to be correct. In a more reasonable crystal–chemical sense, the formula of these crystals should be rewritten as Pb3(IO4(OH)2)2.
Structural commentary
Three Pb2+ cations and two IO4(OH)2 3− octahedra are present in the asymmetric unit. The anions form a slightly distorted hexagonal rod packing with the rods extending parallel to [021]. Cations and anions are linked through common oxygen atoms into a framework structure (Fig. 1 ▶).
Figure 1.

The crystal structure of Pb3(IO4(OH)2)2 in a projection along [021]. Displacement ellipsoids are drawn at the 90% probability level. O atoms bearing the OH function are given in green, the other O atoms are white. Pb—O bonds are omitted for clarity; hydrogen-bonding interactions are shown as green dashed lines.
Each of the Pb2+ cations exhibits a coordination number of eight if Pb—O interactions less than 3.1 Å are considered to be relevant. The resulting [PbO8] polyhedra are considerably distorted [Pb—O distances range from 2.433 (7) to 3.099 (8) Å]. The stereochemical activity of the electron lone pairs in each of the polyhedra appears not to be very pronounced (Fig. 2 ▶).
Figure 2.
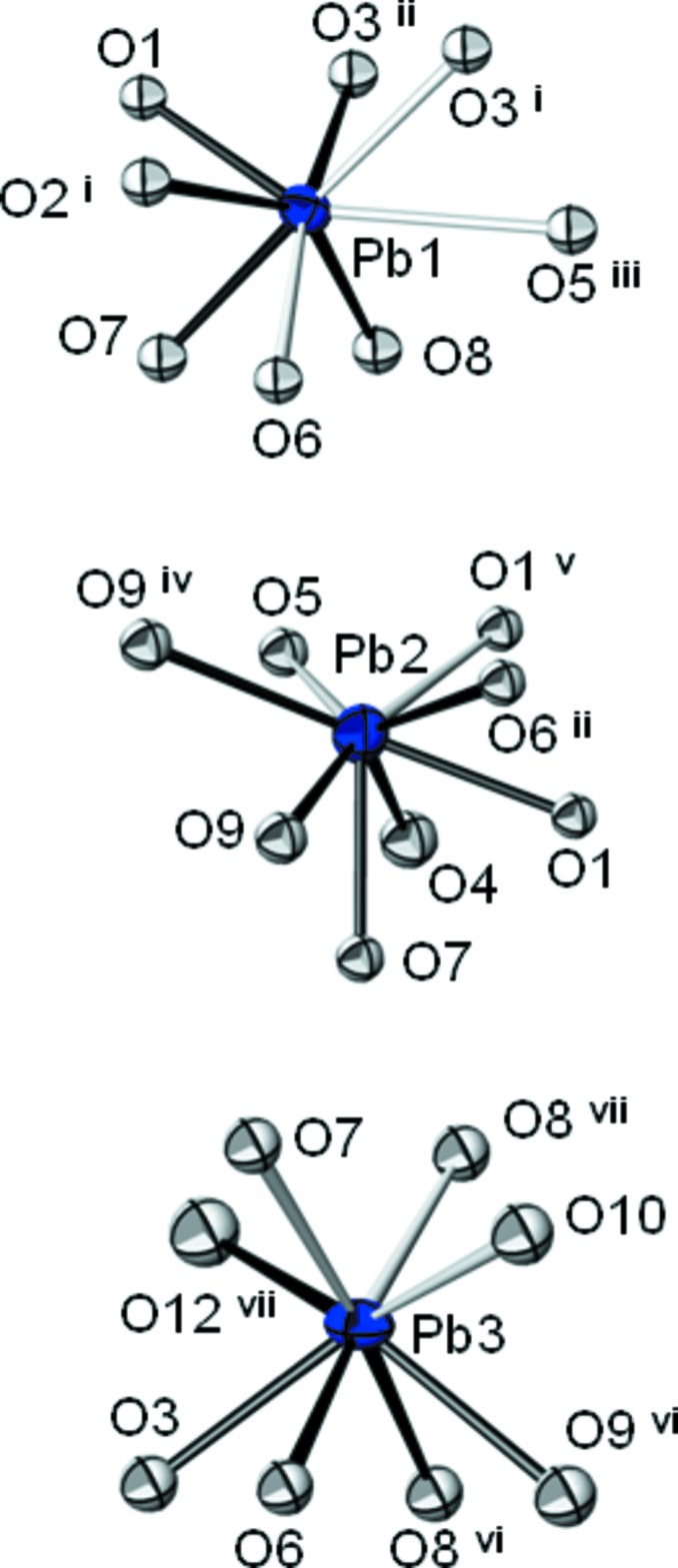
Coordination polyhedra of the three Pb2+ cations in the structure of Pb3(IO4(OH)2)2. Bonds shorter than 2.7 Å are given by solid black lines, longer bonds between 2.7 and 3.1 Å as open black lines. Displacement ellipsoids are drawn at the 90% probability level. [Symmetry codes: (i) −x, y −  , −z + ; (ii) x, −y + , z − ; (iii) x, y − 1, z; (iv) −x + 1, −y + 1, −z; (v) −x, −y + 1, −z; (vi) x, −y + , z + ; (vii) −x + 1, y + , −z + ; (viii) −x, y + , −z + .]
, −z + ; (ii) x, −y + , z − ; (iii) x, y − 1, z; (iv) −x + 1, −y + 1, −z; (v) −x, −y + 1, −z; (vi) x, −y + , z + ; (vii) −x + 1, y + , −z + ; (viii) −x, y + , −z + .]
Compounds and structures containing the periodate anion have been reviewed some time ago by Levason (1997 ▶). The compiled I—O bond lengths are in good agreement with the two IO6 octahedra of the title compound, having a mean I—O distance of 1.884 Å. Very similar mean values are found for comparable periodate compounds with large divalent cations, for example in BaI2O6(OH)4·2H2O (one IO6 octahedron, 1.895 Å; Häuseler, 2008 ▶), in Ba(IO3(OH)3) (one IO6 octahedron, 1.879 Å; Sasaki et al., 1995 ▶), in Hg3(IO4(OH)2)2 (two IO6 octahedra, 1.888 Å; Mormann & Jeitschko, 2000a ▶) or in Sr(IO2(OH)4)2·3H2O (two IO6 octahedra, 1.888 Å; Alexandrova & Häuseler, 2004 ▶).
Results of bond-valence calculations (Brown, 2002 ▶), using the parameters of Brese & O’Keeffe (1991 ▶) for I—O bonds and of Krivovichev & Brown (2001 ▶) for Pb—O bonds, are reasonably close to the expected values (in valence units): Pb1 1.89, Pb2 1.73, Pb3 1.89, I1 6.78, I2 6.90, O1 1.95, O2 1.49, O3 1.90, O4 1.15, O5 1.15, O6 1.92, O7 1.98, O8 1.95, O 9 1.97, O10 1.09, O11 1.34, O12 1.12. The O atoms involved in hydrogen bonding are readily identifiable. The donor O atoms O4, O5, O10 and O12 exhibit the longest I—O bonds and the lowest bond-valence sums. Atom O11 has also a low bond-valence sum, explainable by its role as a twofold acceptor atom of medium-strength hydrogen-bonding interactions (Table 2 ▶) that additionally stabilize the packing of the structure (Fig. 1 ▶).
Table 2. Hydrogen-bond geometry (Å).
| D—H⋯A | D⋯A | D—H⋯A | D⋯A |
|---|---|---|---|
| O4⋯O7 | 2.849 (11) | O10⋯O11iv | 2.675 (11) |
| O4⋯O2i | 2.849 (11) | O12⋯O2iv | 2.852 (11) |
| O5⋯O11iii | 2.634 (11) |
Symmetry codes: (i)  ; (iii)
; (iii)  ; (iv)
; (iv)  .
.
Comparison of the structures of Pb3(IO4(OH)2)2 and of Hg3(IO4(OH)2)2 [P21/c; Z = 4, a = 8.5429 (7), b = 12.2051 (8) Å, c = 9.3549 (8) Å, β = 90.884 (7)°] reveals some close similarities. A ‘true’ isotypic relationship (Lima-de-Faria et al., 1990 ▶) is difficult to derive for the two structures. However, they are isopointal and show the same type of arrangement in terms of the crystal packing. In the mercury compound, the IO4(OH)2 3− octahedra are likewise hexagonally packed in rods (Fig. 3 ▶). The cations are situated in between this arrangement which is further consolidated by O—H⋯O hydrogen bonding.
Figure 3.

The crystal structure of Hg3(IO4(OH)2)2 (Mormann & Jeitschko, 2000a ▶) in a projection along [011]. Colour code as in Fig. 1 ▶. Hg—O and O—H⋯O interactions are omitted for clarity.
Synthesis and crystallization
The preparation conditions described by Willard & Thompson (1934 ▶) were modified slightly. Instead of using NaIO4 as the periodate source, periodic acid was employed.
1.25 g Pb(NO3)2 was dissolved in 25 ml water, acidified with a few drops of concentrated nitric acid and heated until boiling. Then the periodic acid solution (0.85 g in 25 ml water) was slowly added to the lead solution. The addition of the first portion of the periodic acid solution (ca. 3–4 ml) resulted in an off-white precipitate near the drop point that redissolved under stirring. After further addition, the precipitate remained and changed the colour in the still boiling solution from off-white to yellow–orange within half an hour. After filtration of the precipitate, a few colourless crystals of the title compound formed in the mother liquor on cooling. X-ray powder diffraction data of the polycrystalline precipitate are in very good agreement with simulated data based on the refinement of Pb3(IO4(OH)2)2.
Refinement
All investigated crystals were twinned by non-merohedry. Intensity data of the measured crystal could be indexed to belong to two domains, with a refined twin domain ratio of 0.73 (1):0.27 (1). Reflections originating from the minor component as well as overlapping reflections of the two domains (less than 10% of all measured reflections) were separated and excluded. The H atoms of the IO4(OH)2 octahedra could not be located from difference maps and were therefore not considered in the final model. The O atoms were refined with isotropic displacement parameters. The remaining maximum and minimum electron densities are found 0.73 and 0.68 Å, respectively, from atom Pb2. Structure data were finally standardized with STRUCTURE-TIDY (Gelato & Parthé, 1987 ▶). It should be noted that the intensity statistics point to a pronounced C-centred basis cell (space group C2/c with lattice parameters of a ≃ 14.16, b ≃ 9.21, c ≃ 8.97 Å, β ≃ 117.4°) with weak superstructure reflections violating the C-centering.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536814009520/hb0004sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814009520/hb0004Isup2.hkl
CCDC reference: 1004265
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Selected bond lengths (Å).
| I1—O6 | 1.845 (8) | I2—O11 | 1.820 (8) |
| I1—O3 | 1.860 (7) | I2—O9 | 1.850 (8) |
| I1—O2i | 1.861 (7) | I2—O8 | 1.855 (7) |
| I1—O1ii | 1.877 (7) | I2—O7 | 1.874 (8) |
| I1—O5i | 1.920 (8) | I2—O12 | 1.932 (9) |
| I1—O4i | 1.956 (8) | I2—O10 | 1.954 (8) |
Symmetry codes: (i) ; (ii)  .
.
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | Pb3[IO4(OH)2]2 |
| M r | 1071.40 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 296 |
| a, b, c (Å) | 8.9653 (9), 9.2113 (9), 12.8052 (13) |
| β (°) | 101.042 (2) |
| V (Å3) | 1037.90 (18) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 54.55 |
| Crystal size (mm) | 0.06 × 0.06 × 0.05 |
| Data collection | |
| Diffractometer | Siemens SMART CCD |
| Absorption correction | Multi-scan (TWINABS; Bruker, 2008 ▶) |
| T min, T max | 0.253, 0.746 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 3196, 3196, 2587 |
| (sin θ/λ)max (Å−1) | 0.716 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.041, 0.087, 1.07 |
| No. of reflections | 3196 |
| No. of parameters | 94 |
| H-atom treatment | H-atom parameters not refined |
| Δρmax, Δρmin (e Å−3) | 2.88, −1.95 |
Acknowledgments
The X-ray centre of the Vienna University of Technology is acknowledged for providing access to the single-crystal diffractometer.
supplementary crystallographic information
Crystal data
| Pb3[IO4(OH)2]2 | F(000) = 1808 |
| Mr = 1071.40 | Dx = 6.857 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 3673 reflections |
| a = 8.9653 (9) Å | θ = 3.2–30.5° |
| b = 9.2113 (9) Å | µ = 54.55 mm−1 |
| c = 12.8052 (13) Å | T = 296 K |
| β = 101.042 (2)° | Block, colourless |
| V = 1037.90 (18) Å3 | 0.06 × 0.06 × 0.05 mm |
| Z = 4 |
Data collection
| Siemens SMART CCD diffractometer | 3196 independent reflections |
| Radiation source: fine-focus sealed tube | 2587 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.000 |
| ω scans | θmax = 30.6°, θmin = 2.3° |
| Absorption correction: multi-scan (TWINABS; Bruker, 2008) | h = −12→12 |
| Tmin = 0.253, Tmax = 0.746 | k = 0→13 |
| 3196 measured reflections | l = 0→18 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.041 | H-atom parameters not refined |
| wR(F2) = 0.087 | w = 1/[σ2(Fo2) + (0.0319P)2 + 17.8096P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.07 | (Δ/σ)max < 0.001 |
| 3196 reflections | Δρmax = 2.88 e Å−3 |
| 94 parameters | Δρmin = −1.95 e Å−3 |
| 0 restraints |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Pb1 | 0.12192 (5) | 0.13042 (4) | 0.11904 (3) | 0.01318 (10) | |
| Pb2 | 0.25685 (5) | 0.51903 (5) | 0.01540 (4) | 0.01842 (11) | |
| Pb3 | 0.37507 (5) | 0.36874 (5) | 0.38134 (3) | 0.01409 (10) | |
| I1 | 0.00670 (7) | 0.23313 (7) | 0.36046 (5) | 0.00838 (13) | |
| I2 | 0.50186 (7) | 0.24738 (6) | 0.14267 (5) | 0.00735 (13) | |
| O1 | 0.0343 (9) | 0.3332 (8) | 0.0015 (6) | 0.0110 (14)* | |
| O2 | 0.0389 (9) | 0.7922 (8) | 0.2811 (6) | 0.0125 (15)* | |
| O3 | 0.1058 (9) | 0.4046 (8) | 0.4090 (6) | 0.0122 (15)* | |
| O4 | 0.1088 (10) | 0.5549 (9) | 0.1797 (6) | 0.0178 (17)* | |
| O5 | 0.1831 (9) | 0.8100 (8) | 0.1159 (6) | 0.0129 (15)* | |
| O6 | 0.1842 (9) | 0.1429 (8) | 0.3433 (6) | 0.0117 (15)* | |
| O7 | 0.3161 (9) | 0.3260 (8) | 0.1618 (6) | 0.0121 (15)* | |
| O8 | 0.4052 (9) | 0.0832 (8) | 0.0785 (6) | 0.0121 (15)* | |
| O9 | 0.4802 (9) | 0.3391 (8) | 0.0121 (6) | 0.0144 (16)* | |
| O10 | 0.5247 (9) | 0.1474 (8) | 0.2794 (6) | 0.0145 (16)* | |
| O11 | 0.6146 (10) | 0.3912 (8) | 0.2170 (6) | 0.0158 (16)* | |
| O12 | 0.6856 (10) | 0.1562 (9) | 0.1172 (6) | 0.0182 (17)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Pb1 | 0.0134 (2) | 0.01402 (19) | 0.01215 (19) | −0.00236 (15) | 0.00243 (15) | 0.00042 (14) |
| Pb2 | 0.0168 (2) | 0.01424 (19) | 0.0236 (2) | 0.00084 (17) | 0.00251 (16) | 0.00040 (16) |
| Pb3 | 0.0153 (2) | 0.0173 (2) | 0.01036 (18) | −0.00366 (16) | 0.00426 (15) | −0.00050 (14) |
| I1 | 0.0075 (3) | 0.0104 (3) | 0.0072 (3) | −0.0003 (2) | 0.0014 (2) | 0.0006 (2) |
| I2 | 0.0066 (3) | 0.0078 (3) | 0.0076 (3) | −0.0002 (2) | 0.0011 (2) | −0.0005 (2) |
Geometric parameters (Å, º)
| Pb1—O1 | 2.433 (7) | Pb3—O9vi | 2.599 (8) |
| Pb1—O7 | 2.493 (8) | Pb3—O6 | 2.678 (8) |
| Pb1—O2i | 2.577 (8) | Pb3—O12vii | 2.704 (8) |
| Pb1—O3ii | 2.685 (7) | Pb3—O8vii | 2.767 (8) |
| Pb1—O8 | 2.723 (8) | Pb3—O7 | 2.787 (7) |
| Pb1—O6 | 2.821 (7) | Pb3—O10 | 2.886 (8) |
| Pb1—O3i | 2.888 (8) | I1—O6 | 1.845 (8) |
| Pb1—O5iii | 3.004 (7) | I1—O3 | 1.860 (7) |
| Pb2—O7 | 2.564 (7) | I1—O2i | 1.861 (7) |
| Pb2—O9 | 2.606 (8) | I1—O1vi | 1.877 (7) |
| Pb2—O1 | 2.609 (7) | I1—O5i | 1.920 (8) |
| Pb2—O6ii | 2.638 (7) | I1—O4i | 1.956 (8) |
| Pb2—O4 | 2.714 (8) | I2—O11 | 1.820 (8) |
| Pb2—O9iv | 2.777 (8) | I2—O9 | 1.850 (8) |
| Pb2—O1v | 2.915 (7) | I2—O8 | 1.855 (7) |
| Pb2—O5 | 3.099 (8) | I2—O7 | 1.874 (8) |
| Pb3—O8vi | 2.527 (7) | I2—O12 | 1.932 (9) |
| Pb3—O3 | 2.528 (8) | I2—O10 | 1.954 (8) |
| O1—Pb1—O7 | 73.1 (2) | O7—I2—O10 | 90.5 (3) |
| O1—Pb1—O2i | 73.6 (2) | O12—I2—O10 | 90.0 (3) |
| O7—Pb1—O2i | 84.6 (2) | I1ii—O1—Pb1 | 108.2 (3) |
| O1—Pb1—O3ii | 61.5 (2) | I1ii—O1—Pb2 | 103.7 (3) |
| O7—Pb1—O3ii | 102.0 (2) | Pb1—O1—Pb2 | 108.0 (3) |
| O2i—Pb1—O3ii | 129.7 (2) | I1ii—O1—Pb2v | 97.4 (3) |
| O1—Pb1—O8 | 102.0 (2) | Pb1—O1—Pb2v | 125.6 (3) |
| O7—Pb1—O8 | 61.3 (2) | Pb2—O1—Pb2v | 111.1 (2) |
| O2i—Pb1—O8 | 144.9 (2) | I1ii—O1—Pb3ii | 57.6 (2) |
| O3ii—Pb1—O8 | 70.3 (2) | Pb1—O1—Pb3ii | 73.14 (18) |
| O1—Pb1—O6 | 125.2 (2) | Pb2—O1—Pb3ii | 73.13 (17) |
| O7—Pb1—O6 | 75.7 (2) | Pb2v—O1—Pb3ii | 154.3 (2) |
| O2i—Pb1—O6 | 59.4 (2) | I1viii—O2—Pb1viii | 106.0 (3) |
| O3ii—Pb1—O6 | 170.7 (2) | I1viii—O2—Pb2ix | 156.0 (4) |
| O8—Pb1—O6 | 101.0 (2) | Pb1viii—O2—Pb2ix | 97.4 (2) |
| O1—Pb1—O3i | 109.8 (2) | I1viii—O2—Pb1x | 75.1 (2) |
| O7—Pb1—O3i | 174.4 (2) | Pb1viii—O2—Pb1x | 155.4 (3) |
| O2i—Pb1—O3i | 91.7 (2) | Pb2ix—O2—Pb1x | 86.03 (16) |
| O3ii—Pb1—O3i | 83.5 (2) | I1viii—O2—Pb3viii | 62.0 (2) |
| O8—Pb1—O3i | 121.6 (2) | Pb1viii—O2—Pb3viii | 78.83 (19) |
| O6—Pb1—O3i | 98.8 (2) | Pb2ix—O2—Pb3viii | 129.9 (2) |
| O1—Pb1—O5iii | 141.7 (2) | Pb1x—O2—Pb3viii | 80.39 (14) |
| O7—Pb1—O5iii | 126.3 (2) | I1—O3—Pb3 | 104.5 (3) |
| O2i—Pb1—O5iii | 134.1 (2) | I1—O3—Pb1vi | 99.4 (3) |
| O3ii—Pb1—O5iii | 81.0 (2) | Pb3—O3—Pb1vi | 104.8 (3) |
| O8—Pb1—O5iii | 70.2 (2) | I1—O3—Pb1viii | 106.8 (3) |
| O6—Pb1—O5iii | 93.0 (2) | Pb3—O3—Pb1viii | 138.4 (3) |
| O3i—Pb1—O5iii | 54.4 (2) | Pb1vi—O3—Pb1viii | 96.5 (2) |
| O7—Pb2—O9 | 61.2 (2) | I1viii—O4—Pb2 | 102.2 (3) |
| O7—Pb2—O1 | 69.1 (2) | I1viii—O4—Pb3 | 146.5 (3) |
| O9—Pb2—O1 | 99.3 (2) | Pb2—O4—Pb3 | 98.2 (2) |
| O7—Pb2—O6ii | 101.7 (2) | I1viii—O4—Pb1viii | 71.6 (2) |
| O9—Pb2—O6ii | 72.2 (2) | Pb2—O4—Pb1viii | 173.4 (3) |
| O1—Pb2—O6ii | 60.6 (2) | Pb3—O4—Pb1viii | 88.39 (17) |
| O7—Pb2—O4 | 65.3 (2) | I1viii—O4—Pb2v | 68.3 (2) |
| O9—Pb2—O4 | 125.5 (2) | Pb2—O4—Pb2v | 87.5 (2) |
| O1—Pb2—O4 | 69.6 (2) | Pb3—O4—Pb2v | 139.5 (2) |
| O6ii—Pb2—O4 | 129.6 (2) | Pb1viii—O4—Pb2v | 87.94 (18) |
| O7—Pb2—O9iv | 110.9 (2) | I1viii—O4—Pb1 | 143.6 (3) |
| O9—Pb2—O9iv | 67.9 (3) | Pb2—O4—Pb1 | 72.12 (18) |
| O1—Pb2—O9iv | 163.1 (2) | Pb3—O4—Pb1 | 68.44 (14) |
| O6ii—Pb2—O9iv | 103.9 (2) | Pb1viii—O4—Pb1 | 111.3 (2) |
| O4—Pb2—O9iv | 126.5 (2) | Pb2v—O4—Pb1 | 75.48 (15) |
| O7—Pb2—O1v | 115.8 (2) | I1viii—O5—Pb1x | 101.0 (3) |
| O9—Pb2—O1v | 167.4 (2) | I1viii—O5—Pb2 | 90.7 (3) |
| O1—Pb2—O1v | 68.9 (2) | Pb1x—O5—Pb2 | 155.4 (3) |
| O6ii—Pb2—O1v | 97.3 (2) | I1viii—O5—Pb1v | 69.1 (2) |
| O4—Pb2—O1v | 55.7 (2) | Pb1x—O5—Pb1v | 75.90 (16) |
| O9iv—Pb2—O1v | 122.7 (2) | Pb2—O5—Pb1v | 88.49 (18) |
| O7—Pb2—O5 | 109.1 (2) | I1viii—O5—Pb3vii | 163.1 (3) |
| O9—Pb2—O5 | 141.5 (2) | Pb1x—O5—Pb3vii | 92.88 (19) |
| O1—Pb2—O5 | 112.0 (2) | Pb2—O5—Pb3vii | 80.25 (17) |
| O6ii—Pb2—O5 | 142.9 (2) | Pb1v—O5—Pb3vii | 124.4 (2) |
| O4—Pb2—O5 | 53.0 (2) | I1—O6—Pb2vi | 103.6 (3) |
| O9iv—Pb2—O5 | 84.2 (2) | I1—O6—Pb3 | 99.4 (3) |
| O1v—Pb2—O5 | 50.8 (2) | Pb2vi—O6—Pb3 | 103.9 (3) |
| O8vi—Pb3—O3 | 76.1 (3) | I1—O6—Pb1 | 97.6 (3) |
| O8vi—Pb3—O9vi | 61.9 (2) | Pb2vi—O6—Pb1 | 142.8 (3) |
| O3—Pb3—O9vi | 104.1 (2) | Pb3—O6—Pb1 | 102.2 (2) |
| O8vi—Pb3—O6 | 105.1 (2) | I2—O7—Pb1 | 107.0 (3) |
| O3—Pb3—O6 | 62.2 (2) | I2—O7—Pb2 | 103.7 (3) |
| O9vi—Pb3—O6 | 71.7 (2) | Pb1—O7—Pb2 | 107.6 (3) |
| O8vi—Pb3—O12vii | 78.7 (2) | I2—O7—Pb3 | 100.6 (3) |
| O3—Pb3—O12vii | 70.9 (3) | Pb1—O7—Pb3 | 108.2 (3) |
| O9vi—Pb3—O12vii | 140.0 (2) | Pb2—O7—Pb3 | 127.7 (3) |
| O6—Pb3—O12vii | 129.8 (2) | I2—O7—Pb3ii | 56.96 (19) |
| O8vi—Pb3—O8vii | 75.7 (3) | Pb1—O7—Pb3ii | 72.30 (17) |
| O3—Pb3—O8vii | 123.1 (2) | Pb2—O7—Pb3ii | 73.10 (17) |
| O9vi—Pb3—O8vii | 104.4 (2) | Pb3—O7—Pb3ii | 154.9 (3) |
| O6—Pb3—O8vii | 174.5 (2) | I2—O8—Pb3ii | 104.5 (3) |
| O12vii—Pb3—O8vii | 55.7 (2) | I2—O8—Pb1 | 99.1 (3) |
| O8vi—Pb3—O7 | 174.9 (2) | Pb3ii—O8—Pb1 | 103.7 (3) |
| O3—Pb3—O7 | 99.1 (2) | I2—O8—Pb3xi | 104.1 (3) |
| O9vi—Pb3—O7 | 121.4 (2) | Pb3ii—O8—Pb3xi | 104.3 (3) |
| O6—Pb3—O7 | 73.5 (2) | Pb1—O8—Pb3xi | 137.3 (3) |
| O12vii—Pb3—O7 | 98.4 (2) | I2—O9—Pb3ii | 102.0 (3) |
| O8vii—Pb3—O7 | 106.2 (2) | I2—O9—Pb2 | 102.9 (3) |
| O8vi—Pb3—O10 | 127.3 (2) | Pb3ii—O9—Pb2 | 107.1 (3) |
| O3—Pb3—O10 | 134.0 (2) | I2—O9—Pb2iv | 112.6 (4) |
| O9vi—Pb3—O10 | 68.2 (2) | Pb3ii—O9—Pb2iv | 118.5 (3) |
| O6—Pb3—O10 | 72.9 (2) | Pb2—O9—Pb2iv | 112.1 (3) |
| O12vii—Pb3—O10 | 143.8 (2) | I2—O10—Pb3 | 95.4 (3) |
| O8vii—Pb3—O10 | 102.3 (2) | I2—O10—Pb2xi | 148.8 (4) |
| O7—Pb3—O10 | 57.2 (2) | Pb3—O10—Pb2xi | 98.9 (2) |
| O6—I1—O3 | 93.1 (3) | I2—O10—Pb3xi | 79.3 (2) |
| O6—I1—O2i | 92.8 (3) | Pb3—O10—Pb3xi | 167.0 (3) |
| O3—I1—O2i | 94.6 (3) | Pb2xi—O10—Pb3xi | 91.50 (19) |
| O6—I1—O1vi | 90.6 (3) | I2—O10—Pb1 | 67.0 (2) |
| O3—I1—O1vi | 89.3 (3) | Pb3—O10—Pb1 | 78.32 (18) |
| O2i—I1—O1vi | 174.7 (3) | Pb2xi—O10—Pb1 | 143.2 (2) |
| O6—I1—O5i | 174.5 (3) | Pb3xi—O10—Pb1 | 88.65 (17) |
| O3—I1—O5i | 90.9 (3) | I2—O11—Pb3 | 85.6 (3) |
| O2i—I1—O5i | 90.5 (3) | I2—O11—Pb2iv | 88.1 (3) |
| O1vi—I1—O5i | 85.8 (3) | Pb3—O11—Pb2iv | 157.4 (3) |
| O6—I1—O4i | 90.9 (3) | I2—O11—Pb1vii | 171.0 (4) |
| O3—I1—O4i | 174.5 (3) | Pb3—O11—Pb1vii | 95.8 (2) |
| O2i—I1—O4i | 89.0 (3) | Pb2iv—O11—Pb1vii | 93.74 (19) |
| O1vi—I1—O4i | 86.9 (3) | I2—O11—Pb2 | 64.5 (2) |
| O5i—I1—O4i | 84.8 (3) | Pb3—O11—Pb2 | 83.53 (18) |
| O11—I2—O9 | 95.3 (3) | Pb2iv—O11—Pb2 | 74.20 (15) |
| O11—I2—O8 | 172.0 (3) | Pb1vii—O11—Pb2 | 124.5 (2) |
| O9—I2—O8 | 90.7 (3) | I2—O12—Pb3xi | 104.2 (4) |
| O11—I2—O7 | 93.9 (4) | I2—O12—Pb2iv | 85.5 (3) |
| O9—I2—O7 | 90.0 (3) | Pb3xi—O12—Pb2iv | 151.9 (3) |
| O8—I2—O7 | 91.2 (3) | I2—O12—Pb3ii | 68.4 (2) |
| O11—I2—O12 | 89.9 (4) | Pb3xi—O12—Pb3ii | 79.9 (2) |
| O9—I2—O12 | 89.5 (4) | Pb2iv—O12—Pb3ii | 79.42 (16) |
| O8—I2—O12 | 84.9 (3) | I2—O12—Pb1xii | 155.4 (4) |
| O7—I2—O12 | 176.1 (3) | Pb3xi—O12—Pb1xii | 98.2 (2) |
| O11—I2—O10 | 85.5 (3) | Pb2iv—O12—Pb1xii | 79.41 (17) |
| O9—I2—O10 | 179.1 (3) | Pb3ii—O12—Pb1xii | 126.6 (2) |
| O8—I2—O10 | 88.4 (3) |
Symmetry codes: (i) −x, y−1/2, −z+1/2; (ii) x, −y+1/2, z−1/2; (iii) x, y−1, z; (iv) −x+1, −y+1, −z; (v) −x, −y+1, −z; (vi) x, −y+1/2, z+1/2; (vii) −x+1, y+1/2, −z+1/2; (viii) −x, y+1/2, −z+1/2; (ix) x, −y+3/2, z+1/2; (x) x, y+1, z; (xi) −x+1, y−1/2, −z+1/2; (xii) x+1, y, z.
Hydrogen-bond geometry (Å)
| D—H···A | D···A |
| O4···O7 | 2.849 (11) |
| O4···O2i | 2.849 (11) |
| O5···O11vii | 2.634 (11) |
| O10···O11xi | 2.675 (11) |
| O12···O2xi | 2.852 (11) |
Symmetry codes: (i) −x, y−1/2, −z+1/2; (vii) −x+1, y+1/2, −z+1/2; (xi) −x+1, y−1/2, −z+1/2.
References
- Alexandrova, M. & Häuseler, H. (2004). J. Mol. Struct. 706, 7–13.
- Breitinger, D. K. (2004). Cadmium and Mercury, in Comprehensive Coordination Chemistry II, edited by J. A. McCleverty & T. J. Meyer, ch. 6, pp. 1253–1292. Oxford: Elsevier.
- Brese, N. E. & O’Keeffe, M. (1991). Acta Cryst. B47, 192–197.
- Brown, I. D. (2002). In The Chemical Bond in Inorganic Chemistry: The Bond Valence Model Oxford University Press.
- Bruker (2008). SMART, SAINT-Plus and TWINABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Dowty, E. (2006). ATOMS for Windows Shape Software, Kingsport, Tennessee, USA.
- Drátovský, M. & Matějčková, J. (1965a). Chem. Zvesti, 19, 604–610.
- Drátovský, M. & Matějčková, J. (1965b). Chem. Zvesti, 19, 447–455.
- Gelato, L. M. & Parthé, E. (1987). J. Appl. Cryst. 20, 139–143.
- Häuseler, H. (2008). J. Mol. Struct. 892, 1–7.
- Hawthorne, F. C. & Faggiani, R. (1979). Acta Cryst. B35, 717–720.
- Holloway, C. E. & Melnik, M. (1997). Main Group Met. Chem. 20, 583–625.
- Krivovichev, S. V. & Brown, I. D. (2001). Z. Kristallogr. 216, 245–247.
- Levason, W. (1997). Coord. Chem. Rev. 161, 33–79.
- Lima-de-Faria, J., Hellner, E., Liebau, F., Makovicky, E. & Parthé, E. (1990). Acta Cryst. A46, 1–11.
- Losilla, E. R., Aranda, M. A. G., Ramirez, F. J. & Bruque, S. (1995). J. Phys. Chem. 99, 12975–12979.
- Mormann, T. J. & Jeitschko, W. (2000a). Z. Kristallogr. New Cryst. Struct., 215, 315–316.
- Mormann, T. J. & Jeitschko, W. (2000b). Z. Kristallogr. New Cryst. Struct., 215, 471–472.
- Mormann, T. J. & Jeitschko, W. (2001). Z. Kristallogr. New Cryst. Struct., 216, 1–2.
- Sasaki, M., Yarita, T. & Sato, S. (1995). Acta Cryst. C51, 1968–1970.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Weil, M. (2000). Z. Naturforsch. Teil B, 55, 699–706.
- Weil, M. (2004). Acta Cryst. E60, i25–i27.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Willard, H. H. & Thompson, J. J. (1934). J. Am. Chem. Soc. 56, 1828–1830.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536814009520/hb0004sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814009520/hb0004Isup2.hkl
CCDC reference: 1004265
Additional supporting information: crystallographic information; 3D view; checkCIF report