

Abstract
Objective
Vascular cells, particularly endothelial cells, adopt aerobic glycolysis to generate energy to support cellular functions. The effect of endothelial glycolysis on angiogenesis remains unclear. 6-Phosphofructo-2-kinase/fructose-2, 6-bisphosphatase, isoform 3 (PFKFB3), is a critical enzyme for endothelial glycolysis. By blocking or deleting PFKFB3 in endothelial cells, we investigated the influence of endothelial glycolysis on angiogenesis both in vitro and in vivo.
Approach and Results
Under hypoxic conditions or following treatment with angiogenic factors, endothelial PFKFB3 was upregulated both in vitro and in vivo. The knockdown or overexpression of PFKFB3 suppressed or accelerated endothelial proliferation and migration in vitro, respectively. Neonatal mice from a model of oxygen-induced retinopathy showed suppressed neovascular growth in the retina when endothelial PFKFB3 was genetically deleted or when the mice were treated with a PFKFB3 inhibitor. Additionally, tumors implanted in mice deficient in endothelial PFKFB3 grew more slowly and were provided with less blood flow. A lower level of phosphorylated AKT (pAKT) was observed in PFKFB3-knockdown endothelial cells, which was accompanied by a decrease in intracellular lactate. The addition of lactate to PFKFB3-knockdown cells rescued the suppression of endothelial proliferation and migration.
Conclusions
The blockade or deletion of endothelial PFKFB3 decreases angiogenesis both in vitro and in vivo. Thus, PFKFB3 is a promising target for the reduction of endothelial glycolysis and its related pathological angiogenesis.
Keywords: endothelial cells, angiogenesis, glycolysis, hypoxia
Introduction
Angiogenesis is the growth of a new blood vessel from the existing vasculature. This capability is critical for many physiological and pathological processes.1 When the angiogenic process is dysregulated, the formation of new blood vessels can take the form of pathological angiogenesis, leading to the development and progression of various malignant, ischemic, inflammatory and immune diseases.1, 2 Endothelial cells are especially critical in the process of angiogenesis. For example, the migration and proliferation of endothelial cells initiate the formation of capillary networks, which provide a frame for further vascular maturation.3 Much attention has been given to investigating the effect of angiogenic growth factors on endothelial activities during angiogenesis. Indeed, a number of mechanisms have been described for the actions of these angiogenic factors, including vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) among many others.4
Endothelial cells have high glycolytic activity.5–9 The level of glycolysis in endothelial cells is comparable to that of tumor cells and much higher than that of other healthy cells.6 Additionally, glycolytic flux in endothelial cells is more than 200-fold higher than glucose oxidation, fatty acid oxidation and glutamine oxidation, resulting in the generation of over 85% of the total cellular ATP content.5 In glycolytic flux, the conversion of fructose-6-phosphate (F6P) to fructose-1, 6-bisphosphate (F1, 6P2), is one of three rate-limiting checkpoints. 6-Phosphofructo-1 kinase (PFK-1), the enzyme that catalyzes the above reaction, is activated by its allosteric activator, fructose-2, 6-bisphosphate (F2, 6P2).10 In endothelial cells, F2, 6P2, is synthesized by 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase, isoform 3 (PFKFB3).11 A recent study demonstrated that PFKFB3-driven glycolysis is important for the migration of endothelial cells. In response to angiogenic factors, PFKFB3-knockdown endothelial cells exhibit defects in the formation of filopodia and lamellipodia.6 However, it remains unclear whether PFKFB3 is important for angiogenesis.
We were interested in determining whether the inhibition of PFKFB3 could suppress angiogenesis, especially pathological angiogenesis. Using PFKFB3 knockdown, overexpression or inhibition of PFKFB3 activity with an inhibitor, we examined endothelial cell proliferation and tube formation in vitro. Furthermore, we generated floxed PFKFB3 (PFKFB3fl/fl) mice. By breeding these mice with endothelial cell-specific Cre (cdh5-Cre) mice, we generated mice with a deficiency of PFKFB3 specifically in endothelial cells. With these mice and their controls, the role of endothelial PFKFB3 in angiogenesis was evaluated by comparing the size of implanted tumors and the blood supply of these tumors and the severity of retinal neovascularization in the retina of mice from an oxygen-induced retinopathy model. Furthermore, the underlying mechanisms contributing to PFKFB3-associated angiogenesis were explored.
Materials and Methods
Materials and Methods are available in the online-only Supplement.
Results
Hypoxia and angiogenic factors upregulate endothelial PFKFB3
To examine the effect of hypoxia on the expression of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (PFKFB), HUVECs were cultured in an incubator with 0.5% oxygen for 24 h. The mRNA expression levels of PFKFB isoforms 1 to 4 were analyzed with real-time RT-PCR. PFKFB isoforms 1 and 3 were increased 1- and 5-fold, respectively, whereas isoforms 2 and 4 did not show significant changes compared with normoxia controls (Fig. 1A). The upregulation of PFKFB3 protein expression in endothelial cells under hypoxic conditions was confirmed using western blotting (Fig. 1C). The expression of endothelial PFKFB3 in response to VEGF treatment was also examined. PFKFB3 mRNA expression was increased at 12 but not 24 h following VEGF stimulation, and the protein expression was increased at both time points (Fig. 1B and 1D), indicating that in addition to the regulation of PFKFB3 at the transcriptional level, VEGF may also regulate endothelial PFKFB3 at the posttranscriptional level.
Figure 1. Induction of endothelial PFKFB3 by hypoxia and hVEGF.
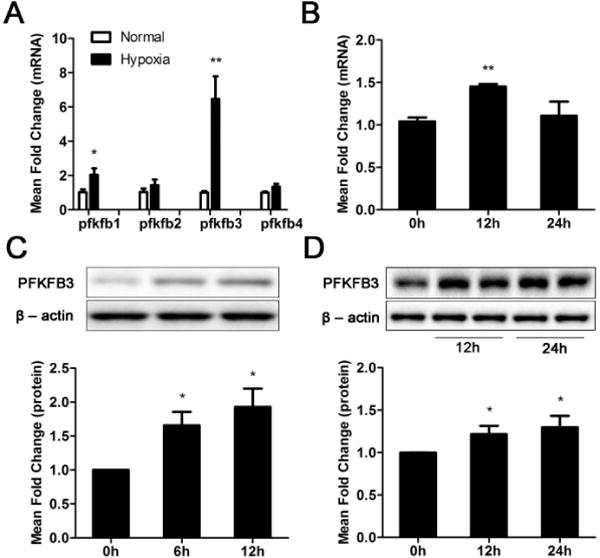
A, Real-time RT-PCR analysis of PFKFB transcripts from total RNA isolated from HUVECs exposed to hypoxia (0.5% oxygen) for 24 h. B, Real-time RT-PCR analysis of PFKFB3 transcripts in HUVECs treated with or without hVEGF (100 ng/ml) for 24 h. C, Immunoblot analysis of PFKFB3 and β-actin (loading control) expression in lysates of HUVECs exposed to hypoxia for 0, 6 and 12 h. D, Immunoblot analysis of PFKFB3 and β-actin expression in HUVECs treated with hVEGF (100 ng/ml) for 0, 12 or 24 h. For C and D, top panels are representative images. For bar graphs (A – D), data are the mean ± SD, n = 3. *, P < 0.05 and **, P < 0.01 for Hypoxia (6 or 12 h) vs. Normal (0 h) (in C), for the same gene (in A) and 12 h or 24 h vs. 0 h (in B and D).
Endothelial PFKFB3 is involved in endothelial proliferation and endothelial tube formation in vitro
To examine whether PFKFB3 affects endothelial proliferation, endothelial cells pretreated with control adenovirus (Ad-shctl) or PFKFB3-knockdown adenovirus (Ad-shpfkfb3) were placed in endothelial growth cell medium under normoxic (21% oxygen, left panel of Fig. 2A) or hypoxic (0.5% oxygen, the middle panel of Fig. 2A) conditions. Infection with PFKFB3-knockdown adenovirus resulted in a decreased expression of PFKFB3 in HUVECs (Supp. Fig. 1A) The growth of PFKFB3-knockdown endothelial cells decreased 50–80% compared with control cells over a period of 96 h (Fig. 2A). In contrast, the growth of endothelial cells infected with a PFKFB3-overexpressing adenovirus increased 35% compared with endothelial cells treated with a control virus (right panel of Fig. 2A). Furthermore, under normoxic conditions the cell cycle was analyzed and BrdU incorporation was evaluated in endothelial cells cultured in a complete growth medium for 24 h after the cells were synchronized by total FBS depletion. Flow cytometry showed that the percentage of cells in S phase was 20–22% in control HUVECs, whereas this percentage decreased to 9–11% for PFKFB3-knockdown HUVECs (Fig. 2B). Immunostaining in the BrdU incorporation assay showed that the percentage of BrdU-positive cells decreased 30–35% in PFKFB3-knockdown cells and increased 85–95% in PFKFB3-overexpressing cells compared with control cells (Fig. 2C). To investigate whether PFKFB3 affected endothelial migration, a tube formation assay was conducted, which consisted of placing cells on growth factor-deprived Matrigel. The number of formed tubes was decreased 40–46% in PFKFB3-knockdown HUVECs compared with control HUVECs (Fig. 2D). In PFKFB3-overexpressing HUVECs, the number of tubes was 52–60% higher than that of control HUVECs (Fig. 2D), indicating that PFKFB3 increases endothelial migration.
Figure 2. Involvement of PFKFB3 in endothelial proliferation and tube formation.
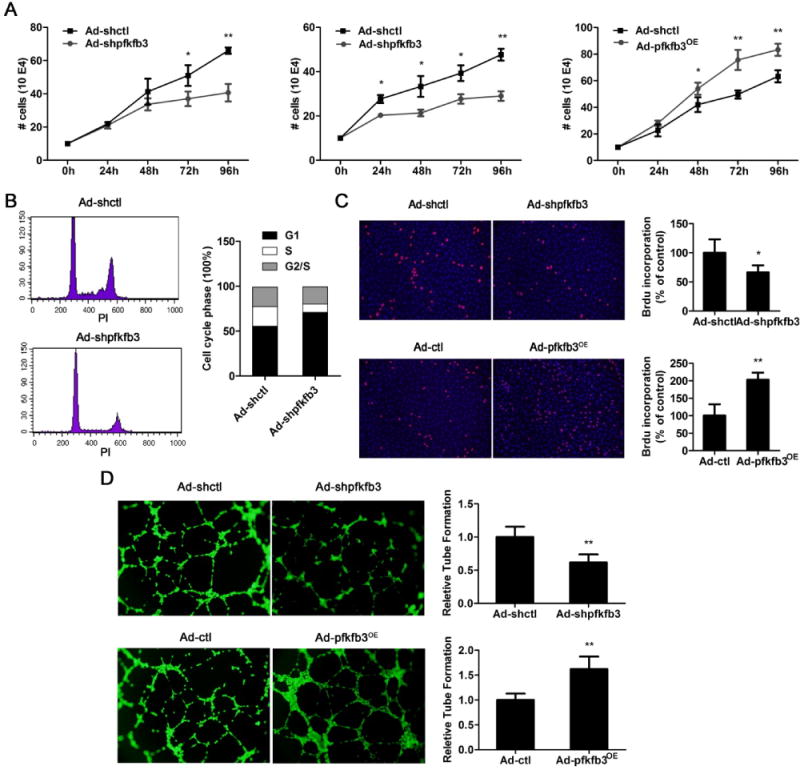
A, Hypoxia-induced cell growth. The number of PFKFB3-knockdown or overexpressing HUVECs was counted after exposure to hypoxia (0.5% oxygen, the middle panel of A) or normal oxygen (the left and right panes of A) for 0, 24, 48, 72 or 96 h. B, Flow cytometric analysis of the cell cycle. PFKFB3-knockdown HUVECs and control cells were synchronized in G0/G1 by total FBS depletion for 16 h and then exposed to complete medium for an additional 24 h. C, BrdU staining of proliferating HUVECs. PFKFB3-knockdown, PFKFB3-overxpressing and control cells were synchronized in G0/G1 by total FBS depletion for 16 h and then incubated with complete medium for an additional 24 h. D, Representative images and quantification of tube formation in control, PFKFB3-knockdown and PFKFB3-overexpressing HUVECs cultured in growth factor-deprived Matrigel. Experiments in B, C and D were performed under normoxic conditions. For bar graphs (A – D), data are mean ± SD, n = 3. *, P < 0.05 and **, P < 0.01 for Ad-shpfkfb3 or Ad-pfkfb3OE vs. Ad-shctl or Ad-ctl (in A, C and D).
Endothelial PFKFB3 participates in pathological angiogenesis in vivo
To investigate the role of endothelial PFKFB3 in angiogenesis in vivo, floxed PFKFB3 mice (PFKFB3WT) were generated (Fig. 3A to 3C) and then bred with cdh5-Cre mice to create mice with deficiency in PFKFB3 in endothelial cells only (PFKFB3VEC-KO, Fig. 3D), and then both the oxygen-induced retinopathy (OIR) model and the tumor implantation model were used in these mice. For mouse pups in normal room air, the expression of retinal PFKFB3 in seven-day-old (P7), P12 and P17 mice was comparable (data not shown). In contrast, in OIR mice, the mRNA expression level of retinal PFKFB3 was decreased 25–30% at P12 and increased over 2-fold at P17 compared with the expression level at P7 (Fig. 4A). This dynamic change in retinal PFKFB3 expression was also observed at the protein level (Fig. 4B). Retinal angiogenesis was evaluated by measuring the vascular area in retinal whole-mounts following isolectin staining. The retinal density was slightly lower in PFKFB3VEC-KO mice than in PFKFB3WT mice at P4, and this difference was insignificant at P7 (Supp. Fig. 2A and 2B). After P7, mice were exposed to 75% oxygen for 5 d. PFKFB3VEC-KO mice at P12 showed an increase in avascular retinal area compared with PFKFB3WT mice, although this increase was not statistically significant (Fig. 4C). At P17, when the neovascular tuft (NVT) reaches its maximum, the NVT area of PFKFB3VEC-KO mice was 50–55% smaller than that of PFKFB3WT mice (Fig. 4D). Similar decreases in NVT area were also observed in control mouse pups treated with the PFKFB3 inhibitor 3PO12 (Fig. 4E). Additionally, the same OIR models were generated in heterozygotes of PFKFB3 deletion mice,13–15 the retinal areas of both avascular and NVT were the same as those in littermate controls (Supp. Fig. 3), indicating that the deleting of one allele of PFKFB3 gene in endothelial cells is not sufficient to alter endothelial angiogenesis.
Figure 3. Generation of mice with a PFKFB3 deficiency in endothelial cells.
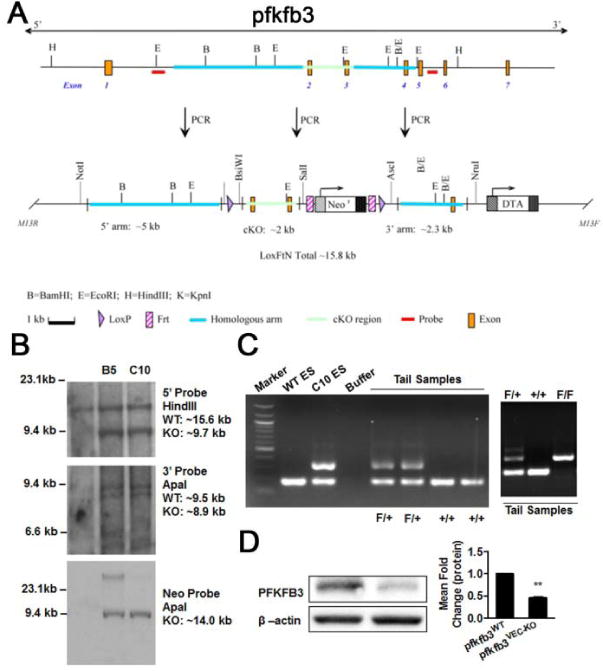
A, LoxP targeting of PFKFB3. The targeting construct to introduce the LoxP sites into the PFKFB3 gene. The top diagram indicates the wild-type PFKFB3 locus; the diagram below that indicates the targeted PFKFB3 locus. cKO, conditional knockout. B, C57BL/6 ES cells were transfected with a PFKFB3-floxed vector and selected with 200 mg/ml G418. Selected positive clones (B5 and C10) were expanded for further analysis. Clone C10 was confirmed for homologous recombination with a single neo integration using the 5′, 3′ Neo probes. C, heterozygous (F/+, PFKFB3-floxed) mice were obtained by breeding the male chimera with wild-type (+/+) C57BL/6 females. D, floxed mice were bred with cdh5-Cre mice to generate mice with a deficiency of PFKFB3 in the endothelial cells. Aortic endothelial cells were isolated from the mice and assayed for the expression of PFKFB3 with western blotting.
Figure 4. Endothelial PFKFB3 participates in pathological angiogenesis in vivo.
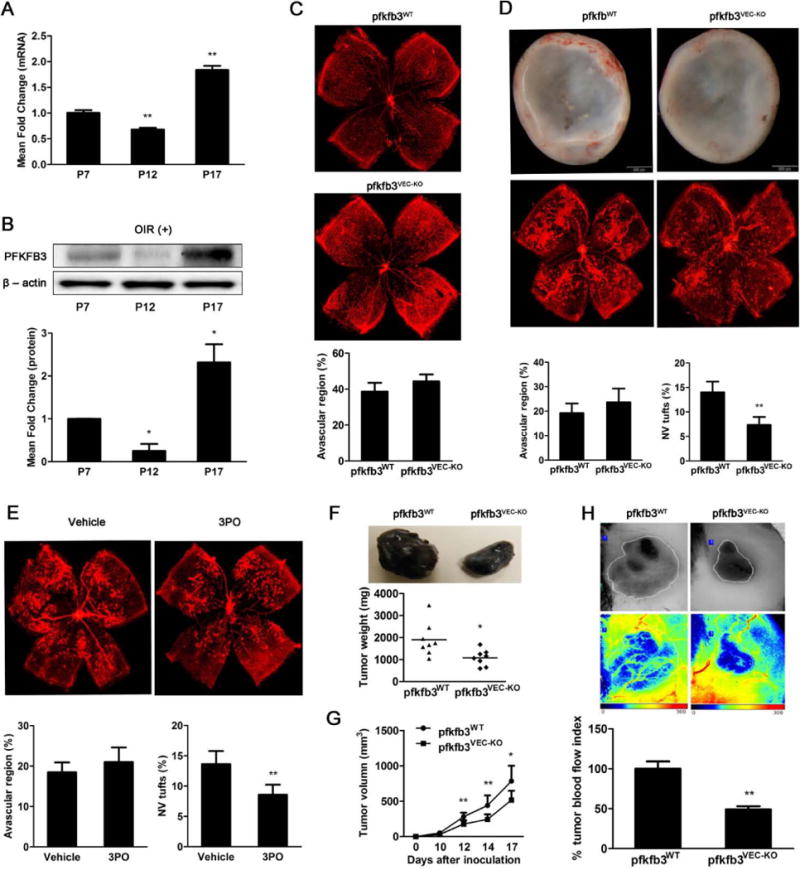
A, Real-time RT-PCR analysis of PFKFB3 mRNA expression levels and immunoblot analysis of PFKFB3 protein expression levels (B) in retinas of OIR mice at P7, P12 and P17. C, Isolectin staining of whole-mount retinas from pfkfb3flox/flox (pfkfb3WT) and pfkfb3flox/floxcdh5cre (pfkfb3VEC-KO) OIR mice at P12 and quantification of the avascular areas (n = 7 mice per genotype). D, Isolectin staining and stereomicroscopy of whole-mount retinas from pfkfb3WT and pfkfb3VEC-KO OIR mice at P17 and quantification of the neovascular tuft (NVT) and avascular areas (n = 9 mice per genotype). E, Isolectin staining and quantification of whole-mount retinas of C57BL/6 mice treated systemically with 3PO or a vehicle at P17 in OIR mice (n = 9 mice per group). F, The appearance of B16 tumors under the skin of pfkfb3WT and pfkfb3VEC-KO mice generated with a dissecting microscope and the quantification of tumor weight (n=8 mice per group). G, Quantification of tumor volume in pfkfb3WT and pfkfb3VEC-KO mice (n = 8 mice per group). H, Laser Doppler imaging (including reconstituted tumor photographs) and quantification of perfusion in tumors in pfkfb3WT and pfkfb3VEC-KO mice at 15 d after implantation (n = 6 mice per group). For bar graphs (A – G), data are the mean ± SD. *, P < 0.05 and **, P < 0.01 for P12 or P17 vs. P7 (in A and B) 3PO vs. vehicle control (in E), or pfkfb3VEC-KO vs. pfkfb3WT (in D, F, G and H).
Tumor implantation models were also used to examine the effect of endothelial PFKFB3 on angiogenesis. Adult female PFKFB3VEC-KO and PFKFB3WT mice were injected with B16 mouse melanoma under the back skin of the mouse. After tumor implantation, the tumor size and weight were increased over a period of 18 d. However, at the same time points the tumors in PFKFB3VEC-KO were much smaller and lighter compared to those in PFKFB3WT (Fig. 4F and 4G). More importantly, the laser speckle contrast imaging showed 55% decreased blood flow in PFKFB3VEC-KO mice as compared to their wild type littermates, PFKFB3WT mice (Fig. 4H).
AKT acts downstream of PFKFB3 in endothelial cells
AKT and its phosphorylation are directly associated with angiogenesis.16, 17 To examine whether AKT and phosphorylated AKT (pAKT) are involved in the effect of PFKFB3 on angiogenesis, the expression levels of AKT and pAKT in PFKFB3-knockdown HUVECs and control cells were examined using Western blotting. There were no significant differences in the AKT expression levels between PFKFB3-knockdown HUVECs and control cells. However, the pAKT expression level in PFKFB3-knockdown cells was decreased compared with control cells under both normoxic and hypoxic conditions (Fig. 5A), and in response to hVEGF treatment (Supp. Fig. 4). Consistent with the in vitro results from cultured HUVECs, the expression level of retinal pAKT was decreased by 50–60% in OIR PFKFB3VEC-KO mice compared with OIR PFKFB3WT mice at P17 (Fig. 5B). To further examine the correlation between the expression levels of pAKT and PFKFB3, HUVECs were treated with an adenovirus encoding PFKFB3 (Ad-PFKFB3). Treatment of HUVECs with Ad-PFKFB3 resulted in overexpression of PFKFB3 (Supp. Fig. 1B) and increased expression of pAKT in HUVECs (Fig. 5C), suggesting that the expression level of PFKFB3 stimulates pAKT expression in endothelial cells. To determine the effect of decreased pAKT on endothelial proliferation and migration in PFKFB3-knockdown, PFKFB3-knockdown HUVECs were treated with an AKT activator to elevate the level of pAKT. Over a period of 72 h, control HUVECs cultured in complete growth cell medium with or without the addition of the AKT activator II exhibited similar levels of proliferation. However, the addition of the AKT activator II increased the proliferation of PFKFB3-knockdown HUVECs to levels similar to those observed in control HUVECs grown in a complete growth cell medium with or without the addition of the AKT activator II (Fig. 5D). Compared with control HUVECs, PFKFB3-knockdown HUVECs exhibited a defect in tube formation. However, the addition of the AKT activator II improved tube formation in both control and PFKFB3-knockdown HUVECs (Fig. 5E), resulting in no differences in tube formation between control and PFKFB3-knockdown HUVECs. These results indicate that a decrease in pAKT in PFKFB3-knockdown HUVECs is a major cause of the observed defects in angiogenesis (Fig. 5E).
Figure 5. AKT acts downstream of PFKFB3 in endothelial cells.
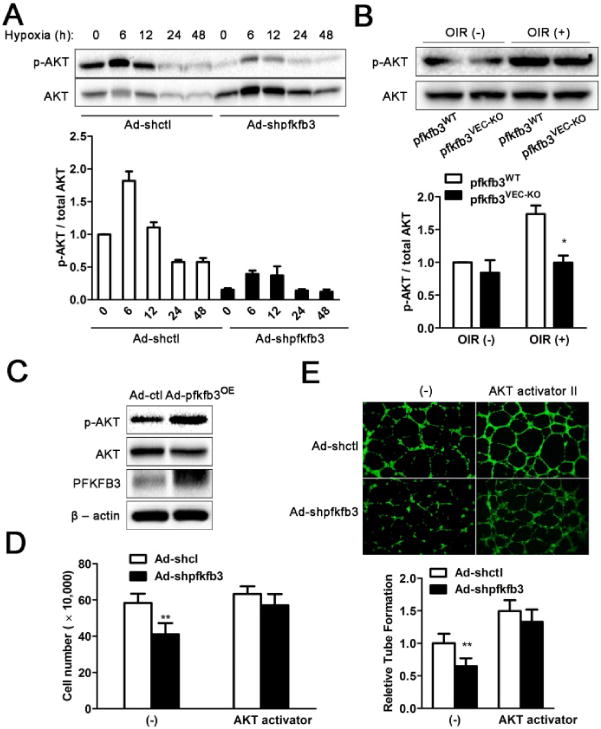
A, Western blot analysis of pAKT and AKT protein expression in control and PFKFB3-knockdown HUVECs under hypoxia for 0, 6, 12, 24 or 48 h. B, Western blot analysis of pAKT and AKT protein expression in retinas from pfkfb3WT and pfkfb3VEC OIR mice at P17. C, Western blot analysis of pAKT and AKT protein expression in control and PFKFB3-overexpressing HUVECs. D, Quantification of the number of control and PFKFB3-knockdown HUVECs exposed to vehicle or the AKT activator II (4ug/ml) for 72 h. E, Representative images and quantification of tube formation in control and PFKFB3-knockdown HUVECs treated with vehicle or the AKT activator II (4ug/ml) on growth factor-deprived Matrigel. All images shown are representative, and data are the mean ± SD (n = 3 independent experimental groups). *, P < 0.05 and **, P < 0.01 for Ad-shpfkfb3 vs. Ad-shctl under the time point (in A) or same condition (in D and E) or for pfkfb3VEC-KO vs. pfkfb3WT for the same condition (in B).
Lactate is involved in PFKFB3-mediated endothelial proliferation and tube formation
Recent studies have indicated the involvement of lactate in angiogenesis. To examine whether lactate plays a role in PFKFB-associated endothelial proliferation and migration, the levels of lactate were measured. Consistent with the differences observed in the expression level of pAKT in PFKFB3-knockdown, PFKFB3-overexpressing, and/or control HUVECs (Fig. 5A and 5C), the levels of intracellular lactate and lactate in the cell medium were decreased in PFKFB3-knockdown cells and increased in PFKFB3-overexpressing cells compared with their respective levels in control cells under both normoxic (Fig. 6A) and hypoxic conditions (Supp. Fig. 5). To determine the relationship between the expression level of pAKT and lactate, lactate was added to the cell medium. The levels of pAKT in PFKFB3-knockdown HUVECs cultured in a complete growth cell media were increased to levels similar to those in control HUVECs with the addition of lactate (Fig. 6B). Additionally, Ad-shpfkfb3-transduced endothelial cells were pretreated with lactate and then exposed to hypoxia conditions. The responses of pAKT to hypoxia after correcting the basal pAKT by lactate were dramatically inhibited in PFKFB3-knockdown cells compared with control cells (Supp. Fig. 6). The addition of lactate to the complete growth cell medium in which control HUVECs were cultured did not significantly enhance the expression of pAKT (Fig. 6B). Similarly, over a period of 72 h, the culture of control HUVECs in complete growth cell medium with the addition of lactate did not alter proliferation. In contrast, the culture of PFKFB3-knockdown HUVECs in complete growth cell medium with the addition of lactate increased the proliferation to levels similar to that of control HUVECs grown in a complete growth cell medium with or without the addition of lactate (Fig. 6C). As shown in Fig. 2D and 5E, compared with control HUVECs, PFKFB3-knockdown HUVECs exhibited a defect in tube formation. The addition of lactate to the medium improved tube formation in both PFKFB3-knockdown HUVECs and control cells (Fig. 6D). Furthermore, the addition of lactate to PFKFB3-knockdown HUVECs rescued the observed decrease in tube formation, indicating that a decrease in lactate in PFKFB3-knockdown HUVECs is a major cause of the defects in tube formation observed in this group of cells.
Figure 6. Lactate is involved in PFKFB3-mediated endothelial proliferation and tube formation.
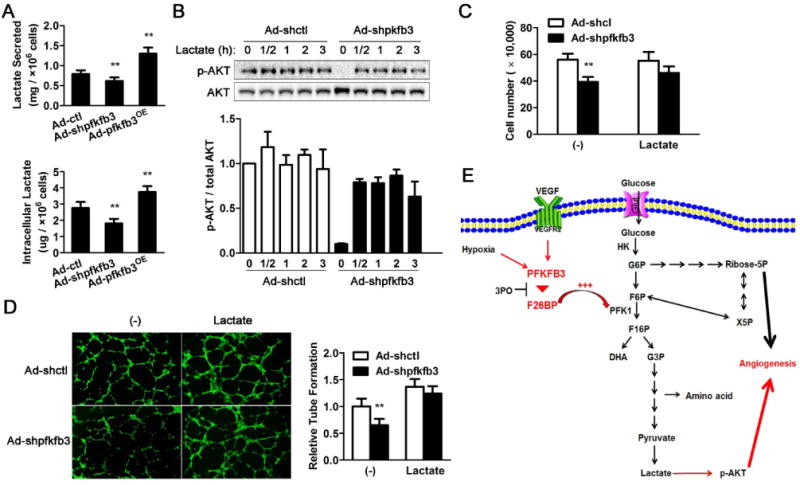
A, Quantification of secreted lactate and intracellular lactate in control and PFKFB3-knockdown HUVECs after 72 h incubation. B, Immunoblot analysis of pAKT and total AKT in control and PFKFB3-knockdown HUVECs upon lactate (10mM) treatment for 0, 0.5, 1, 2 or 3 h. C, Quantification of the number of control and PFKFB3-knockdown HUVECs treated with lactate (10mM) for 72 h. D, Representative images and quantification of tube formation in control and PFKFB3-knockdown HUVECs treated with lactate (10mM) for 8 h on growth factor-deprived Matrigel. E, Schematic of the proposed role of PFKFB3 in angiogenesis. For bar graphs (A – E), data are the mean ± SD (n = 3). *, P < 0.05 and **, P < 0.01 for Ad-shpfkfb3 or Ad-pfkfb3OE vs. Ad-shctl (in A) under the same condition (in C and D) or for the same time point (in B).
Discussion
Angiogenic factors upregulate endothelial PFKFB3. Endothelial cells are highly glycolytic even under resting conditions.6, 9 A recent study indicated that the glycolysis level in endothelial cells is much higher than in any other healthy cells, including cardiomyocytes, hepatocytes, fibroblasts and macrophages, and is even similar to that of many tumor cells. The majority of intracellular ATP is generated from glycolysis in cultured endothelial cells. PFKFB3 is critical in endothelial cell glycolysis. The knockdown of PFKFB3 protein expression in endothelial cells (76–84%) results in a decrease in glycolysis (35–40%), indicating that PFKFB3 plays a significant role in the physiological activity of endothelial cells. PFKFB3 is also critical for glycolysis under pathological conditions. A recent study demonstrated that following VEGF stimulation, PFKFB3 protein expression was significantly upregulated.6 We have studied the expression of all of the isoforms of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase, and found that the PFKFB3 isoform is the most significantly upregulated in stimulated endothelial cells. This upregulation was also observed in vivo, as evidenced by OIR retinal PFKFB3 expression. Compared with the other isoforms of PFKFB, 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase encoded by PFKFB3 is an enzyme with a kinase activity over 700-fold higher than its phosphatase activity, thereafter signaling to achieve a high level of glycolysis in proliferative endothelial cells.
Endothelial PFKFB3 participates in pathological angiogenesis. In cancers of the breast, colon, lung, pancreas, prostate and ovary, PFKFB3 exhibited increased protein and mRNA expression as well as increases of its phosphorylated form compared with healthy controls.10, 11, 18 This increased expression leads to high rates of aerobic glycolysis in tumor cells, a phenomenon known as the Warburg effect.19 In addition to general tumor cells, vascular cells in tumors also exhibit an increased expression and activation of PFKFB3, suggesting that endothelial PFKFB3 is involved in the Warburg effect.11, 20 In our study, the tumor blood supply and tumor size were decreased in mice deficient in endothelial PFKFB3 compared with control mice, demonstrating that PFKFB3 in endothelial cells is indeed critical for tumor angiogenesis. The role of endothelial PFKFB3 in pathological angiogenesis was further supported by its effect on neovascularization in OIR mouse retinas. In this model, both the selective deletion of endothelial PFKFB3 and the application of a PFKFB3 inhibitor to mice dramatically suppressed retinal neovascularization. Additionally, consistent with the data from Katrien et al.,6 we found that the density of retinal vessels in newborn mouse pups deficient in endothelial PFKFB3 was lower than that in control pups (Supp. Fig. 2A). Furthermore, in OIR mice, the vascular regrowth in the avascular area of the retina was also relatively delayed in mice deficient in endothelial PFKFB3 compared with control mice, implying that endothelial PFKFB3 may play a role in physiological angiogenesis.
The underlying mechanisms for the role of endothelial PFKFB3 in pathological angiogenesis arise from the influence of this gene (enzyme) in endothelial migration and proliferation. PFKFB3 affects endothelial migration by regulating the formation of filopodia and lamellipodia and directional migration. One possible mechanism underlying this effect is that PFKFB3 compartmentalizes with F-actin in motile protrusions to provide ATP.6 Thus, deficiency in or knockdown of PFKFB3 impairs tip cell formation. Consistent with these results, we also observed a significant defect in the formation of lamellipodia in PFKFB3-knockdown endothelial cells under both normoxic and hypoxic conditions (Supp. Fig. 7). In addition, an analysis of the endothelial cell tube formation indicated that endothelial tubes are formed much more rapidly in control endothelial cells than in PFKFB3-knockdown cells. The tube formation assay was performed within 6–8 h after the endothelial cells were placed on the Matrigel, indicating that the compromised tube formation is mainly due to a defect in migration. PFKFB3 is also important for endothelial proliferation.21–23 In both HeLa cells and lymphocytes, a temporal expression pattern of PFKFB3 has been observed during the cell cycle progression. PFKFB3 appears in mid-to-late G1 and is vital for cell division.22 Silencing PFKFB3 dramatically reduced T lymphocyte proliferation.21 Consistent with these findings, in the current study, the knockdown of endothelial PFKFB3 prevented endothelial cells from proceeding to the S phase, resulting in a much lower proliferative rate compared with control endothelial cells under both normoxic and hypoxic conditions (Fig. 2B and Supp. Fig. 8). Additionally, we have found that under hypoxic condition, knockdown of PFKFB3 accelerated endothelial apoptosis, indicating that, additional to endothelial proliferation and migration, a PFKFB3-knockdown-associated decrease in angiogenesis may involve other mechanisms (Supp. Fig. 9).
Lactate-induced AKT phosphorylation is involved in PFKFB3-driven endothelial angiogenesis. Katrien et al. examined PFKFB3-knockdown cells and found no significant differences in ATP levels, cell death, oxidative stress, glucose oxidation, and fatty acid oxidation or respiration between these cells and wild-type controls.6 Therefore, due to the cellular adaptation to PFKFB3 silencing, cellular energy distress does not arise, and thus, energy distress is not responsible for the compromised angiogenesis observed in PFKFB3-knockdown endothelial cells. Due to a high glycolytic activity, activated endothelial cells produce a substantial amount of lactate. In endothelial cells, lactate functions as a signaling molecule for angiogenesis.24–29 Lactate acts through AxI, Tie2 and vascular endothelial growth factor receptor 2 to activate PI3K/AKT.28 Likely, Pim1 and foxo transcription factors are also involved in the lactate-mediated effect since it has been demonstrated that these molecules are tied to AKT in mediating proliferation and anti-apoptotic action of glycolysis.30, 31 The effect of lactate on angiogenesis has been demonstrated in a few models, including pulmonary microvascular endothelial cell proliferation and the healing of superficial and ischemic wounds. The knockdown or overexpression of PFKFB3 in endothelial cells decreased or increased the levels of lactate, respectively, while the latter closely correlated with the levels of pAKT. The addition of lactate to PFKFB3-knockdown endothelial cells increased pAKT levels and rescued impaired endothelial migration. Thus, as summarized in Fig. 6E, endothelial PFKFB3 knockdown led to a low level of glycolysis and a low level of intracellular lactate and secreted lactate, which caused a decrease in pAKT and a failure to signal properly for the generation of a strong angiogenic response. In addition, low levels of lactate and glycolysis in PFKFB3-knockdown endothelial cells are not able to provide sufficient substrates for the generation of macromolecules, including DNA, RNA and relevant lipids, which lead to the impairment of endothelial proliferation19, 20 (Fig. 6E).
As a key enzyme of the pentose phosphate pathway, glucose-6-phosphate dehydrogenase (G6PD) critically regulates glucose metabolism during angiogenesis and tissue repair.32, 33 Deficiency of G6PD decreases endothelial proliferation, migration and tube formation, whereas activation or over-expression of G6PD increases angiogenesis as well as cardiac repair in diabetes. The effect of G6PD on angiogenesis and tissue repair involves Flk-1, AKT and Pim-1.32, 33 However, pentose phosphate pathway was increased during PFKFB3 knockdown.34 Given this, PFKFB3 appears to not act through G6PD-associated pathway to alter endothelial angiogenesis; although PFKFB3 knockdown and G6PD deficiency share many similarities in the effect and mechanisms.
This study has indicated that the knockdown or inhibition of PFKFB3 leads to significant suppression of pathological angiogenesis. Additional to tumors/cancers, many other diseases are also associated with pathological angiogenesis. For instance, retina angiogenesis is an important cause for vision loss in retinopathy of prematurity, diabetic retinopathy and age-related macular degeneration.35 Also, angiogenesis in atherosclerotic lesion is an important contributor to the formation and the progression of atherosclerosis whereas some anti-angiogenic approaches suppress atherosclerosis.36 Thus, PFKFB3 is a promising target for the treatment of pathological angiogenesis and its associated diseases.
Supplementary Material
Significance.
The glycolytic activity of endothelial cells is critical for angiogenesis. Fructose-2, 6-bisphosphate (F2, 6P2), a product of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase, isoform 3 (PFKFB3), is an activator of 6-Phosphofructo-1 kinase (PFK-1), one rate-limiting checkpoint of glycolysis. Using both chemical and genetic approaches, we demonstrated that the knockdown or inhibition of endothelial PFKFB3 reduced endothelial glycolysis, leading to significant suppression of endothelial proliferation in vitro and pathological angiogenesis in vivo in murine models of tumor growth and oxygen-induced retinopathy. Furthermore, we found that lactate-associated Akt phosphorylation contributed to PFKFB3-modulated angiogenesis. Many diseases are associated with pathological angiogenesis, including tumors/cancers, retinopathies (retinopathy of prematurity, diabetic retinopathy and age-related macular degeneration), and the formation and the progression of atherosclerosis. This study indicates that PFKFB3 is a promising target for the treatment of pathological angiogenesis and its associated diseases.
Acknowledgments
Sources of Funding
This work was supported by grants from the National Key Basic Research Program of China (2012CB910402 to X.A. and Y.H.), the American Diabetes Association (1-10-BS-76 to Y.H., 1-10-JF-54 to C.W.), the American Heart Association (10GRNT4400005 to Y.H., 12BGIA9050003 to C.W.) and the National Institutes of Health (HL108922 and HL095556 to Y.H., R01DK095828 and R01DK095862 to C.W.).
Nonstandard Abbreviations and Acronyms
- PFKFB3
6-Phosphofructo-2-kinase/fructose-2, 6-bisphosphatase, isoform 3
- VEGF
vascular endothelial growth factor
- HUVEC
human umbilical vein endothelial cell
- Ad
adenovirus
- OIR
oxygen-induced retinopathy
- NVT
neovascular tuft
- pAKT
phosphorylated AKT
- G6PD
glucose-6-phosphate dehydrogenase
Footnotes
Disclosures
The authors have no conflicts of interest to declare.
References
- 1.Carmeliet P. Angiogenesis in health and disease. Nature medicine. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 2.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 3.Eilken HM, Adams RH. Dynamics of endothelial cell behavior in sprouting angiogenesis. Current opinion in cell biology. 2010;22:617–625. doi: 10.1016/j.ceb.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Bock K, Georgiadou M, Carmeliet P. Role of endothelial cell metabolism in vessel sprouting. Cell metabolism. 2013;18:634–647. doi: 10.1016/j.cmet.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 6.De Bock K, Georgiadou M, Schoors S, et al. Role of pfkfb3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 7.Merchan JR, Kovacs K, Railsback JW, Kurtoglu M, Jing Y, Pina Y, Gao N, Murray TG, Lehrman MA, Lampidis TJ. Antiangiogenic activity of 2-deoxy-d-glucose. PloS one. 2010;5:e13699. doi: 10.1371/journal.pone.0013699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5379–5384. doi: 10.1073/pnas.0601026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson SM, Duchen MR. Endothelial mitochondria: Contributing to vascular function and disease. Circulation research. 2007;100:1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- 10.Ros S, Schulze A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer & metabolism. 2013;1:8. doi: 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, Yoshioka N, Bucala R, Koike T. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/pfkfb3 family of glycolytic regulators in human cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:5784–5792. doi: 10.1158/1078-0432.CCR-05-0149. [DOI] [PubMed] [Google Scholar]
- 12.Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, Lane A, Trent JO, Chesney J. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Molecular cancer therapeutics. 2008;7:110–120. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 13.Guo X, Li H, Xu H, Halim V, Thomas LN, Woo SL, Huo Y, Chen YE, Sturino JM, Wu C. Disruption of inducible 6-phosphofructo-2-kinase impairs the suppressive effect of ppargamma activation on diet-induced intestine inflammatory response. The Journal of nutritional biochemistry. 2013;24:770–775. doi: 10.1016/j.jnutbio.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo X, Xu K, Zhang J, Li H, Zhang W, Wang H, Lange AJ, Chen YE, Huo Y, Wu C. Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of peroxisome proliferator-activated receptor gamma activation in mice. The Journal of biological chemistry. 2010;285:23711–23720. doi: 10.1074/jbc.M110.123174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huo Y, Guo X, Li H, Wang H, Zhang W, Wang Y, Zhou H, Gao Z, Telang S, Chesney J, Chen YE, Ye J, Chapkin RS, Wu C. Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. The Journal of biological chemistry. 2010;285:3713–3721. doi: 10.1074/jbc.M109.058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. Akt1/protein kinase balpha is critical for ischemic and vegf-mediated angiogenesis. The Journal of clinical investigation. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nature medicine. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolanos JP. Adapting glycolysis to cancer cell proliferation: The mapk pathway focuses on pfkfb3. The Biochemical journal. 2013;452:e7–9. doi: 10.1042/BJ20130560. [DOI] [PubMed] [Google Scholar]
- 19.Lunt SY, Vander Heiden MG. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annual review of cell and developmental biology. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 20.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colombo SL, Palacios-Callender M, Frakich N, De Leon J, Schmitt CA, Boorn L, Davis N, Moncada S. Anaphase-promoting complex/cyclosome-cdh1 coordinates glycolysis and glutaminolysis with transition to s phase in human t lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18868–18873. doi: 10.1073/pnas.1012362107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tudzarova S, Colombo SL, Stoeber K, Carcamo S, Williams GH, Moncada S. Two ubiquitin ligases, apc/c-cdh1 and skp1-cul1-f (scf)-beta-trcp, sequentially regulate glycolysis during the cell cycle. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5278–5283. doi: 10.1073/pnas.1102247108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, Brock E, Siow D, Wattenberg B, Telang S, Chesney J. Nuclear targeting of 6-phosphofructo-2-kinase (pfkfb3) increases proliferation via cyclin-dependent kinases. The Journal of biological chemistry. 2009;284:24223–24232. doi: 10.1074/jbc.M109.016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunt TK, Aslam RS, Beckert S, Wagner S, Ghani QP, Hussain MZ, Roy S, Sen CK. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxidants & redox signaling. 2007;9:1115–1124. doi: 10.1089/ars.2007.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parra-Bonilla G, Alvarez DF, Alexeyev M, Vasauskas A, Stevens T. Lactate dehydrogenase a expression is necessary to sustain rapid angiogenesis of pulmonary microvascular endothelium. PloS one. 2013;8:e75984. doi: 10.1371/journal.pone.0075984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parra-Bonilla G, Alvarez DF, Al-Mehdi AB, Alexeyev M, Stevens T. Critical role for lactate dehydrogenase a in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. American journal of physiology. Lung cellular and molecular physiology. 2010;299:L513–522. doi: 10.1152/ajplung.00274.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porporato PE, Payen VL, De Saedeleer CJ, Preat V, Thissen JP, Feron O, Sonveaux P. Lactate stimulates angiogenesis and accelerates the healing of superficial and ischemic wounds in mice. Angiogenesis. 2012;15:581–592. doi: 10.1007/s10456-012-9282-0. [DOI] [PubMed] [Google Scholar]
- 28.Ruan GX, Kazlauskas A. Lactate engages receptor tyrosine kinases axl, tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/akt and promote angiogenesis. The Journal of biological chemistry. 2013;288:21161–21172. doi: 10.1074/jbc.M113.474619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter mct1 supports an nf-kappab/il-8 pathway that drives tumor angiogenesis. Cancer research. 2011;71:2550–2560. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- 30.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of foxo transcription factors in angiogenesis and postnatal neovascularization. The Journal of clinical investigation. 2005;115:2382–2392. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zippo A, De Robertis A, Bardelli M, Galvagni F, Oliviero S. Identification of flk-1 target genes in vasculogenesis: Pim-1 is required for endothelial and mural cell differentiation in vitro. Blood. 2004;103:4536–4544. doi: 10.1182/blood-2003-11-3827. [DOI] [PubMed] [Google Scholar]
- 32.Katare R, Oikawa A, Cesselli D, Beltrami AP, Avolio E, Muthukrishnan D, Munasinghe PE, Angelini G, Emanueli C, Madeddu P. Boosting the pentose phosphate pathway restores cardiac progenitor cell availability in diabetes. Cardiovascular research. 2013;97:55–65. doi: 10.1093/cvr/cvs291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leopold JA, Walker J, Scribner AW, Voetsch B, Zhang YY, Loscalzo AJ, Stanton RC, Loscalzo J. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. The Journal of biological chemistry. 2003;278:32100–32106. doi: 10.1074/jbc.M301293200. [DOI] [PubMed] [Google Scholar]
- 34.Schoors S, De Bock K, Cantelmo AR, et al. Partial and transient reduction of glycolysis by pfkfb3 blockade reduces pathological angiogenesis. Cell metabolism. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 35.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nature reviews. Drug discovery. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 36.Herrmann J, Lerman LO, Mukhopadhyay D, Napoli C, Lerman A. Angiogenesis in atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1948–1957. doi: 10.1161/01.ATV.0000233387.90257.9b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.