

Abstract
Engineering enzymes capable of modes of activation unprecedented in nature will increase the range of industrially important molecules that can be synthesized through biocatalysis. However, low activity for a new function is often a limitation in adopting enzymes for preparative scale synthesis, reaction with demanding substrates, or when a natural substrate is also present. By mutating the proximal ligand and other key active site residues of the cytochrome P450 from Bacillus megaterium (P450-BM3), we have engineered a highly active His-ligated variant of P450-BM3, BM3-Hstar, that can be employed for the enantioselective synthesis of the levomilnacipran core. This enzyme catalyzesthe cyclopropanation of N,N-diethyl-2-phenylacrylamide (1) with an estimated initial rate of over 1000 turnovers per minute and can be used under an aerobic environment. Cyclopropanation activity is highly dependent on the electronics of the P450 proximal ligand, which can be used to tune this non-natural enzyme activity.
Keywords: biocatalysis, cytochrome P450, cyclopropanation
Enzymes in nature catalyze only a small subset of industrially-relevant chemical transformations.1 Thus, increasing the number of activation modes accessible to enzymes is an important goal in biocatalysis, green chemistry, and sustainable synthesis.2 Drawing an analogy between the mechanism of monooxygenation catalyzed by cytochrome P450 and transition metal-catalyzed carbene insertions, we hypothesized that an iron-carbenoid intermediate could be generated at the enzyme’ sheme prosthetic group in the presence of diazo compounds. Staring from this hypothesis, we have developed methods for intermolecular, enantioselective cyclopropanation of styrenes and N-H insertion of anilines catalyzed by variants of P450-BM3.3 These enzymatic methods are attractive alternatives to transition metal catalysiswith Rh-or Cu-complexes4 for cyclopropanation because cytochrome P450s are genetically encoded, can be very stereoselective, and use inexpensive and nontoxic Fe as the catalytic center.
One challenge to using enzymes for non-natural chemistry is that the activity is often low.3,5 Wild-type P450-BM3 has very low cyclopropanation activity, for example, and even the best reported variants are still 10–100 times less active for styrene cyclopropanation than for epoxidation or hydroxylation of their preferred fatty acid substrates.6,7 Although introducing mutations at amino acid residues critical for native monooxygenation readily eliminates the competing P450-catalyzed styrene epoxidation reaction, molecular oxygen severely inhibits the desired cyclopropanation reaction.8 As a result, anaerobic procedures must be employed to favor carbenoid transfer reactions. Another consequence of the relatively low activity for cyclopropanation is that the transformation is limited to electron-neutral and electron-rich alkenes.3a Increasing the activity of P450s toward these non-natural reactions would greatly expand their utility as cyclopropanation catalysts for bench top synthesis of biologically active molecules and even large scale industrial use.
Recently, we reported that mutation of the proximal Cys at position 400 in P450-BM3 to Ser enabled cyclopropanation by whole cells expressing these proteins (Figure 1a).8 The Cys to Ser mutation increased the protein redox potential by 140 mV, allowing efficient reduction from the inactive Fe(III)-heme resting state to the Fe(II)-heme active catalyst in vivo.9 However, in addition to modulating redox potential, we reasoned that the axial ligand also affects the electronics of the proposed iron-carbenoid intermediate and thus the inherent rate of P450-catalyzedcyclopropanation. In particular, we hypothesized that by varying the axial coordinating ligand we might discover a more active enzyme, which would enable cyclopropanation of a broader range of substrates.
Figure 1.

(a) Ser-ligated heme in BM3enablescyclopropanationin vivo.3b (b) Proposed Ala-, Met-, His-, and Tyr-ligated heme in AxX variants of BM3.
To test this, we have initially focused on variants of P450-BM3 bearing His, Met, Tyr, or Ala at the proximal position (Figure 1b). This set represents a range of possible coordinating heteroatom ligands, and His, Met and Tyr are found at the axial position of naturally occurring heme proteins like horseradish peroxidase (HRP), cytochrome c, and catalase.10 We chose Alaas well because it is anarchetypal small amino acid and may allow a water or hydroxide molecule to coordinate to the Fe center.11 To examine how the different axial ligands affect cyclopropanation activity, we introduced all four axial mutations into the wild-type holoenzyme containing one additional T268A mutation. This mutation was previously found to be highly beneficial for cyclopropanation.3a Four variants, T268A-AxX(where “X” denotes the single letter amino acid code of each axial variant), were expressed as the His-tagged heme domains, purified and characterized by UV-Vis spectroscopy. All four bound CO efficiently and provided distinct Fe(II)-CO absorbances in the 414–422 nm range (Figure S1).
When we monitored the reactions of the four variants with styrene and ethyl diazoacetate (EDA), we found thatT268A-AxH was highly active and catalyzed the cyclopropanation of styrene to greater than 50% conversion within 30 min (Figure 2). Both the Cys-ligated T268A and the Ser-ligated T268A-AxS (enzymes having the AxS mutation were previously denoted as “P411s” due to the characteristic Fe(II)-CO absorbance at 411 nm of AxS proteins3b) exhibited much slower kinetics at the same protein concentration. T268A is not expected to be highly active for cyclopropanation of styrene in vivo because its redox potential in the absence of native substrates is more negative than that of biological reductants (−410 mV versus −310 mV for NAD(P)H). Thus, the Fe(III) resting state cannot be easily reduced to the Fe(II) active catalyst. However, the higher activity of T268A-AxM and T268A-AxH mutants relative to T268A-AxSsuggests that, among variants that can be reduced to Fe(II) by biological reductants, those that have more electron-donating axial ligands are more active in vivo cyclopropanation catalysts.
Figure 2.

Reaction progress of in vivo cyclopropanation of styrene with EDA catalyzed by axial variants of T268A.
Our finding that the cyclopropanation activity of BM3 variants increases when natural Cys-ligation is replaced with His-or Ser-coordination contrasts what has been previously observed for P450-catalyzed monooxygenation.12 Mutation of the proximal Cys to Ser or His causes the monooxygenation activity of the enzyme to fall precipitously.13 We have also observed that mutation of other highly conserved amino acids in the P450 active site, such as the distal Thron the I-helix (T268 in P450-BM3) that facilitates multiple proton transfers en route to compound I, also produces more active cyclopropanation catalysts. Thus conserved amino acids required for activation of molecular oxygen are not necessarily required for activation of diazo compounds. Indeed, mutation of the axial residue and other conserved positions in heme proteins may allow us to develop new enzymes that are better suited for carbenoid or nitrenoid modes of reaction.14
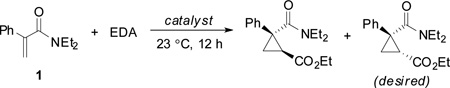
To showcase the improved activity of the T268A-AxX mutants, we wanted to develop a method for cyclopropanation of electron-deficient olefins which have been challenging substrates for transition metal catalysts.15 In particular, we hypothesized that an enantioselectiveBM3 catalyst could be used for the cyclopropanation of 1 with EDA in an expedient synthesis of the levomilnacipran core (Scheme 1). Levomilnacipran (Fetzima™) is a selective serotonin and norepinephrine reuptake inhibitor that was recently approved by the Food and Drug Administration for treatment of clinical depression.16 The more active (1R,2S)-isomer of milnacipran,17 levomilnacipran is sold in enantiopure form. While multiple syntheses of milnacipran and levomilnacipran have been described,18 none have used intermolecular enantioselective cyclopropanation to construct the cyclopropane core of the molecule. The most high yielding and efficient of the reported methods requires a series of alkylations and thermal rearrangements that require strong alkali bases and operate at temperatures in excess of 80 °C.15a,b A chemoenzymatic synthesis could constitute a mild and energy-efficient alternative that is concise and highly convergent.
Scheme 1.

Proposed formal synthesis of levomilnacipran using P450-catalyzed enantioselective cyclopropanation in the key ring-forming step.
When we combined 10 mM 1 with 10 mM EDA in the presence of whole cells expressing the axial variants, we found that T268A-AxH catalyzed the reaction to 81% yield with 6:94diastereoselectivity and 42% enantioselectivity for the desired product (Table 1, entry 2). T268A and hemin failed to provide the desired product in synthetically useful yields (entry 6 and 7, respectively). Although variants T268A-AxA and T268A-AxS also showed appreciable activity, they were less active and less enantioselective than the His mutant when normalized for catalyst expression level. T268A-AxH is also more active when used as the the purified holoenzyme (Table S3). Interestingly, horseradish peroxidase, which is naturally ligated by an axial His, is a poor catalyst for this reaction (Table 1, entry 8). This suggests that heme His-ligation alone is not sufficient for catalysis and that theP450 fold is privileged for this transformation. The changes in diastereo-and enantioselectivity observed for the different axial mutants suggest that the amino acid that replaces the Cys axial ligand can affect active site geometry as well as reactivity. When whole-cell catalysts expressing T268A-AxH were purged with CO prior to addition of both substrates, no catalysis was observed (entry 9). Furthermore, control cells transformed with pET-22b(+) vector without the BM3 gene also failed to catalyze the reaction (entry 10).These controls indicate that a folded His-ligated BM3 enzyme is the active catalyst.
Table 1.
Reaction of 1 with EDA catalyzed by T268A-AxX mutants in vivo.
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst[a] | Yield (%)[b] | TTN[c] | trans:cis | ee (%)[d] |
| 1 | T268A-AxA | 19 | 1600 | 18:82 | 21 |
| 2 | T268A-AxH | 81 | 7100 | 6:94 | 42 |
| 3 | T268A-AxM | 12 | 1000 | 16:84 | 4 |
| 4 | T268A-AxY | 12 | 1000 | 17:83 | 5 |
| 5 | T268A-AxS | 18 | 1500 | 16:84 | 17 |
| 6 | T268A | 0.8 | 80 | 16:84 | n/a[e] |
| 7 | Hemin (5 µM)[f] | 3.0 | 50 | 18:82 | 0 |
| 8 | HRP (10 µM)[f] | 0.4 | 3 | n/a | n/a |
| 9 | T268A-AxH + CO | 0 | n/a | n/a | n/a |
| 10 | E. coli w/o BM3 | 0 | n/a | n/a | n/a |
For whole cells expressing T268A-AxX, protein concentrations determined by CO-assay. Cell densities were normalized such that all whole cell samples has [BM3] = 1.0 µM.
Reactions were performed on 0.5 mL scale with final concentrations of 8.7 mM EDA and 10 mM 1. Yields were determined by gas chromatography using phenylethanol as an internal standard.
Turnover numbers are determined by dividing the millimolar product yield by the catalyst concentration (1 µM for BM3 variants and 5 µM for hemin).
Enantioselectivity was determined by chiral supercritical fluid chromatography with CO2.
Could not be determined due to low product yield.
Used in vitro as isolated protein or complex.
To create a catalyst more enantioselective than T268A-AxH, we performed site saturation mutagenesis at four active site positions that had been shown previously to affect selectivity in cyclopropanantion or monooxygenation: F87, I263, L437 and T438. The libraries were screened in 96-well plate format using whole cells and an oxygen quenching system containing glucose oxidase and catalase in sealed plates (see Supporting Information for a detailed description of the screening process). The enantioselectivity of each reaction was determined using chiral supercritical fluid chromatography. Mutagenesis at positions F87 and I263 failed to produce variants with higher selectivity than the parent T268A-AxH. The libraries at L437 and T438, however, yielded variants T268A-AxH-L437W and T268A-AxH-T438W which catalyzed the reaction to 69 and 68% enantioselectivity, respectively. Unfortunately, combining both Trp mutations at position 437 and 438 significantly reduced the yield and decreased the enantioselectivity to 23%.
As T268A-AxH-L437W showed higher activity than T268A-AxH-T438W (Table S4), we chose the former as parent for a second round of site saturation at two sites, V78 and L181. These two positions are located in the same region of the BM3 active site as L437 but are not located on the same helix or loop as L437. We identified two variants, T268A-AxH-L437W-V78M and T268A-AxH-L437W-L181V,that showed improved enantioselectivity relative to T268A-AxH-L437W (87% and 75%, respectively) without loss of reactivity. V78M and L181V were combined in variant T268A-AxH-L437W-V78M-L181V (named “BM3-Hstar”) which provided the desired product in greater than 92% yield with 92% enantioselectivity and 98:2 diastereoselectivity.
When the reaction of 1 and EDA catalyzed by BM3-Hstar in E. coli whole cells was monitored by gas chromatography, we found that the reaction reached greater than 80% conversion within the first 10 min. This is much faster than what we had observed for cyclopropanation of 1 with T268A-AxS (Figure S3). The reaction catalyzed by parent T268A-AxH was also complete within the first 15 min, which shows that the AxH mutation alone dramatically increases the rate of olefin cyclopropanation. An estimate of the initial rate based on the first five minutes of reaction shows that catalysis proceeds at over 1000 turnovers per minute. Indeed, even underan aerobic environment, BM3-Hstar is able to catalyze the reaction of 1 and EDA to 90% yield (compared to 92–98% for an anaerobic control performed in parallel, Eq. 1) when cells expressing BM3-Hstar to 2.0µM are used. This observation suggests that cyclopropanation of 1 out-competes catalyst inhibition or deactivation by molecular oxygen. No other BM3 variant has demonstrated this tolerance to atmospheric oxygen in these non-natural reactions.
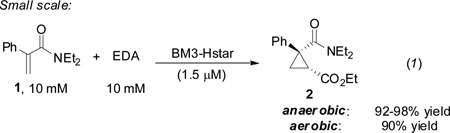 |
(1) |
The reaction of 1 with EDA could be performed on preparative scale under aerobic conditions to provide the levomilnacipran precursor in 93% conversion and 86% isolated yield (204 mg) using preparative HPLC, a diastereomeric ratio of 98:2 and 92% enantioselectivity. Cyclopropane 2, which could not be separated from unreacted 1 by column chromatography, was readily reduced to the corresponding alcohol in the presence of 1.5 equivalents of LiBH4 to provide alcohol 3 in 88% isolated yield, without loss of enantioselectivity (Scheme 1). Elaboration of this key intermediate to levomilnacipran has been well-documented and can be achieved in two facile and high-yielding steps.18c
Mutating the amino acid at the axial site of P450-BM3, we have identified a highly active and enantioselective cyclopropanation catalyst for a challenging electron-deficient olefin. Starting from a parent BM3 variant with His in position 400, we engineered BM3-Hstar through two rounds of site saturation mutagenesis and recombination to catalyze the reaction of 1 and EDA to 92% yield, 92% enantioselectivity and 98:2 diastereomeric ratio. Remarkably, a single mutation at the axial position dramatically increases the reaction rate such that these new variants can be used for cyclopropanation of 1 with EDA in an aerobic environment with minimal loss of yield. Three additional mutations identified through site saturation mutagenesis, L437W, L181V, and V78M, greatly enhanced the enantioselectivity of the transformation and further improved the activity of the catalyst, demonstrating that the cyclopropanation reaction of P450-BM3 scaffold can be readily tuned for a desired substrate. The tolerance of P450-catalyzed cyclopropanation to amino acid substitution at the axial position contrasts with the strict requirement for axial Cys for monooxygenation activity. The P450 protein’s ability to accept other amino acids at this key position and still bind heme will allow us to tune the electronics of the catalytic center and thereby access a broader range of carbenoid-based transformations.
Supplementary Material
Figure 3.
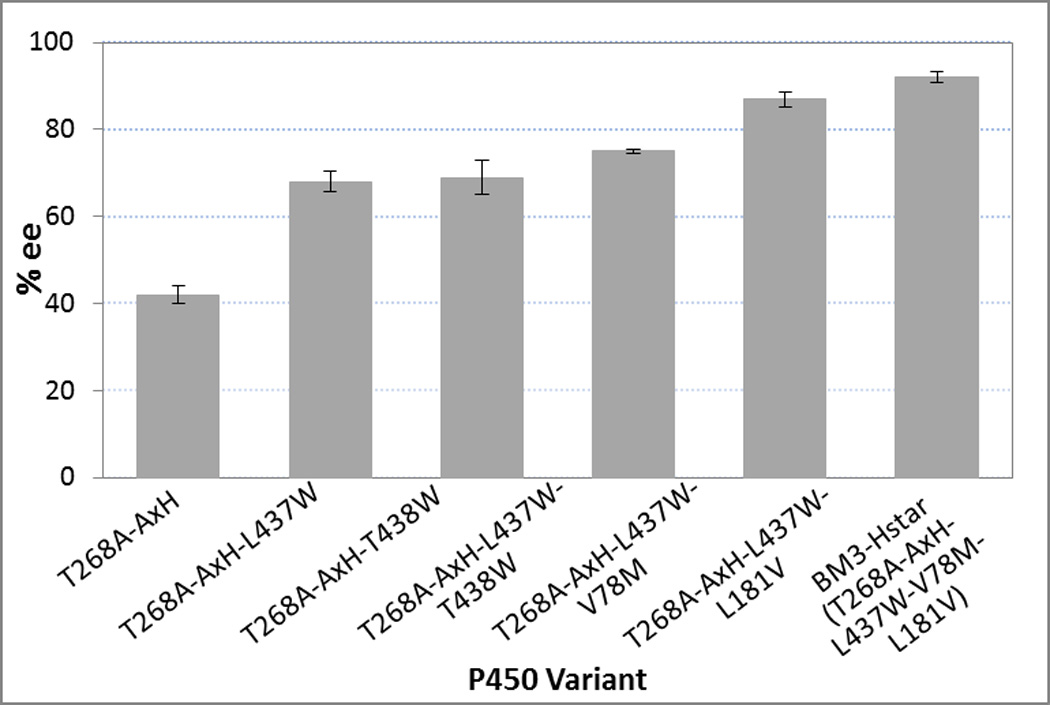
Sequential site saturation mutagenesis at key active site positions in T268A-AxH variant led to increased enantioselectivity for cyclopropanation of 1.
Footnotes
We thank Dr. S. Virgil and Center for Catalysis and Chemical Synthesis (3CS) at Caltech for assistance with HPLC, Dr. J. McIntosh and Dr. T. Heel for helpful discussions, and R. Kitto for help during preparative scale reactions. This work was supported by the Gordon and Betty Moore Foundation through Grant GBMF2809 to the Caltech Programmable Molecular Technology Initiative. Z. J. Wang was supported by a Ruth L. Kirschstein Fellowship from the National Institutes of Health, award number F32EB015846-01.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.Huisman GW, Collier SJ. Curr. Op. in Biotech. 2013;17:284–292. doi: 10.1016/j.cbpa.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 2.Wohlgemuth R. Curr. Op. in Biotech. 2010;21:713–724. doi: 10.1016/j.copbio.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 3.a) Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; b) Wang ZJ, Peck NE, Renata H, Arnold FH. Chem. Sci. 2014;5:598–601. doi: 10.1039/C3SC52535J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For reviews, see: Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem. Rev. 2003;103:977–1050. doi: 10.1021/cr010007e. Doyle M, Forbes DC. Chem. Rev. 1998;98:911–935. doi: 10.1021/cr940066a. For selected examples, see: Aratani T. Pure & Appl. Chem. 1985;57:1839–1844. Evans D, Woerpel KA, Hinman MA, Faul MM. J. Am. Chem. Soc. 1991;113:726–728. Davies HM, Venkataramani C. Org. Lett. 2003;5:1403–1406. doi: 10.1021/ol034002a. For recent examples, see: Marcoux D, Azzi S, Charette AB. J. Am. Chem. Soc. 2009;131:6970–6972. doi: 10.1021/ja902205f. Qin C, Boyarskikh V, Hansen JH, Hardcastle KI, Musaev DG, Davies HML. J. Am. Chem. Soc. 2011;133:19198–19204. doi: 10.1021/ja2074104.
- 5.a) Purkarthofer T, Gruber K, Gruber-Khadjawi M, Waich K, Mink D, Griengl H. Angew. Chem. Int. Ed. 2006;45:3454–3456. doi: 10.1002/anie.200504230. [DOI] [PubMed] [Google Scholar]; b) Johnson DV, Zabelinskaja-Mackova AA, Griengl H. Curr. Op. Chem. Bio. 2000;4:103–109. doi: 10.1016/s1367-5931(99)00059-9. [DOI] [PubMed] [Google Scholar]; c) McLoughlin SY, Copley SD. Proc. Nat. Aca. Sci. 2008;105:13497–13502. doi: 10.1073/pnas.0804804105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yun C-H, Kim K-H, Kim D-H, Jung H-C, Pan J-G. Trends in Biochem. Sci. 2007;25:289–298. [Google Scholar]
- 7.a) Whitehouse CJC, Bell SG, Wong LL. Chem. Soc. Rev. 2012;47:1218–1260. doi: 10.1039/c1cs15192d. [DOI] [PubMed] [Google Scholar]; b) Munro AW, Leys DG, Mclean KJ, Marshall KR, Ost TWB, Daff S, Miles CS, Chapman SK, Lysek DA, Moser CC, Page CC, Dutton PL. Trends in Biochem. Sci. 2002;27:250–257. doi: 10.1016/s0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- 8.Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM. Nat. Chem. Bio. 2013;9:485–487. doi: 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For report and characterization of a Ser-ligated CYP2B4, see: Vatsis KP, Peng H-M, Coon MJ. J. Inorg. Biochem. 2002;91:542–553. doi: 10.1016/s0162-0134(02)00438-5. Perera R, Sono M, Voegtle HL, Dawson JH. Arch. Biochem. And Biophys. 2011;507:119–125. doi: 10.1016/j.abb.2010.12.010.
- 10.a) Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Nat. Struct. Bio. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]; b) Murthy MRN, Reid TJ, III, Sicignano A, Tanaka N, Rossmann MG. J. Mol. Bio. 1981;152:487–499. doi: 10.1016/0022-2836(81)90254-0. [DOI] [PubMed] [Google Scholar]; c) Takano T, Dickerson RE. Proc. Natl. Acad. Sci. 1980;77:6371–6375. doi: 10.1073/pnas.77.11.6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Lu Y, Casimiro DR, Bren KL, Richards JH, Gray HB. Proc. Nat. Acad. Sci. 1993;90:11456–11459. doi: 10.1073/pnas.90.24.11456. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Banci L, Bertini I, Bren KL, Gray HB, Sompornpisut P, Turano P. Biochemistry. 1995;34:11385–11398. doi: 10.1021/bi00036a011. [DOI] [PubMed] [Google Scholar]; c) Raphael AL, Gray HB. J. Am. Chem. Soc. 1991;113:1038–1040. [Google Scholar]
- 12.Green MT. Curr. Opin. Chem. Bio. 2009;13:84–88. doi: 10.1016/j.cbpa.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 13.a) Auclair K, Moёnne-Loccoz P, Ortiz de Montellano PR. J. Am. Chem. Soc. 2001;123:4877–4885. doi: 10.1021/ja0040262. [DOI] [PubMed] [Google Scholar]; b) Perera R, Sono M, Voegtle HL, Dawson JH. Arch. Biochem. Biophys. 2011;507:119–125. doi: 10.1016/j.abb.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vatsis KP, Peng H-M, Coon MJJ. Inorg. Biochem. 2002;91:542–553. doi: 10.1016/s0162-0134(02)00438-5. [DOI] [PubMed] [Google Scholar]
- 14.a) McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, Arnold FH. Angew. Chem. Int. Ed. 2013;52:9309–9312. doi: 10.1002/anie.201304401. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Singh R, Bordeaux M, Fasan R. ACS Catal. 2014;4:546–552. doi: 10.1021/cs400893n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Guptill DM, Varela-Alvarez A, Masaev DG, Davies HML. Chem. Sci. 2013;4:2844–2850. doi: 10.1039/C3SC50425E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asnis GM, Bose A, Gommoll CP, Chen C, Greenberg WM. J. Clin. Psychiatry. 2013;74:242–248. doi: 10.4088/JCP.12m08197. [DOI] [PubMed] [Google Scholar]
- 17.Viazzo P, Alphand V, Furstoss R. Tett. Lett. 1996;37:4519–4522. [Google Scholar]
- 18.For syntheses of milnacipran, see Shuto S, Ono S, Hase Y, Kamiyama N, Takada H, Yamasihita K, Matsuda A. J. Org. Chem. 1996;61 915-; Bonnaud B, Cousse H, Mouzin G, Briley M, Stenger A, Fauran F, Couzinier J-P. J. Med. Chem. 1987;30:318–325. doi: 10.1021/jm00385a013. Zhang M-X, Eaton P. Angew. Chem. Int. Ed. 2002;41:2169–2170. Forsyntheses of levomilnacipran, see: Alliot J, Gravel E, Pillon F, Buisson D-A, Nicolas M, Doris E. Chem. Commun, 2012;48:8111–8113. doi: 10.1039/c2cc33743f. Doyle MP, Davies SB, Hu W. Org. Lett. 2000;2:1145–1147. doi: 10.1021/ol005730q.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.