

Abstract
The title compound, [Pd(NH3)4]Cl2·4NH3, was crystallized in liquid ammonia from the salt Pd(en)Cl2 (en is ethylenediamine) and is isotypic with [Pt(NH3)4]Cl2·4NH3 [Grassl & Korber (2014 ▶). Acta Cryst. E70, i31]. The Pd2+ cation is coordinated by four ammonia molecules, exhibiting a square-planar geometry. The chloride anions are surrounded by nine ammonia molecules. These are either bound in the palladium complex or solvent molecules. The packing of the ammonia solvent molecules enables the formation of an extended network of N—H⋯N and N—H⋯Cl interactions with nearly ideal hydrogen-bonding geometry.
Related literature
For weak intermolecular interactions such as hydrogen bonds and their application in crystal engeneering, see: Desiraju (2002 ▶); Desiraju (2007 ▶); Steiner (2002 ▶). For the structure of tetraamminepalladium(II) chloride monoydrate and complexation of palladium by carbohydrates, see: Bell et al. (1976 ▶); Ahlrichs et al. (1998 ▶). The structure of the platinum analogue is given by Grassl & Korber (2014 ▶)
Experimental
Crystal data
[Pd(NH3)4]Cl2·4NH3
M r = 313.58
Monoclinic,
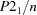
a = 7.6856 (5) Å
b = 10.1505 (7) Å
c = 8.7170 (6) Å
β = 100.384 (7)°
V = 668.90 (8) Å3
Z = 2
Mo Kα radiation
μ = 1.76 mm−1
T = 123 K
0.32 × 0.29 × 0.23 mm
Data collection
Agilent Xcalibur (Ruby, Gemini ultra) diffractometer
Absorption correction: analytical [CrysAlis PRO (Agilent, 2012 ▶), using a multi-faceted crystal model based on expressions derived by Clark & Reid (1995 ▶)] T min = 0.649, T max = 0.741
2418 measured reflections
1266 independent reflections
1076 reflections with I > 2σ(I)
R int = 0.034
Refinement
R[F 2 > 2σ(F 2)] = 0.027
wR(F 2) = 0.058
S = 1.06
1266 reflections
100 parameters
All H-atom parameters refined
Δρmax = 0.45 e Å−3
Δρmin = −0.55 e Å−3
Data collection: CrysAlis PRO (Agilent, 2012 ▶); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: OLEX2.solve (Bourhis et al., 2014 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg & Putz, 2012 ▶); software used to prepare material for publication: OLEX2 (Dolomanov et al., 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814012355/pk2523sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814012355/pk2523Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814012355/pk2523Isup3.mol
CCDC reference: 1005539
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1A⋯Cl1 | 0.86 (4) | 2.49 (4) | 3.351 (3) | 175 (3) |
| N1—H1B⋯Cl1i | 0.85 (3) | 2.49 (4) | 3.328 (3) | 171 (3) |
| N2—H2A⋯N3ii | 1.03 (4) | 2.02 (4) | 3.025 (5) | 163 (3) |
| N1—H1C⋯N4 | 0.96 (4) | 2.02 (5) | 2.975 (5) | 170 (3) |
| N2—H2B⋯Cl1i | 0.73 (3) | 2.66 (3) | 3.384 (4) | 172 (3) |
| N2—H2C⋯Cl1iii | 0.88 (5) | 2.62 (5) | 3.463 (3) | 162 (4) |
| N3—H3A⋯Cl1 | 0.83 (3) | 2.83 (3) | 3.563 (4) | 148 (3) |
| N4—H4A⋯Cl1iv | 0.91 (4) | 2.65 (4) | 3.563 (4) | 173 (3) |
| N4—H4B⋯Cl1v | 1.00 (5) | 2.61 (5) | 3.606 (4) | 174 (4) |
| N3—H3B⋯Cl1vi | 0.89 (5) | 2.71 (5) | 3.578 (4) | 163 (3) |
| N3—H3C⋯Cl1vii | 1.09 (5) | 2.48 (5) | 3.535 (4) | 162 (4) |
Symmetry codes: (i) 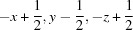 ; (ii)
; (ii) 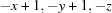 ; (iii)
; (iii) 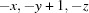 ; (iv)
; (iv) 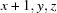 ; (v)
; (v) 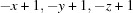 ; (vi)
; (vi) 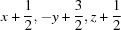 ; (vii)
; (vii) 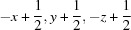 .
.
supplementary crystallographic information
S1. Comment
The crystal structure of the title compound was determined in the course of investigations into the reactivity of carbohydrates towards metal cations in liquid ammonia.
As in the platinum compound, the palladium cation forms a homoleptic ammine complex with a square-planar coordination geometry. Pd—N bond lengths are 2.032 (3) Å and 2.048 (3) Å, respectively, while the angles N—Pd—N are 88.59 (13)° and 91.41 (13)°. Ammonia ligands opposite to each other within the complex cation have staggered hydrogen atom positions (Fig. 1).
The chloride anion exhibits nine contacts to hydrogen atoms of ammonia molecules which are either bound in the complex or solvate molecules, forming a network of hydrogen bonds (Fig. 2 and Fig. 3). Bond angles (N—H···Cl) are between 148 (3)° and 175 (3)° whereas N—H···Cl bond lengths are observed with values between 2.48 (5) Å and 2.83 (3) Å. The two N—H···N bridges are close to 180°, with bond angles of 163 (3)° and 170 (3)° and bond lengths significantly less than the sum of the van der Waals radii of nitrogen and hydrogen (2.02 (4) Å and 2.02 (5) Å). These observations give strong evidence that a significant energy contribution from the hydrogen bond network drives the arrangement of the overall structure.
S2. Experimental
0.25 g (1.05 mmol) Pd(en)Cl2 and 0.188 g (1.05 mmol) D-(+)-glucono-1,5-lactone were placed under argon atmosphere in a reaction flask and 50 ml of dry liquid ammonia were condensed. This mixture was stored in a refrigerator at 237 K for one week to ensure that all substances were completely dissolved. The flask was then stored at 161 K for five months. After that period of time, clear colorless crystals of the title compound were found on the wall of the reaction vessel.
S3. Refinement
The crystal structure does not show any features where special refinement procedures had to be applied. All hydrogen atoms were located in difference maps and both bond angle/bond length and isotropic displacement parameters were refined.
Figures
Fig. 1.

: Crystal structure of the title compound with labeling and displacement ellipsoids drawn at the 50% probability level. Symmetry code: (i) 1 - x, 1 - y, - z.
Fig. 2.

: The chloride anion is shown with its surrounding molecules. The predominant bond type is hydrogen bonding. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 3.

: Extended network of hydrogen bonds in the crystal structure. The solvent ammonia molecules are oriented to optimize the hydrogen bond geometry. Displacement ellipsoids are drawn at the 50% probability level.
Crystal data
| [Pd(NH3)4]Cl2·4NH3 | F(000) = 320 |
| Mr = 313.58 | Dx = 1.557 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 7.6856 (5) Å | Cell parameters from 1429 reflections |
| b = 10.1505 (7) Å | θ = 3.1–29.3° |
| c = 8.7170 (6) Å | µ = 1.76 mm−1 |
| β = 100.384 (7)° | T = 123 K |
| V = 668.90 (8) Å3 | Block, clear colourless |
| Z = 2 | 0.32 × 0.29 × 0.23 mm |
Data collection
| Agilent Xcalibur (Ruby, Gemini ultra) diffractometer | 1266 independent reflections |
| Radiation source: fine-focus sealed tube | 1076 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.034 |
| phi and ω scans | θmax = 25.7°, θmin = 3.1° |
| Absorption correction: analytical [CrysAlis PRO (Agilent, 2012), using a multi-faceted crystal model based on expressions derived by Clark & Reid (1995)] | h = −7→9 |
| Tmin = 0.649, Tmax = 0.741 | k = −12→12 |
| 2418 measured reflections | l = −10→10 |
Refinement
| Refinement on F2 | Primary atom site location: iterative |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.027 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.058 | All H-atom parameters refined |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0075P)2] where P = (Fo2 + 2Fc2)/3 |
| 1266 reflections | (Δ/σ)max < 0.001 |
| 100 parameters | Δρmax = 0.45 e Å−3 |
| 0 restraints | Δρmin = −0.55 e Å−3 |
Special details
| Experimental. Absorption correction: CrysAlisPro, Agilent Technologies, Version 1.171.35.21 Analytical numeric absorption correction using a multifaceted crystal model based on expressions derived by R.C. Clark & J.S. Reid. (Clark & Reid, 1995) |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Pd1 | 0.5000 | 0.5000 | 0.0000 | 0.01592 (13) | |
| Cl1 | 0.09460 (10) | 0.66086 (7) | 0.23005 (10) | 0.0236 (2) | |
| N1 | 0.4602 (4) | 0.4827 (3) | 0.2233 (3) | 0.0200 (6) | |
| N2 | 0.3507 (4) | 0.3356 (3) | −0.0659 (4) | 0.0219 (6) | |
| N3 | 0.5125 (5) | 0.8226 (3) | 0.3132 (4) | 0.0358 (8) | |
| N4 | 0.7709 (4) | 0.5695 (4) | 0.4561 (4) | 0.0353 (8) | |
| H1A | 0.364 (5) | 0.525 (3) | 0.229 (5) | 0.038 (12)* | |
| H1B | 0.449 (4) | 0.402 (3) | 0.246 (4) | 0.024 (10)* | |
| H2A | 0.382 (4) | 0.294 (3) | −0.166 (5) | 0.036 (10)* | |
| H1C | 0.556 (6) | 0.521 (3) | 0.296 (5) | 0.046 (12)* | |
| H2B | 0.358 (4) | 0.292 (3) | 0.001 (4) | 0.018 (11)* | |
| H2C | 0.238 (6) | 0.355 (4) | −0.097 (5) | 0.064 (14)* | |
| H3A | 0.403 (5) | 0.818 (3) | 0.287 (4) | 0.017 (9)* | |
| H4A | 0.861 (5) | 0.594 (3) | 0.406 (4) | 0.028 (10)* | |
| H4B | 0.817 (6) | 0.507 (4) | 0.541 (6) | 0.066 (15)* | |
| H3B | 0.554 (6) | 0.815 (4) | 0.415 (6) | 0.055 (14)* | |
| H4C | 0.751 (5) | 0.646 (4) | 0.517 (5) | 0.062 (14)* | |
| H3C | 0.493 (6) | 0.925 (5) | 0.277 (6) | 0.086 (16)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Pd1 | 0.01442 (19) | 0.0156 (2) | 0.0177 (2) | −0.00015 (13) | 0.00265 (14) | 0.00067 (14) |
| Cl1 | 0.0235 (4) | 0.0204 (4) | 0.0260 (4) | 0.0014 (4) | 0.0024 (3) | −0.0023 (4) |
| N1 | 0.0242 (16) | 0.0182 (16) | 0.0186 (16) | −0.0026 (14) | 0.0069 (13) | 0.0011 (13) |
| N2 | 0.0226 (16) | 0.0219 (16) | 0.0210 (17) | −0.0041 (14) | 0.0031 (14) | 0.0023 (15) |
| N3 | 0.041 (2) | 0.035 (2) | 0.031 (2) | 0.0050 (17) | 0.0056 (17) | 0.0004 (17) |
| N4 | 0.0296 (17) | 0.042 (2) | 0.0320 (19) | −0.0026 (17) | −0.0002 (15) | 0.0059 (18) |
Geometric parameters (Å, º)
| Pd1—N1i | 2.032 (3) | N2—H2B | 0.73 (3) |
| Pd1—N1 | 2.032 (3) | N2—H2C | 0.88 (5) |
| Pd1—N2i | 2.048 (3) | N3—H3A | 0.83 (3) |
| Pd1—N2 | 2.048 (3) | N3—H3B | 0.89 (5) |
| N1—H1A | 0.86 (4) | N3—H3C | 1.09 (5) |
| N1—H1B | 0.85 (3) | N4—H4A | 0.91 (4) |
| N1—H1C | 0.96 (4) | N4—H4B | 1.00 (5) |
| N2—H2A | 1.03 (4) | N4—H4C | 0.97 (4) |
| N1—Pd1—N1i | 179.999 (1) | Pd1—N2—H2A | 111.2 (18) |
| N1—Pd1—N2i | 88.59 (13) | Pd1—N2—H2B | 109 (3) |
| N1i—Pd1—N2i | 91.41 (13) | Pd1—N2—H2C | 112 (3) |
| N1i—Pd1—N2 | 88.59 (13) | H2A—N2—H2B | 115 (3) |
| N1—Pd1—N2 | 91.41 (13) | H2A—N2—H2C | 102 (3) |
| N2—Pd1—N2i | 180.00 (10) | H2B—N2—H2C | 108 (4) |
| Pd1—N1—H1A | 107 (3) | H3A—N3—H3B | 115 (4) |
| Pd1—N1—H1B | 110 (2) | H3A—N3—H3C | 84 (3) |
| Pd1—N1—H1C | 112 (3) | H3B—N3—H3C | 112 (4) |
| H1A—N1—H1B | 110 (3) | H4A—N4—H4B | 109 (3) |
| H1A—N1—H1C | 108 (3) | H4A—N4—H4C | 105 (3) |
| H1B—N1—H1C | 109 (3) | H4B—N4—H4C | 100 (4) |
Symmetry code: (i) −x+1, −y+1, −z.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1A···Cl1 | 0.86 (4) | 2.49 (4) | 3.351 (3) | 175 (3) |
| N1—H1B···Cl1ii | 0.85 (3) | 2.49 (4) | 3.328 (3) | 171 (3) |
| N2—H2A···N3i | 1.03 (4) | 2.02 (4) | 3.025 (5) | 163 (3) |
| N1—H1C···N4 | 0.96 (4) | 2.02 (5) | 2.975 (5) | 170 (3) |
| N2—H2B···Cl1ii | 0.73 (3) | 2.66 (3) | 3.384 (4) | 172 (3) |
| N2—H2C···Cl1iii | 0.88 (5) | 2.62 (5) | 3.463 (3) | 162 (4) |
| N3—H3A···Cl1 | 0.83 (3) | 2.83 (3) | 3.563 (4) | 148 (3) |
| N4—H4A···Cl1iv | 0.91 (4) | 2.65 (4) | 3.563 (4) | 173 (3) |
| N4—H4B···Cl1v | 1.00 (5) | 2.61 (5) | 3.606 (4) | 174 (4) |
| N3—H3B···Cl1vi | 0.89 (5) | 2.71 (5) | 3.578 (4) | 163 (3) |
| N3—H3C···Cl1vii | 1.09 (5) | 2.48 (5) | 3.535 (4) | 162 (4) |
Symmetry codes: (i) −x+1, −y+1, −z; (ii) −x+1/2, y−1/2, −z+1/2; (iii) −x, −y+1, −z; (iv) x+1, y, z; (v) −x+1, −y+1, −z+1; (vi) x+1/2, −y+3/2, z+1/2; (vii) −x+1/2, y+1/2, −z+1/2.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: PK2523).
References
- Agilent (2012). CrysAlis PRO Agilent Technologies, Yarnton, England.
- Ahlrichs, R., Ballauff, M., Eichkorn, K., Hanemann, O., Kettenbach, G. & Klüfers, P. (1998). Chem. Eur. J. 4, 835–844.
- Bell, J. D., Bowles, J. C., Cumming, H. J., Hall, D. & Holland, R. V. (1976). Acta Cryst. B32, 634–636.
- Bourhis, L. J., Dolomanov, O. V., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2014). In preparation.
- Brandenburg, K. & Putz, H. (2012). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Clark, R. C. & Reid, J. S. (1995). Acta Cryst. A51, 887–897.
- Desiraju, G. R. (2002). Acc. Chem. Res. 35, 565–573. [DOI] [PubMed]
- Desiraju, G. R. (2007). Angew. Chem. Int. Ed. 46, 8342–8356. [DOI] [PubMed]
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Grassl, T. & Korber, N. (2014). Acta Cryst. E70, i31. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Steiner, T. (2002). Angew. Chem., 114, 50–80.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814012355/pk2523sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814012355/pk2523Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814012355/pk2523Isup3.mol
CCDC reference: 1005539
Additional supporting information: crystallographic information; 3D view; checkCIF report