

Abstract
Genetics plays a crucial role in human aging with up to 30% of those living to the mid-80s being determined by genetic variation. Survival to older ages likely entails an even greater genetic contribution. There is increasing evidence that genes implicated in age-related diseases, such as cancer and neuronal disease, play a role in affecting human life span. We have selected the 10 most promising late-onset Alzheimer’s disease (LOAD) susceptibility genes identified through several recent large genome-wide association studies (GWAS). These 10 LOAD genes (APOE, CLU, PICALM, CR1, BIN1, ABCA7, MS4A6A, CD33, CD2AP, and EPHA1) have been tested for association with human aging in our dataset (1385 samples with documented age at death [AAD], age range: 58–108 years; mean age at death: 80.2) using the most significant single nucleotide polymorphisms (SNPs) found in the previous studies. Apart from the APOE locus (rs2075650) which showed compelling evidence of association with risk on human life span (p = 5.27 × 10−4), none of the other LOAD gene loci demonstrated significant evidence of association. In addition to examining the known LOAD genes, we carried out analyses using age at death as a quantitative trait. No genome-wide significant SNPs were discovered. Increasing sample size and statistical power will be imperative to detect genuine aging-associated variants in the future. In this report, we also discuss issues relating to the analysis of genome-wide association studies data from different centers and the bioinformatic approach required to distinguish spurious genome-wide significant signals from real SNP associations.
Keywords: Lifespan, Late onset Alzheimer’s disease, GWAS, Aging, Genes
1. Introduction
Human aging is affected by both environmental and genetic factors (Cutler and Mattson, 2006). The heritability of aging is estimated to be 20%–30% to reach mid-80s estimated from twin studies. Survival to older ages likely involves an even greater genetic contribution (Herskind et al., 1996; Iachine et al., 1998; McGue et al., 1993). Furthermore, previous studies have shown that siblings of centenarians have an approximately 4-fold higher chance of survival to their early 90s compared with siblings of individuals who die at 73 years of age (Perls et al., 1998). A larger study conducted by the same group has shown an even higher increase (8–18-fold) in the “risk” of successful aging for siblings of centenarians compared with random controls (US 1900 birth cohort) (Perls et al., 2002). Evidence indicates strong familial aggregation toward aging.
The molecular genetics that underlie the human aging process is complex and it is suggested that successful aging is likely due to numerous genes and environmental factors, each exerting a small effect (Kenyon, 2010; Lescai et al., 2009; Plomin et al., 2009).
GenAge is a database of genes related to aging (genomics.senescence.info). To date there are over 250 genes that have been recorded by the GenAge database based on extensive literature reviews. All of these genes have shown possible association with human aging (de Magalhães et al., 2009). Most of these genes play critical parts in a variety of biological pathways in humans, and a significant number of these genes (>100) are related to severe human diseases. It is generally believed that genes and biomarkers implicated in age-related diseases such as coronary artery disease (CAD), cerebrovascular disease (CVD), and Alzheimer’s disease (AD) have a role in successful aging (Panza et al., 2009). Identification of genuine aging genes may uncover “master genes” that increase our understanding of many age-related diseases.
There are a number of biological pathways that have been reported important in human aging, including lipid/cholesterol metabolism [GO:0006629; GO:0008203] (APOE, PON1, CETP) (Barzilai et al., 2003; de Chaves and Narayanaswami, 2008; Efrat and Aviram, 2010), immune system processes [GO:0002376] (IL6 and IL10) (Jylhävä and Hurme, 2009), drug metabolism [KEGG:hsa00982] (GSTT1) (Glatt et al., 2007; Taioli et al., 2001), energy metabolism in mitochondria (SIRT3) (Polito et al., 2010), and insulin receptor signaling pathway [GO:0008286] (IGF1R, FOXO3A, KLOTHO) (Arking et al., 2005; Suh et al., 2008; Willcox et al., 2008).
Insights into human aging have been gained from studying model organisms. Extension of lifespan can be achieved by manipulating a few genes in laboratory animals, such as flies, worms, and mice (Kenyon, 2010). The insulin/insulin-like growth factor 1 (IGF-1) signaling pathway has a well-established role in influencing lifespan within model organisms with large effects (Clancy et al., 2001; Holzenberger et al., 2003; Kenyon, 2010;). Genetic inactivation of the daf-2 gene (encoding the IGF-1 receptor homolog in C. elegans) increases the lifespan of C. elegans by approximately 100% (Sebastiani et al., 2009). Interestingly, there is emerging evidence that genes such as IGF-1/IGF1R (the orthologs of which play a major part in aging in animals) can play a role in human life span. Loss of function mutations in IGF1R have been found to be overrepresented in Ashkenazi Jewish centenarians compared with controls (Suh et al., 2008).
Aging genes in humans may not only increase the life span but also postpone age-related diseases. A previous study has indicated a significant decreased prevalence of age related diseases in offspring of long-lived parents (hypertension by 23%, diabetes mellitus by 50%, heart attacks by 60%, and no incidences of stroke) compared with several age-matched control groups (Atzmon et al., 2004).
Characterizing various genetic and environmental factors influencing human life span is currently one of the world’s major scientific challenges (Jylhava and Hurme, 2010). To date, genome-wide association studies (GWAS) are one of the most widely used approaches for identifying common genetic variations associated with human diseases. It has been suggested that with increasing sample size, promising signals of association between human traits and genetic variants can be revealed (McCarthy, et al., 2008). Tan and Christensen have shown that increasing sample age from nonagenarians to centenarians further increases the power to discover variants associated with human aging (Tan et al., 2010).
The aim of this study was to (1) investigate whether the “known” late-onset Alzheimer’s disease (LOAD) genes play a role in human aging. This could address the question if these genes are directly associated with AD or indirectly by allowing successful aging; and (2) search for candidate genetic risk factors associated with human survival and aging, which may merit further study.
2. Methods
Through collaborative efforts, we were able to draw on combined sample GWAS datasets and analyze subject-level genotype data from 1385 subjects (1047 LOAD cases and 338 controls) with documented age at death (AAD) (Table 1). The AAD histogram follows a normal distribution (Fig. 1), with mean AAD of 80.2 years of age (SD = 8.9 years). All of these data were subject to subsequent quality control (QC) procedures and analysis. The data analysis was performed using PLINK software package (pngu.mgh.harvard. edu/~purcell/plink) release 1.06 (Purcell et al., 2007).
Table 1.
Summary of sample information
| Dataset | Sample size | AD status (AD/controls) | Gender (male/female) | Genotyping chip | Mean AAD (AD/controls) | Origin |
|---|---|---|---|---|---|---|
| Mayo | 434 | 220/214 | 246/188 | Illumina 300 | 73.5/71.7 | USA |
| NIMH | 46 | 46/0 | 12/34 | Illumina 610 | 78.1/- | USA |
| WashU | 332 | 294/38 | 140/192 | Illumina 610 | 84.1/86.1 | USA |
| Belfast | 235 | 213/22 | 99/136 | Illumina 610 | 82.2/83.1 | UK |
| Bristol | 59 | 43/16 | 21/38 | Illumina 610 | 82.5/81.6 | UK |
| London | 238 | 194/44 | 83/155 | Illumina 610 | 86.1/83.0 | UK |
| Manchester | 1 | 1/0 | 0/1 | Illumina 610 | 79.0/- | UK |
| Nottingham | 39 | 35/4 | 18/21 | Illumina 610 | 83.6/79.5 | UK |
| Oxford | 1 | 1/0 | 0/1 | Illumina 610 | 79.5/- | UK |
| Pooled | 1385 | 1047/338 | 619/766 | — | 81.5/76.1 | — |
Genome-wide association studies (GWAS) data were obtained from a total of 9 centers, 3 from the USA (NIMH, WashU, and Mayo) and 6 from the UK (Nottingham, Bristol, Manchester, Belfast, Oxford, and London). Sample size, number of cases and controls, and gender for each cohort are as indicated together with details of the mean age at death in years for cases and controls and the genotyping chip used in each study. “-” means the information is not applicable.
Key: AAD, age at death; AD, Alzheimer’s disease; NIMH, National Institute of Mental Health; WashU, Washington University.
Fig. 1.
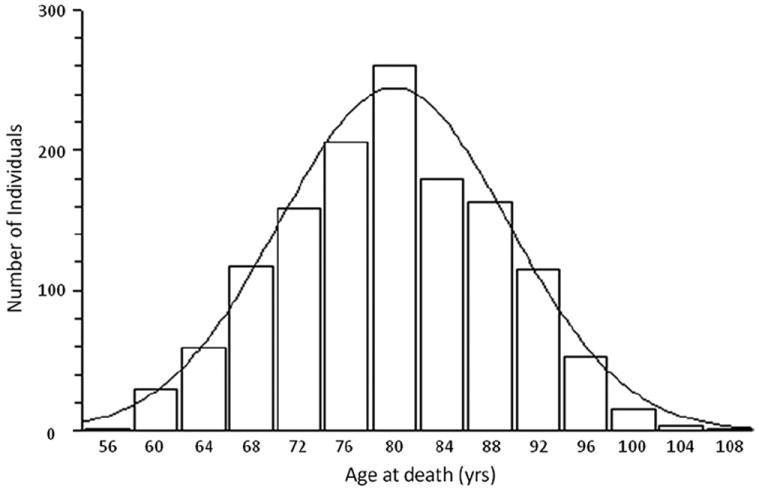
Histogram plot representing the spread of age at death (AAD) of samples included in this study. The x- and y-axis represent AAD in years and number of individuals, respectively. This graph follows a normal distribution, with mean AAD = 80.2 years (n = 1385).
2.1. Datasets; pooling and QC
We have obtained data from 9 research centers, 3 from the USA and 6 from the UK (GERAD1 Consortium). The GERAD1 sample comprised up to 3941 AD cases and 7848 controls. A subset of this sample has been used in this study, comprising 3333 cases and 1225 elderly screened controls genotyped at the Sanger Institute on the Illumina (San Diego, CA, USA) 610-quad chip. These samples were recruited by the Medical Research Council (MRC) Genetic Resource for AD (Cardiff University; Kings College, London; Cambridge University; Trinity College, Dublin), the Alzheimer’s Research Trust (ART) Collaboration (University of Nottingham; University of Manchester; University of Southampton; University of Bristol; Queen’s University, Belfast; the Oxford Project to Investigate Memory and Ageing [OPTIMA], Oxford University); Washington University, St. Louis, MO, USA; MRC PRION Unit, University College London, London; and the South East Region AD project (LASER-AD), University College, London; Competence Network of Dementia (CND) and Department of Psychiatry, University of Bonn, Germany; and the National Institute of Mental Health (NIMH) AD Genetics Initiative. All AD cases met criteria for either probable (National Institute of Neurology and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (now known as the Alzheimer’s Association) [NINCDS-ADRDA], Diagnostic and Statistical Manual of Mental Disorders, 4th Ed [DSM-IV]) or definite (Consortium to Establish a registry for Alzheimer’s Disease [CERAD]) AD. All elderly controls screened for dementia using the Mini Mental State Examination (MMSE) or Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog), were determined to be free from dementia at neuropathological examination or had a Braak score of 2.5 or lower. All studies used the Illumina 610 QuadChip, except the Mayo study which used the Illumina HumanHap300 chip (Carrasquillo et al., 2009). The Illumina 610 QuadChip includes all single nucleotide polymorphisms (SNPs) present on the Illumina 300 chip, and enabled us to merge all the datasets.
Individual data characterized as “AUT - autopsy” or having AAD information were extracted from the Mayo dataset (Carrasquillo et al., 2009) using the “--keep” and “--make-bed” command in PLINK. This was repeated for samples from the Alzheimer’s Research UK (ARUK) data (which encompassed centers in Nottingham, Bristol, Manchester, Belfast, Oxford, and London), NIMH, and Washington University (WashU) where possible. All GWAS datasets were transformed into the same PLINK format (1 and 2 coding in PLINK.bed.fam and .bim format). Any samples which overlapped between GWAS datasets were removed. Each sample was checked individually for discrepancies between AAD and age at sampling (AAS). Samples with age at sampling greater than AAD were removed from further analysis. Data merging was performed using “--bmerge” in PLINK in “Consensus call” mode.
QC procedures were undertaken for the merged data to account for population stratification and differences in the Illumina chip versions. Data merging and QC procedures are illustrated in Fig. 2. The merged data were separated into 2 GWAS datasets (“A” and “B”) using the “--geno 0.05” command in PLINK.
Fig. 2.
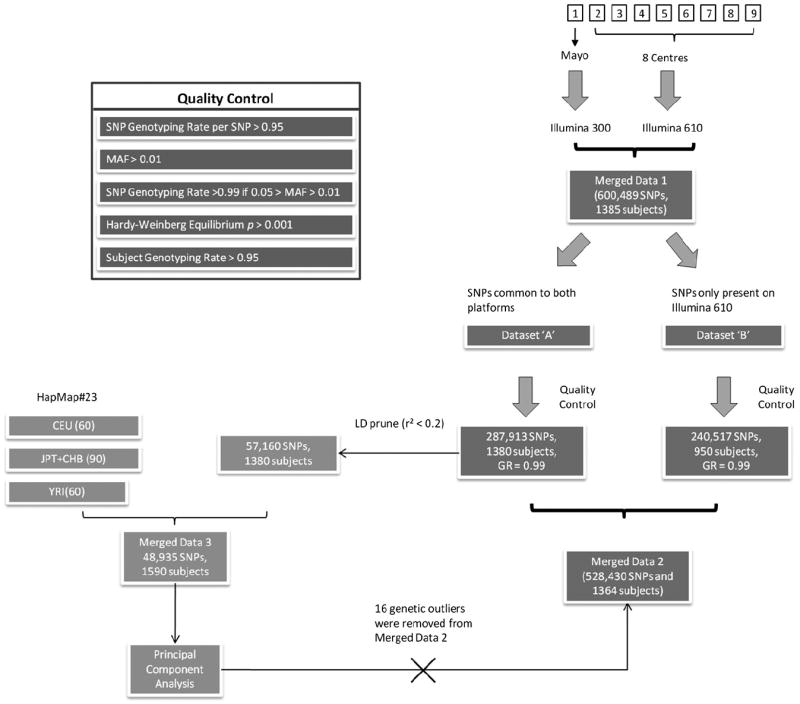
Genome-wide association studies (GWAS) data quality control (QC) and data merging strategy. Flow diagram summarizing the processes undertaken for data preparation and QC prior to subsequent analyses. Each GWAS dataset is represented by numbered squares (top right corner). The data were then merged together under PLINK “Consensus call” mode (Merged Data 1). These data were split into 2 groups (Dataset “A” and “B”) according to genotyping rate (single nucleotide polymorphisms [SNPs] which had > 95% genotyping rate for all samples [both chips] and the rest of the SNPs with genotyping rate > 95% for samples typed on the Illumina 610 chip). Both of these groups were subject to QC separately (see Quality Control panel at top left corner). The 2 datasets were then merged (Merged Data 2). Dataset “A” (which contained SNPs common to both platforms) was linkage disequilibrium (LD) pruned and merged with HapMap data (European [CEU], Asian [CHB/JPT], and Yoruba [YRI]) to form “Merged Data 3”. This was then used in a principal components analysis which revealed 16 individuals as genetic outliers. These were removed from “Merged Data 2”. Abbreviation: GR, genotyping rate.
For both of the GWAS datasets, the following QC procedures were carried out in order.
SNPs with genotyping rate less than 0.95 (--geno 0.05) were excluded from further analysis.
SNPs with a minor allele frequency (MAF) less than 0.01 (--maf 0.01) were excluded from further analysis.
A list of SNPs with MAF between 0.01 and 0.05 was generated (--freq). Within this short list, SNPs with a genotyping rate less than 0.99 (--geno 0.01) were excluded (--exclude) from further analysis.
SNPs with a Hardy-Weinberg Equilibrium p value less than 0.001 (--hwe 0.001 --hwe-all) were excluded from further analysis, irrespective of status (AD cases or controls).
Individuals with a genotyping rate less than 0.95 (--mind 0.05) were excluded from further analysis.
- Using the QC’d GWAS dataset “A”, a linkage disequilibrium (LD) pruned subset of 57,160 SNPs common to all arrays and HapMap data (--indep-pairwise) was generated. No 2 SNPs within this list had an LD r2 value greater than 0.2 across sliding windows (window size of 1500 SNPs and 150 SNPs to shift the window). This subset of SNPs was used by EIGENSTRAT (Price et al., 2006) for the following calculations:
- to detect genetic outliers defined by any individual whose ancestry is at least 6 SD from the mean on 1 of the top 10 axes of variation;
- to calculate principal components (PCs),
- To generate a population stratification plot HapMap data were used as the reference dataset—we used a filtered version of HapMap data (European [CEU], Asian [CHB/JPT], Yoruba [YRI]) release 23 in PLINK binary format (.bed.bim and .fam) from the PLINK web site (Purcell et al., 2007).
EIGENSTRAT was used to calculate the genomic control inflation factor (λ). This was performed iteratively using the GWAS dataset “A” with AAD including between 0 to 10 PCs. Each calculation generated a single λ value, thus a total of 11 λ values were generated. The number of PCs to be included as covariates was determined when the lowest λ value was acquired after comparison of all 11 λ values.
Extraction of PCs from EIGENSTRAT results.
After the above QC, dataset “A” consisted of SNPs common to both Illumina 300 and Illumina 610 chips, whereas da-taset “B” consisted of SNPs only on the Illumina 610 chip. The 2 GWAS datasets were merged using the same methods (--bmerge) as described above (shown as merged data 2 in Fig. 2).
We sought to find SNPs which showed bias in allele frequency due to interchip and intercohort differences, as they can cause an inflation of the type I error rate. A box and whisker plot was drawn using StatsDirect (version 2.7.8) (Fig. 3). Only 2 centers (Mayo and NIMH) showed significant differences in AAD range compared with the rest of the data. Therefore, we carried out 2 logistic regression tests using the WashU data as a control and the Mayo and NIMH data as cases. The test incorporated the top 6 PC (described previously) and AAD as covariates. For each comparison, a Q-Q plot of χ2 of observed versus expected p values was generated using “estlambda” function in GenABEL (version 1.6.4) (Fig. 4) (Aulchenko et al., 2007).
Fig. 3.
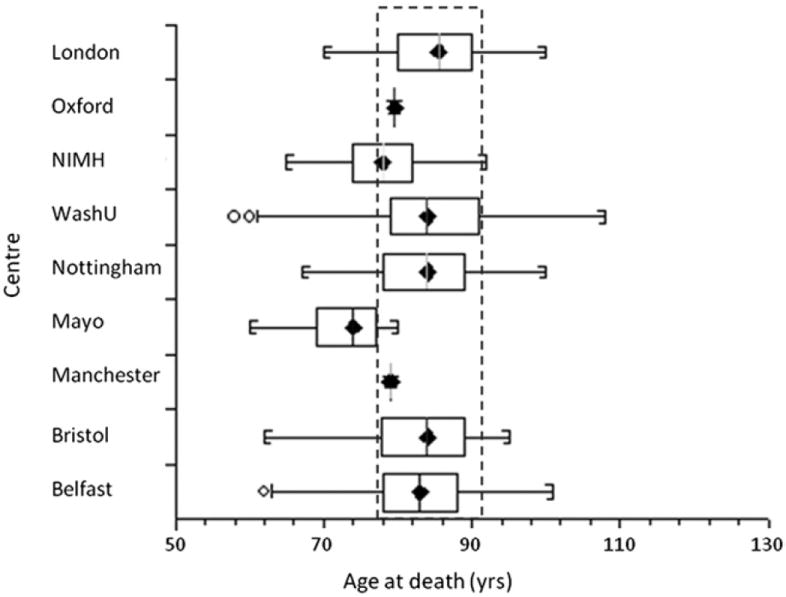
Box and whisker plot, showing the age at death (AAD) distribution for each center. The central box represents the distance between the first and third quartiles with the median marked with a diamond. The circles indicate that an individual’s AAD is outside 2 times the interquartile range. The dashed rectangle highlights that the majority of the data have a similar range of AAD with the exception of the National Institute of Mental Health (NIMH) and Mayo data.
Fig. 4.
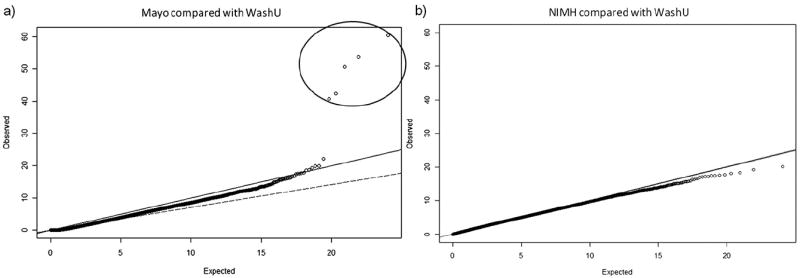
Q-Q plot of χ2-χ2 p values to determine bias in single nucleotide polymorphism (SNP) frequencies observed in Mayo (a), and National Institute of Mental Health (NIMH) (b) versus Washington University (WashU) data. (a) Logistic regression (Mayo versus WashU samples) adjusted for the top 6 principal components (PCs) and age at death (AAD). Five SNPs (circled) showed significant bias in the Mayo compared with WashU data taking into account population stratification and AAD. (b) Logistic regression comparing NIMH data versus WashU data adjusted for the top 6 PCs and AAD. No bias was observed in NIMH compared with WashU. Solid line represents expected under null hypothesis, i.e., no difference (or no association); open circles represent data points; dashed line the fitted slope of all data points. The diagram was drawn using GenABEL in R (version 2.12.1).
2.2. Quantitative trait analysis
Ten known LOAD susceptibility genes (APOE, CLU, PICALM, CR1, BIN1, ABCA7, MS4A6A, CD33, CD2AP, and EPHA1) were tested for association with human aging using the most significant SNPs found in the previous GWAS (Table 2). The best proxy (with the highest LD; using HapMap data) was used to inform the effect of SNPs if they were not present in our data.
Table 2.
SNP selection
| SNP used in our study | Gene | SNP cited in literature | LD (r2) | Literature |
|---|---|---|---|---|
| rs2075650 | APOE | rs2075650 | — | Harold et al. (2009) |
| rs11136000 | CLU | rs11136000 | — | Harold et al. (2009) |
| rs3851179 | PICALM | rs3851179 | — | Harold et al. (2009) |
| rs3818361 | CR1 | rs3818361 | — | Hollingworth et al. (2011) |
| rs744373 | BIN1 | rs744373 | — | Hollingworth et al. (2011) |
| rs3764650 | ABCA7 | rs3764650 | — | Hollingworth et al. (2011) |
| rs610932 | MS4A6A | rs610932 | — | Hollingworth et al. (2011) |
| rs3865444 | CD33 | rs3865444 | — | Hollingworth et al. (2011) |
| rs1485780 | CD2AP | rs9349407 | 0.913 | Hollingworth et al. (2011) |
| rs11767557 | EPHA1 | rs11767557 | — | Hollingworth et al. (2011) |
The most significant single nucleotide polymorphisms (SNPs) found in previous late-onset Alzheimer’s disease (LOAD) genome-wide association studies (GWAS) were selected for testing for their effects on human life span. The SNPs, associated genes, and the GWAS are as indicated. “-” indicates the original index SNP was used rather than a proxy.
Key: LD, linkage disequilibrium.
Quantitative trait (QT) analysis of all SNPs was performed using multivariable linear regression (--linear) adjusted for AD status, gender, and the top 6 PC. AAD was used as a continuous trait in this analysis thus giving maximum statistical power. Manhattan plots were drawn using Haploview (version 4.1) to visualize GWAS results (Fig. 5) (Barrett et al., 2005). A histogram of AAD from all individuals that passed QC was drawn using StatsDirect software (version 2.7.8) (Fig. 1).
Fig. 5.
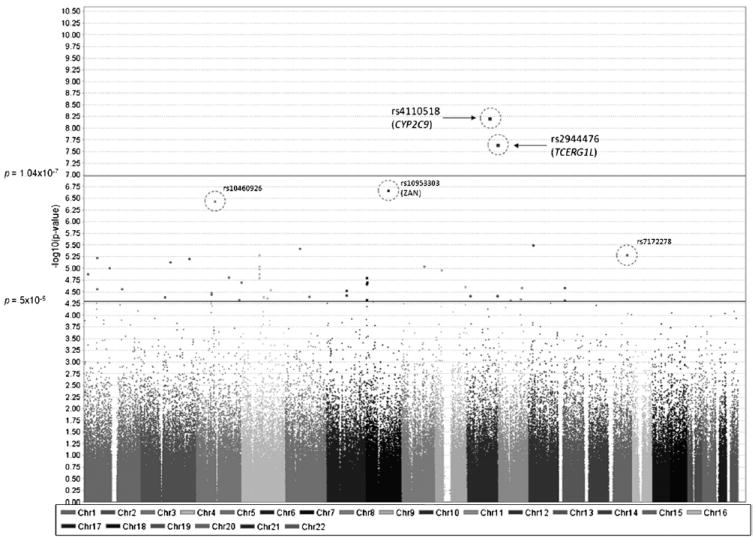
Manhattan plot of genome-wide association studies (GWAS) in human aging. Chromosomal position is shown on the x-axis versus -log10 GWAS p value on the y-axis. The threshold for genome-wide significance (p = 1.04 × 10−7) and p value threshold (p = 5 × 10−5) are indicated by the horizontal lines. Single nucleotide polymorphisms (SNPs) between these thresholds show “suggestive” associations. The 5 SNPs (highlighted by circles) exhibit significant differences in allele frequencies between samples from Mayo and Washington University (WashU) (see Fig. 4). Two of the 5 SNPs (rs4110518 and rs2944476) showed spurious genome-wide significant signals as a result of this bias.
2.3. Power calculation
Power calculations were undertaken using QUANTO version 1.2.4. The required sample size was estimated using an additive model created by the software.
2.4. MAF analysis of SNPs responsible for LOAD
The full range of AAD (58–108 years) was separated into 5 AAD categories, the boundaries of which were selected to ensure each group contains an approximately equal number of samples. This ensures SNP minor allele frequencies across each of the age categories are directly comparable. This was carried out using “Grouping = > Categorize” function in StatsDirect. For each of the LOAD susceptibility gene loci, the allele frequency was calculated and stratified by AAD category. The effect of the minor allele of each SNP involved in life span was as illustrated (Fig. 6).
Fig. 6.
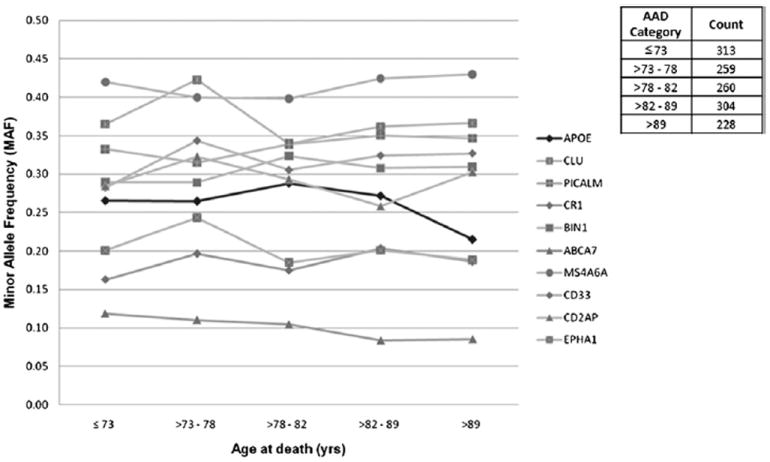
Minor allele frequency (MAF) analysis for 10 late-onset Alzheimer’s disease (LOAD) genes with respect to aging. The figure shows the relationship between single nucleotide polymorphism (SNP) MAFs and human aging, where age at death (AAD) is separated into 5 categories. Each AAD category contains roughly equal amounts of samples to avoid bias in sample sizes. All 10 known LOAD genes are shown together with the APOE locus highlighted in bold (all other loci in gray). The APOE locus (rs2075650) showed significant association with aging, with MAF = 0.27, AAD ≤ 89 years of age (n = 1136), and MAF = 0.21, AAD > 89 years of age (n = 228). None of the other gene loci were significantly associated with aging.
The separation into 5 AAD categories were used only to facilitate visualization of MAF of candidate SNPs in the different age ranges, and was not used to generate p values.
3. Results
3.1. Dataset composition and QC
The combined GWAS dataset has a sample size 1385 before QC. After QC, 4 participants were removed for low genotyping rate (--mind 0.05) from the Mayo GWAS (Carrasquillo et al., 2010). A single sample from Bristol was removed due to discrepancies between AAD and age at onset (AAO). An additional 16 samples were removed as genetic outliers by PCs analysis using EIGENSTRAT. This included 14 samples from the Mayo data, 1 from NIMH, and 1 from Belfast. The mean AAD in the pooled dataset (post QC) was greater than 80 years of age (Table 1).
The multidimensional scaling (MDS) plot showed 3 distinct clusters. As expected each cluster represents different population ancestry—CEU, middle right; CHB/JPT, bottom left; and YRI, top left (Fig. 7). UK, USA, and HapMap_ CEU samples formed a single cluster. On closer inspection, slight deviation between UK and USA samples exists and this was accounted for by including PCs as covariates.
Fig. 7.
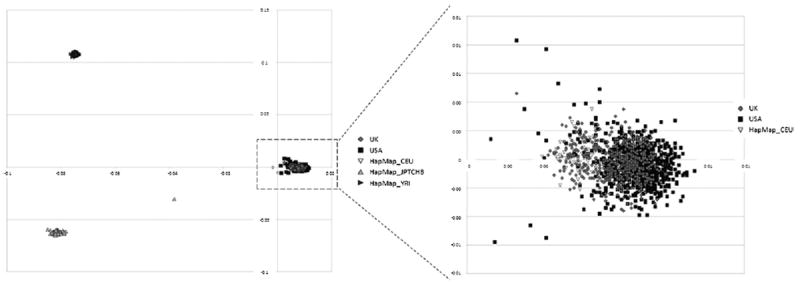
Multidimensional scaling (MDS) plot depicting the principal component analysis of Merged Data 3. Population stratification was tested using HapMap data #23 as reference. UK and USA and HapMap CEU samples formed a single cluster (shown inside the dashed rectangle). Up-pointing triangles represent Asian (CHB/JPT) population (bottom left), and right-pointing triangles represent Yoruba (YRI) population (top left). One HapMap individual from the Asian samples appears to have dual ethnicity. The diagram on the right shows a magnified section including UK, USA, and HapMap CEU samples.
Genomic control inflation factor (λ) was calculated using EIGENSTRAT by iteratively including 0 to 10 PC (Price et al., 2006). We found that including 6 PC generated the lowest genomic control inflation factor (λ = 1.003).
It was noted that there is a difference in AAD between LOAD cases (mean AAD = 81.63 years) and controls (mean AAD = 76.09 years), and similarly between male (mean AAD = 77.93 years) and female (mean AAD = 82.17 years). Anaysis of variance (ANOVA) tests of the variance of AAD between the groups (LOAD cases vs. controls and male vs. female) were found to be significant (p < 0.001). This confirmed that AD status and gender were appropriate covariates.
After stringent QC, there were 1364 samples (1031 LOAD cases and 333 controls, 608 male and 756 female) and 528,430 SNPs remaining for further analysis.
3.2. Analysis and results
Assessment of the 10 LOAD susceptibility genes yielded compelling evidence of association of APOE locus (rs2075650) with human aging (uncorrected p = 5.27 × 10−4), which withstood multiple testing after Bonferroni correction for 10 independent tests (Table 3A).
Table 3.
Summary of results
| SNP [major/minor] | CHR | BP | Gene | p | Direction of effect |
|---|---|---|---|---|---|
| (A) | |||||
| rs2075650 [T/C] | 19 | 50087459 | APOE | 5.27E-04 | — |
| rs3764650 [A/C] | 19 | 997520 | ABCA7 | 1.35E-01 | — |
| rs610932 [C/A] | 11 | 59695883 | MS4A6A | 1.76E-01 | + |
| rs3851179 [G/A] | 11 | 85546288 | PICALM | 2.27E-01 | + |
| rs11767557 [A/G] | 7 | 142819261 | EPHA1 | 2.67E-01 | — |
| rs3865444 [C/A] | 19 | 56419774 | CD33 | 4.42E-01 | + |
| rs3818361 [G/A] | 1 | 205851591 | CR1 | 5.63E-01 | + |
| rs1485780 [A/C] | 6 | 47664589 | CD2AP | 6.04E-01 | — |
| rs744373 [A/G] | 2 | 127611085 | BIN1 | 6.89E-01 | + |
| rs11136000 [G/A] | 8 | 27520436 | CLU | 9.75E-01 | — |
| (B) | |||||
| rs987839 [T/C] | 12 | 21266105 | SLCO1B1 | 3.19E-06 | + |
| rs17205854 [G/A] | 5 | 64458658 | 3.74E-06 | + | |
| rs17811551 [T/C] | 5 | 64462993 | ADAMTS6 | 3.74E-06 | + |
| rs1857821 [A/G] | 4 | 77101003 | NAAA | 5.12E-06 | + |
| rs7525717 [G/A] | 1 | 56700226 | 5.76E-06 | — | |
| rs4673651 [A/G] | 2 | 212712848 | ERBB4 | 6.08E-06 | — |
| rs17049647 [G/T] | 2 | 130093855 | 7.12E-06 | + | |
| rs10518142 [G/T] | 4 | 77061898 | NAAA | 8.89E-06 | + |
| rs2444861 [A/G] | 8 | 99170108 | C8orf47 | 8.99E-06 | + |
| rs1418425 [G/A] | 1 | 111270409 | 9.64E-06 | — | |
| rs2271528 [C/T] | 4 | 77107860 | SDAD1 | 1.02E-05 | + |
| rs1555453 [A/C] | 9 | 27316780 | MOBKL2B | 1.07E-05 | + |
| rs13111494 [A/G] | 4 | 77204512 | ART3 | 1.28E-05 | + |
| rs12740413 [C/T] | 1 | 16388466 | ARHGEF19 | 1.30E-05 | — |
| rs3210458 [C/T] | 3 | 142494320 | ACPL2 | 1.52E-05 | + |
| rs4859571 [G/A] | 4 | 77076333 | NAAA | 1.56E-05 | + |
| rs7803143 [T/C] | 7 | 6211011 | PSCD3 | 1.57E-05 | — |
| rs7622678 [T/C] | 3 | 198798845 | BDH1 | 1.93E-05 | — |
| rs4720752 [G/A] | 7 | 7735965 | RPA3 | 1.93E-05 | — |
| rs6962026 [C/T] | 7 | 6213048 | PSCD3 | 2.07E-05 | — |
| rs10901296 [C/T] | 9 | 132755477 | ABL1 | 2.37E-05 | + |
| rs680109 [C/A] | 11 | 105255919 | GRIA4 | 2.52E-05 | + |
| rs1537438 [G/T] | 13 | 26806973 | 2.53E-05 | + | |
| rs11206814 [C/T] | 1 | 56690636 | 2.67E-05 | — | |
| rs12562047 [A/C] | 1 | 164095780 | UCK2 | 2.70E-05 | + |
| rs2710548 [A/G] | 4 | 126492980 | FAT4 | 2.79E-05 | — |
| rs6454676 [C/T] | 6 | 88934174 | 2.90E-05 | + | |
| rs17047650 [C/T] | 3 | 68547103 | FAM19A1 | 3.19E-05 | + |
| rs10433502 [C/T] | 3 | 68569792 | FAM19A1 | 3.49E-05 | + |
| rs10485170 [T/C] | 6 | 88939371 | 3.67E-05 | + | |
| rs12257410 [A/C] | 10 | 13832528 | FRMD4A | 3.77E-05 | + |
| rs3125524 [C/T] | 10 | 133104931 | 3.83E-05 | + | |
| rs4280854 [A/G] | 5 | 105923201 | 3.85E-05 | + | |
| rs1037381 [A/G] | 2 | 105669675 | 3.99E-05 | — | |
| rs6532496 [A/G] | 4 | 95799427 | PDLIM5 | 4.00E-05 | — |
| rs17618813 [G/A] | 4 | 114153483 | ANK2 | 4.16E-05 | + |
| rs7103504 [G/A] | 11 | 99006474 | CNTN5 | 4.39E-05 | — |
| rs10085518 [C/T] | 7 | 6252040 | PSCD3 | 4.59E-05 | — |
| rs4686837 [G/A] | 3 | 188222371 | ST6GAL1 | 4.60E-05 | + |
| rs6491207 [T/C] | 13 | 26828900 | 4.63E-05 | + | |
| rs7952321 [G/T] | 11 | 55539349 | OR5AS1 | 4.68E-05 | — |
Results of the analysis of (A) the 10 documented late-onset Alzheimer’s disease (LOAD) susceptibility loci (APOE, CLU, PICALM, CR1, BIN1, ABCA7, MS4A6A, CD33, CD2AP, and EPHA1) with human aging. (B) Single-nucleotide polymorphisms (SNPs) (rs number [major/minor allele]) from our analysis with p values < 5 × 10−5. Chromosome number (CHR), base pair position (BP), and gene name is shown together with p value and direction of effect: “+” indicates the minor allele of any given SNP is protective, whereas “−” means the minor allele of the SNP has a detrimental effect on aging. In (B), the gene name is shown if the SNP is within 20 kilobase of a known gene.
In addition to examining the association of these 10 LOAD susceptibility genes with aging, we performed a genome-wide analysis which includes all SNPs on the Illumina platforms after QC. The genome-wide significance threshold was calculated (p = 1.04 × 10−7) using Bonferroni correction for the number of independent tests (n = 483,066), which was estimated using the method we have previously described (Shi et al., 2010).
No variants appear to be associated with aging with a genome-wide level of significance (p < 1.04 × 10−7). There were 41 SNPs with p value ≤ 5 × 10−5. These SNPs span the genome, representing 35 distinct signals (pairwise r2 ≤ 0.8) across 13 chromosomes. Twenty-four of them are located within 20 kilobase (kb) of known human genes with a wide range of functions. SNPs with p values < 5 × 10−5 are shown in Table 3B. These signals are at best tentative but may merit study in larger sample sets.
Without conducting logistic regression comparison, initial analysis of the association study suggested 2 genome-wide significant SNPs—rs4110518 (p = 5.96 × 10−9) and rs2944476 (p = 2.19 × 10−8). Comparing SNPs between NIMH and WashU data showed no significant difference in allele frequency, whereas 5 SNPs showed significant difference in allele frequency comparing Mayo data with WashU data after taking into account population stratification (i.e., PCs) and AAD. These 5 SNPs are rs4110518, rs2944476, rs10460926, rs10953303, and rs7172278 (Figs. 4 and 5).
It is perhaps unsurprising that the 2 SNPs which showed genome-wide level of significance overlap with the 5 SNPs that showed significant bias between Mayo and WashU data, as the AAD of the Mayo data are significantly younger (as previously described). It is not possible to correct for center, as the spread of the AAD is considered crucial in detecting genuine aging-associated variants. The difference in allele frequency due to samples with young AAD in Mayo and old AAD in WashU may well represent genuine associations. Including center as a covariate would abolish our ability to detect this effect.
The Manhattan plot shown in Fig. 5 represented a scenario before removal of these 5 false positive SNPs.
3.3. Power calculation
Power calculations indicate that a sample size approximately between 3000 and 15,000 is required in order to have 80% power to detect an association with a MAF ranging above 0.05. Approximately 4000 to 19,000 samples will be needed if 95% power is required.
This estimation should be interpreted with caution as it is based on a number of assumptions (such as effect size and mode of inheritance), and gene-environment interaction (G×E) has not been taken into account.
4. Discussion
It is known that advancing age is one of the biggest risk factors for LOAD. The prevalence of LOAD has been estimated ranging from 0.6% in persons aged 65 to 69 years to 22.2% in persons aged 90 and older (Lobo et al., 2000). Because age is one of the biggest risk factors for LOAD, it is important to understand whether genes involved in LOAD play a role in successful aging.
In this study, we have performed an association test of the top GWAS LOAD genes (APOE, CLU, PICALM, CR1, BIN1, ABCA7, MS4A6A, CD33, CD2AP, and EPHA1) with human aging using the most significant SNPs found in previous studies. Apart from the well-documented association between APOE and LOAD, the association with the other 9 genes was identified recently through large GWAS, each with a sample size of over 10,000 (Harold et al., 2009; Hollingworth et al., 2011; Lambert et al., 2009; Naj et al., 2011).
The results of our analysis provided compelling evidence of association between APOE locus (rs2075650) and human aging (p = 5.27 × 10−4) (Table 3A) with risk effect based on the analysis of 1364 samples using AAD as a continuous trait. The minor allele frequency plot (Fig. 6) shows that the MAF of this SNP significantly decreases in the old AAD category (MAF = 0.21; AAD > 89 years of age; n = 228) compared with the other 4 younger AAD categories (MAF = 0.27; AAD ≤ 89; n = 1136). Individuals homozygous for the minor allele “G” showed significantly lowered AAD (p = 0.002) compared with individuals homozygous for the major allele “A”. No effect was seen for the individuals carrying “AG” genotype (p = 0.891).
APOE has been extensively examined with respect to human aging due to its role in AD and vascular disease. A longitudinal study following subjects for 18 years using 1094 individuals aged 75 and older showed that the risk of mortality was affected by the APOE gene. Risk was increased by 22% in those carrying the APOE ε4 allele, decreased by 28% in those carrying the APOE ε2 allele, and individuals carrying APOE ε3 allele showed no significant difference in risk (Lewis and Brunner, 2004; Rosvall et al., 2009). The association between the APOE ε2 variant and aging has been investigated in Finish centenarians, where a trend of association was observed—9%, 21%, and 25% in people aged 100 to 101, 102 to 103, and 104 years and older, respectively (Frisoni et al., 2001). SNP rs2075650 is known to be in tight LD with the APOE ε4 allele (Yu et al., 2007). The direction of the effect of this SNP in our study is compatible with previous findings for the APOE ε4 allele (Christensen et al., 2006). Apolipoprotein E (APOE) is a major transporter of cholesterol and has been implicated in multiple age-related diseases including LOAD and vascular diseases (Panza et al., 2007).
We observed no evidence of association with the remaining LOAD genes implying that these genes are genuine LOAD genes with no detectable effects on human aging. However we cannot rule out the possibility that these genes may have a weak effect on aging that our dataset was not sufficiently large enough to detect.
We subsequently analyzed all SNPs on the Illumina chips after stringent QC procedures. The mean AAD for the samples analyzed was older than 80 years. This minimized the possibility of early death (prior to age 40 years) as a result of underlying nongenetic factors or highly penetrant genetic factors affecting our analysis (McGue et al., 1993).
In an assessment of all SNPs on the chip, none were found to approach genome-wide significance as calculated for this study (p < 1.04 × 10−7). The inability to detect any novel aging associated variants is likely the result of a lack of power. The calculation of power using QUANTO (version 1.2.4) has suggested a much larger sample size is required in order to detect an association with common variants. With rare exceptions, common variants are known to exert only small to moderate effects, according to previous studies of many complex disorders and traits (Bodmer and Bonilla, 2008). Presenting our data on genes that lie between 10−5 and 10−8, while not making genome wide significance, may enable groups to identify genes for future study especially if there is overlap with other studies. Additionally these data could be used as part of a larger meta-analysis.
GWAS provide a method of identifying common genetic variations associated with disease or phenotype in an unbiased manner. However, it comes with a price of multiple testing given that many thousands of SNPs are tested simultaneously, and a very stringent significance threshold (p < 5 × 10−8) is often used to infer a genome-wide significant association and to avoid a large number of false positives (Bertram and Tanzi, 2008).
We have conducted an analysis using AAD as a quantitative trait; this is believed to provide more power compared with the traditional case/control approach. The advantage of statistical power gained compared with the case/control analysis is dependent upon the design of the study. For example, dichotomizing the AAD distribution into cases and controls would give less power than comparing the low and high extremes of the quantitative trait (Plomin et al., 2009). Increasing statistical power by including more samples is imperative to elucidate genuine genetic associations in our study. Including more samples with the extreme phenotypes (e.g., exceptional longevity—nonagenarian and centenarians) would give more power than addition of samples of average AAD (Plomin et al., 2009; Tan et al., 2010).
Domestic and international collaborations are often required to raise sufficiently large sample sizes in order to have adequate power to detect genuine disease associations. This is especially true for SNPs with a small effect size. However, such combined analysis can in some instances generate new problems. For instance, interchip and interco-hort differences could create spurious genome-wide significant associations. More importantly, these SNPs may pass all conventional QC filtering (e.g., Hardy-Weinberg equilibrium p value, minor allele frequency, and genotyping rate threshold) increasing the likelihood of generating false positive results, which are not corrected for by principal component analysis.
As shown in Figs. 4 and 5, ignoring comparison of data from different centers (Mayo and WashU) would give spurious genome-wide significant associations (rs4110518, p = 5.96 × 10−9 and rs2944476, p = 2.19 × 10−8). Therefore, an extra caution should be made when performing GWAS analysis which utilizes data from multiple centers.
Including center as a covariate has been widely used to solve such problems raised by centers and this is largely effective. However this is not always possible, especially in circumstances where the number of cases and controls are significantly different between centers. In our study, correcting for center was not possible due to the AAD bias in centers sampled. The overall spread of AAD is crucial to our analysis and the difference in allele frequency between individuals with relatively young AAD (Mayo) and relatively old (WashU) may well represent genuine associations.
Furthermore, we have employed samples from both LOAD cases and controls in this study which is intended to achieve maximum power to detect novel aging-associated variants. Ideally this test is better performed using only control samples, as the AD group is likely to be affected by different environmental and genetic factors in addition to factors associated with aging. We have addressed this by correcting for AD status. However, it is possible that APOE association with age in the AD group could be due to its involvement in duration and severity of AD (Dal Forno et al., 2002). We do not however have clinical pathological features for these samples, and therefore cannot explore these endophenotypes.
Considering that “pure controls” where individuals die without experiencing any age-related diseases probably do not exist, we considered it valid to undertake an analysis using both sets. However, due to the large number of AD cases that have been used relative to the number of controls (about three-quarters of the total), any association with human aging implicated in the study may be biased and specific to the AD population. We acknowledge this as a limitation. However, using just the control population would result in insufficient numbers for a valid analysis. Follow-up studies using only control samples will be required to confirm these associations.
Acknowledgments
We thank all of our collaborators who provided data for this study (ARUK and GERAD1 consortia), together with the Alzheimer’s disease centers that collected the samples, as well as the subjects and their families, whose help and participation made this work possible. This work was supported by Alzheimer’s Research UK and the Big Lottery Fund.
Genetic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD1) Collaborators
Denise Harold, Rebecca Sims, Amy Gerrish, Jade Chapman, Valentina Moskvina, Richard Abraham, Paul Hollingworth, Marian Hamshere, Jaspreet Singh Pahwa, Kimberley Dowzell, Amy Williams, Nicola Jones, Charlene Thomas, Alexandra Stretton, and Angharad Morgan, Medical Research Council (MRC) Centre for Neuropsychiatric Genetics and Genomics, Neurosciences and Mental Health Research Institute, Department of Psychological Medicine and Neurology, School of Medicine, Cardiff University, Cardiff, UK; Simon Lovestone, John Powell, Petroula Proitsi, and Michelle K. Lupton, Department of Neuroscience, Institute of Psychiatry, Kings College London, London, UK; Carol Brayne, Institute of Public Health, University of Cambridge, Cambridge, UK; David C. Rubinsztein, Cambridge Institute for Medical Research, University of Cambridge, Cambridge, UK; Michael Gill, Brian Lawlor, and Aoibhinn Lynch, Mercer’s Institute for Research on Aging, St. James’ Hospital and Trinity College, Dublin, Ireland; Kevin Morgan, and Kristelle Brown, Institute of Genetics, Queen’s Medical Centre, University of Nottingham, Nottingham, UK; Peter Passmore, David Craig, Bernadette McGuinness, and Stephen Todd, Ageing Group, Centre for Public Health, School of Medicine, Dentistry and Biomedical Sciences, Queen’s University Belfast, Belfast, UK; Clive Holmes, Division of Clinical Neurosciences, School of Medicine, University of Southampton, Southampton, UK; David Mann, Clinical Neuroscience Research Group, Greater Manchester Neurosciences Centre, University of Manchester, Salford, UK; A. David Smith, Oxford Project to Investigate Memory and Ageing (OPTIMA), University of Oxford, Level 4, John Radcliffe Hospital, Oxford, UK;Seth Love, and Patrick G. Kehoe, Dementia Research Group, University of Bristol Institute of Clinical Neurosciences, Frenchay Hospital, Bristol, UK; John Hardy, Department of Molecular Neuroscience and Reta Lilla Weston Laboratories, Institute of Neurology, London, UK; Simon Mead, MRC Prion Unit, Department of Neurodegenerative Disease, UCL Institute of Neurology, London, UK; Nick Fox, and Martin Rossor, Dementia Research Centre, Department of Neurodegenerative Diseases, University College, London, Institute of Neurology, London, UK; John Collinge, MRC Prion Unit, Department of Neurodegenerative Disease, UCL Institute of Neurology, London, UK; Wolfgang Maier, Frank Jessen, Reiner Heun, and Heike Kölsch, Department of Psychiatry, University of Bonn, Bonn, Germany; Britta Schürmann, Department of Psychiatry, and Institute for Molecular Psychiatry, University of Bonn, Bonn, Germany;Hendrik van den Bussche, Institute of Primary Medical Care, University Medical Center Hamburg-Eppendorf, Germany; Isabella Heuser, Department of Psychiatry, Charité, Berlin, Germany; Johannes Kornhuber, Department of Psychiatry, University of Erlangen, Nürnberg, Germany; Jens Wiltfang, LVR-Hospital Essen, Department of Psychiatry and Psychotherapy, University Duisburg-Essen, Germany; Martin Dichgans, Institute for Stroke and Dementia Research, and Department of Neurology, Klinikum der Universität München, Munich, Germany; Lutz Frölich, Central Institute of Mental Health, Medical Faculty Mannheim, University of Heidelberg, Germany; Harald Hampel, Department of Psychiatry, Psychosomatic Medicine and Psychotherapy, Goethe University, Frankfurt, Germany; Michael Hüll, Centre for Geriatric Medicine and Section of Gerontopsychiatry and Neuropsychology, Medical School, University of Freiburg, Germany; Dan Rujescu, Alzheimer Memorial Center and Geriatric Psychiatry Branch, Department of Psychiatry, Ludwig-Maximilian University, Munich, Germany; Alison Goate, Departments of Psychiatry, Neurology and Genetics, Washington University School of Medicine, St. Louis, MO, USA; John S.K. Kauwe, Department of Biology, Brigham Young University, Provo, UT, USA; Carlos Cruchaga, Petra Nowotny, John C. Morris, and Kevin Mayo, Departments of Psychiatry, Neurology and Genetics, Washington University School of Medicine, St. Louis, MO, USA; Gill Livingston, Nicholas J. Bass, Hugh Gurling, and Andrew McQuillin, Department of Mental Health Sciences, University College London, London, UK; Rhian Gwilliam, and Panagiotis Deloukas, The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK; Markus M. Nöthen, Department of Genomics, Life and Brain Center, University of Bonn, Bonn, Germany; Peter Holmans, Michael O’Donovan, Michael J. Owen, and Julie Williams, Medical Research Council (MRC) Centre for Neuropsychiatric Genetics and Genomics, Neurosciences and Mental Health Research Institute, Department of Psychological Medicine and Neurology, School of Medicine, Cardiff University, Cardiff, UK.
GERAD1 acknowledgements
Cardiff University was supported by the Wellcome Trust, Medical Research Council (MRC), Alzheimer’s Research Trust (ART) and the Welsh Assembly Government. ART supported sample collections at the Kings College, London, the South West Dementia Bank, Universities of Cambridge, Nottingham, Manchester, and Belfast. The Belfast group acknowledges support from the Alzheimer’s Society, Ulster Garden Villages, N. Ireland R&D Office, and the Royal College of Physicians/Dunhill Medical Trust. The MRC and Mercer’s Institute for Research on Ageing supported the Trinity College group. The South West Dementia Brain Bank acknowledges support from Bristol Research into Alzheimer’s and Care of the Elderly. The Charles Wolfson Charitable Trust supported the OPTIMA group. Washington University was funded by NIH grants, Barnes Jewish Foundation, and the Charles and Joanne Knight Alzheimer’s Research Initiative. Patient recruitment for the MRC Prion Unit/UCL Department of Neurodegenerative Disease collection was supported by the UCLH/UCL Biomedical Centre. LASER-AD was funded by Lundbeck SA. The Bonn group was supported by the German Federal Ministry of Education and Research (BMBF), Competence Network Dementia, and Competence Network Degenerative Dementia, and by the Alfried Krupp von Bohlen und Halbach-Stiftung. The GERAD1 Consortium also used samples ascertained by the NIMH AD Genetics Initiative.
Footnotes
Disclosure statement
The authors declare that there are no conflicts of interest.
Approval was obtained from the ethics committee or institutional review board of each institution responsible for the ascertainment and collection of samples. Written informed consent was obtained for all individuals who participated in this study.
References
- Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC. Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ Res. 2005;96:412–418. doi: 10.1161/01.RES.0000157171.04054.30. [DOI] [PubMed] [Google Scholar]
- Atzmon G, Schechter C, Greiner W, Davidson D, Rennert G, Barzilai N. Clinical phenotype of families with longevity. J Am Geriatr Soc. 2004;52:274–277. doi: 10.1111/j.1532-5415.2004.52068.x. [DOI] [PubMed] [Google Scholar]
- Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Barzilai N, Atzmon G, Schechter C, Schaefer EJ, Cupples AL, Lipton R, Cheng S, Shuldiner AR. Unique lipoprotein phenotype and genotype associated with exceptional longevity. Jama. 2003;290:2030–2040. doi: 10.1001/jama.290.15.2030. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo MM, Belbin O, Zou F, Allen M, Ertekin-Taner N, Ansari M, Wilcox SL, Kashino MR, Ma L, Younkin LH, Younkin SG, Younkin CS, Dincman TA, Howard ME, Howell CC, Stanton CM, Watson CM, Crump M, Vitart V, Hayward C, Hastie ND, Rudan I, Campbell H, Polasek O, Brown K, Passmore P, Craig D, McGuinness B, Todd S, Kehoe PG, Mann DM, Smith AD, Beaumont H, Warden D, Holmes C, Heun R, Kölsch H, Kalsheker N, Pankratz VS, Dickson DW, Graff-Radford NR, Petersen RC, Wright AF, Morgan K, Morgan K. Concordant association of insulin degrading enzyme gene (IDE) variants with IDE mRNA, Abeta, and Alzheimer’s disease. PLoS One. 2010;5:e8764. doi: 10.1371/journal.pone.0008764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo MM, Zou F, Pankratz VS, Wilcox SL, Ma L, Walker LP, Younkin SG, Younkin CS, Younkin LH, Bisceglio GD, Ertekin-Taner N, Crook JE, Dickson DW, Petersen RC, Graff-Radford NR, Younkin SG. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer’s disease. Nat Genet. 2009;41:192–198. doi: 10.1038/ng.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen K, Johnson TE, Vaupel JW. The quest for genetic determinants of human longevity: challenges and insights. Nat Rev Genet. 2006;7:436–448. doi: 10.1038/nrg1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Mattson MP. The adversities of aging. Ageing Res Rev. 2006;5:221–238. doi: 10.1016/j.arr.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Dal Forno G, Carson KA, Brookmeyer R, Troncoso J, Kawas CH, Brandt J. APOE genotype and survival in men and women with Alzheimer’s disease. Neurology. 2002;58:1045–1050. doi: 10.1212/wnl.58.7.1045. [DOI] [PubMed] [Google Scholar]
- de Chaves EP, Narayanaswami V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008;3:505–530. doi: 10.2217/17460875.3.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhães JP, Budovsky A, Lehmann G, Costa J, Li Y, Fraifeld V, Church GM. The Human Ageing Genomic Resources: online databases and tools for biogerontologists. Aging Cell. 2009;8:65–72. doi: 10.1111/j.1474-9726.2008.00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efrat M, Aviram M. Paraoxonase 1 interactions with HDL, anti-oxidants and macrophages regulate atherogenesis - a protective role for HDL phospholipids. Adv Exp Med Biol. 2010;660:153–166. doi: 10.1007/978-1-60761-350-3_14. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Louhija J, Geroldi C, Trabucchi M. Longevity and the epsilon2 allele of apolipoprotein E: the Finnish Centenarians Study. J Gerontol A Biol Sci Med Sci. 2001;56:M75–M78. doi: 10.1093/gerona/56.2.m75. [DOI] [PubMed] [Google Scholar]
- Glatt SJ, Chayavichitsilp P, Depp C, Schork NJ, Jeste DV. Successful aging: from phenotype to genotype. Biol Psychiatry. 2007;62:282–293. doi: 10.1016/j.biopsych.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskind AM, McGue M, Holm NV, Sørensen TI, Harvald B, Vaupel JW. The heritability of human longevity: a population-based study of 2872 Danish twin pairs born 1870-1900. Hum Genet. 1996;97:319–323. doi: 10.1007/BF02185763. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, Destefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Géloën A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Iachine IA, Holm NV, Harris JR, Begun AZ, Iachina MK, Laitinen M, Kaprio J, Yashin AI. How heritable is individual susceptibility to death? The results of an analysis of survival data on Danish, Swedish and Finnish twins. Twin Res. 1998;1:196–205. doi: 10.1375/136905298320566168. [DOI] [PubMed] [Google Scholar]
- Jylhava J, Hurme M. Gene variants as determinants of longevity: focus on the inflammatory factors. Pflugers Arch. 2010;459:239–246. doi: 10.1007/s00424-009-0726-3. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lescai F, Marchegiani F, Franceschi C. PON1 is a longevity gene: results of a meta-analysis. Ageing Res Rev. 2009;8:277–284. doi: 10.1016/j.arr.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Lewis SJ, Brunner EJ. Methodological problems in genetic association studies of longevity—the apolipoprotein E gene as an example. Int J Epidemiol. 2004;33:962–970. doi: 10.1093/ije/dyh214. [DOI] [PubMed] [Google Scholar]
- Lobo A, Launer LJ, Fratiglioni L, Andersen K, Di Carlo A, Breteler MM, Copeland JR, Dartigues JF, Jagger C, Martinez-Lage J, Soininen H, Hofman A. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group Neurology. 2000;54:S4–S9. [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- McGue M, Vaupel JW, Holm N, Harvald B. Longevity is moderately heritable in a sample of Danish twins born 1870-1880. J Gerontol. 1993;48:B237–B244. doi: 10.1093/geronj/48.6.b237. [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St, George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, Decarli C, Dekosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F, D’Introno A, Capurso C, Colacicco AM, Seripa D, Pilotto A, Santamato A, Capurso A, Solfrizzi V. Lipoproteins, vascular-related genetic factors, and human longevity. Rejuvenation Res. 2007;10:441–458. doi: 10.1089/rej.2007.0537. [DOI] [PubMed] [Google Scholar]
- Panza F, Frisardi V, Capurso C, D’Introno A, Colacicco AM, Seripa D, Pilotto A, Vendemiale G, Capurso A, Solfrizzi V. Apolipoprotein E, dementia, and human longevity. J Am Geriatr Soc. 2009;57:740–742. doi: 10.1111/j.1532-5415.2009.02211.x. [DOI] [PubMed] [Google Scholar]
- Perls TT, Bubrick E, Wager CG, Vijg J, Kruglyak L. Siblings of centenarians live longer. Lancet. 1998;351:1560. doi: 10.1016/S0140-6736(05)61126-9. [DOI] [PubMed] [Google Scholar]
- Perls TT, Wilmoth J, Levenson R, Drinkwater M, Cohen M, Bogan H, Joyce E, Brewster S, Kunkel L, Puca A. Life-long sustained mortality advantage of siblings of centenarians. Proc Natl Acad Sci U S A. 2002;99:8442–8447. doi: 10.1073/pnas.122587599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, Haworth CM, Davis OS. Common disorders are quantitative traits. Nat Rev Genet. 2009;10:872–878. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- Polito L, Kehoe PG, Forloni G, Albani D. The molecular genetics of sirtuins: association with human longevity and age-related diseases. Int J Mol Epidemiol Genet. 2010;1:214–225. [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosvall L, Rizzuto D, Wang HX, Winblad B, Graff C, Fratiglioni L. APOE-related mortality: effect of dementia, cardiovascular disease and gender. Neurobiol Aging. 2009;30:1545–1551. doi: 10.1016/j.neurobiolaging.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Sebastiani P, Montano M, Puca A, Solovieff N, Kojima T, Wang MC, Melista E, Meltzer M, Fischer SE, Andersen S, Hartley SH, Sedgewick A, Arai Y, Bergman A, Barzilai N, Terry DF, Riva A, Anselmi CV, Malovini A, Kitamoto A, Sawabe M, Arai T, Gondo Y, Steinberg MH, Hirose N, Atzmon G, Ruvkun G, Baldwin CT, Perls TT. RNA editing genes associated with extreme old age in humans and with lifespan in C. elegans PLoS One. 2009;4:e8210. doi: 10.1371/journal.pone.0008210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Medway C, Bullock J, Brown K, Kalsheker N, Morgan K. Analysis of Genome-Wide Association Study (GWAS) data looking for replicating signals in Alzheimer’s disease (AD) Int J Mol Epidemiol Genet. 2010;1:53–66. [PMC free article] [PubMed] [Google Scholar]
- Suh Y, Atzmon G, Cho MO, Hwang D, Liu B, Leahy DJ, Barzilai N, Cohen P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci U S A. 2008;105:3438–3442. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taioli E, Mari D, Franceschi C, Bonafè M, Monti D, Bertolini S, Marinelli D, Garte S. Polymorphisms of drug-metabolizing enzymes in healthy nonagenarians and centenarians: difference at GSTT1 locus. Biochem Biophys Res Commun. 2001;280:1389–1392. doi: 10.1006/bbrc.2001.4280. [DOI] [PubMed] [Google Scholar]
- Tan Q, Zhao JH, Li S, Kruse TA, Christensen K. Power assessment for genetic association study of human longevity using offspring of long-lived subjects. Eur J Epidemiol. 2010;25:501–506. doi: 10.1007/s10654-010-9465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, Seltman H, Peskind ER, Galloway N, Zhou PX, Rosenthal E, Wijsman EM, Tsuang DW, Devlin B, Schellenberg GD. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: patterns of linkage disequilibrium and disease/marker association. Genomics. 2007;89:655–665. doi: 10.1016/j.ygeno.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]