

Abstract
Emerging evidence provide credible support in favor of the potential role of bioactive products derived from ingesting cruciferous vegetables such as broccoli, brussel sprouts, cauliflower and cabbage. Among many compounds, 3,3′-Diindolylmethane (DIM) is generated in the acidic environment of the stomach following dimerization of Indole-3-Carbinol (I3C) monomers present in these classes of vegetables. Both I3C and DIM have been investigated for their use in preventing, inhibiting, and reversing the progression of cancer- as a chemopreventive agent. In this review, we summarize an updated, wide-ranging pleiotropic anti-tumor and biological effects elicited by DIM against tumor cells. It is unfeasible to point one single target as basis of cellular target of action of DIM. We emphasize key cellular and molecular events that are effectively modulated in the direction of inducing apoptosis and suppressing cell proliferation. Collectively, DIM orchestrates signaling through Ah receptor, NF-κB/Wnt/Akt/mTOR pathways impinging on cell cycle arrest, modulation of key cytochrome P450 enzymes, altering angiogenesis, invasion, metastasis and epigenetic behaviors of cancer cells. The ability of DIM to selectively induce tumor cells to undergo apoptosis has been observed in preclinical models, and thus it has been speculated in improving the therapeutic efficacy of other anticancer agents that have diverse molecular targets. Consequently, DIM has moved through preclinical development into phase-I clinical trials, thereby suggesting that DIM could be a promising and novel agent either alone or as an adjunct to conventional therapeutics such as chemo-radio therapy, and targeted therapies. An important development has been the availability of DIM formulation with superior bioavailability for humans. Therefore, DIM appears to be a promising chemopreventive agent or chemo-radio-sensitizer for the prevention of tumor recurrence and/or for the treatment of human malignancies.
Keywords: 3,3′- Diindolylmethane; Brassica sps; Prevention; Therapy
Introduction
Despite advancements in our understanding on cancer prevention and treatment through multi-modality approach including targeted therapies, cancer remains as the leading cause of death worldwide. In United States out of the 1,529,560 new cases diagnosed in the year 2010, it is estimated that 569,490 deaths will occur [1]. This grim statistics underscores the realm of an effective strategy to improvise current prevention and therapeutic modalities towards this malady. Here we review and highlight current knowledge and perspective integrating use of a natural “bioactive phytochemical” with promising inhibitory and therapeutic potential on tumorigenesis. This agent is found in substantial amounts in vegetables that are commonly consumed from the family Cruciferae particularly the genus Brassica which include broccoli, cauliflower, kale, cabbage brussels sprouts, turnips, kohlrabi, bok choy, and radishes, etc. The anticancer properties of cruciferous vegetables were first recognized by the Roman statesman, Cato and Elder (234-149 BC), who in his treatise of medicine wrote: “If a cancerous ulcer appears on the breasts, apply a crushed cabbage leaf and it will make it well.” It is now well established that Cruciferous vegetables contain a precursor phytochemical - Glucosinolate, that undergoes hydrolysis by the plant enzyme myrosinase, yielding a bioactive compound known as Indole 3-carbinol (I3C). I3C is chemically unstable in aqueous and gastric acidic environment, and is rapidly converted to numerous condensation products. A major condensation product of I3C in vivo is 3,3′-diindolylmethane (DIM; Fig-1). DIM has distinct pleiotropic effects on cancer cells resulting in inactivating survival signaling and simultaneously activating multiple death pathways.
Figure - 1. Molecular structure of 3,3′-Diindolylmethane (DIM).
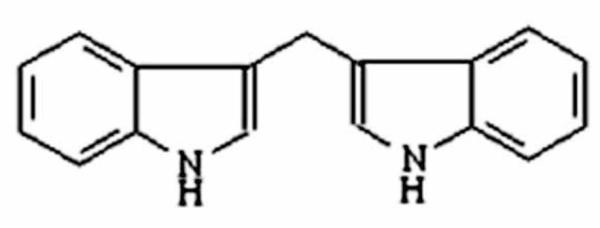
DIM is dimeric bioactive product of Indole 3-carbinol generated in acid environment of the stomach following consumption of diet rich in cruciferous vegetables.
Other relevant information includes:
Molecular formula: C17H14N2
Molecular weight - 246.31
Emerging preclinical evidence reveals an alteration in the urinary estrogen metabolite associated with reduced risk of estrogen dependent cancers in women such as breast, cervical and endometrial cancers following high consumption of cruciferous vegetables. Additional proof of reduced cancer risk inferred from laboratory studies suggest that I3C and its dimeric product DIM contributes to chemoprotection by multiple mechanisms involving modulation of xenobiotic metabolizing enzyme system in the liver and extrahepatic tissues diminishing the bioavailability of genotoxic and active metabolites of hazardous xenobiotic compounds including dietary carcinogens (such as heterocyclic amines) in experimental animals. Additionally, DIM has been confirmed to effectively inhibit the growth of human cancer cells of prostate, breast, colon, cervix and pancreas origin. This inhibition is mediated by abrogation of multiple signaling pathways leading to the impairment of cell proliferation and cell cycle arrest restricting tumor cell migration, invasion, and metastasis and stimulating apoptotic cell death.
One reported study confirmed DIM as the only I3C-derived compound detected in the plasma of women ingesting I3C, corroborating that DIM is the predominant bioactive compound that mediates the biological effects of dietary Brassica [2]. It should be noted that DIM, but not I3C, is safe in humans. Administration of DIM to human volunteers resulted in adequate serum levels that may probably be biological noteworthy [3-5]. Most studies reported to date regarding DIM, relate primary objective pertaining to the investigation and subsequent events associated with the information gained as to whether the effect is proapoptotic, antiangiogenic, antimetastatic or antiinvasive in context of prevention and/ or therapy of cancer.
Here, we summarize the effect of DIM from a mechanistic perspective as well as its bioavailability for unbiased appraisal in cancer prevention strategy and therapeutic relevance. Nevertheless, it is understood any promising forthcoming effects may also be influenced by individual genetic polymorphism. Research findings from our laboratory along with Drs LF Bjeldanes and GL Firestone from the University of California, Berkeley, and S Safe from A&M University, Texas have contributed significantly in our understanding on the molecular mechanism of action of DIM as presented and discussed in this article.
Cellular effects of DIM
DIM and phase-I and II carcinogen and xenobiotic metabolism
The first step in the metabolic biotransformation of lipohilic xenobiotics and endogenous hydrophobic substances including steroids to inactivate hydrophilic excretable metabolites is mediated by the superfamily of heme-containing monooxygenases (cytochrome P450s (CYP) enzymes), and phase-II enzyme systems (GST isoenzymes, UDP-glucuronosyltransferase, sulfotransferase, and catechol O-methyltransferase) [6;7] (Fig-2). These enzymes are involved in functionalization and conjugation reactions. and three families (CYP1, CYP2, and CYP3) catalyze the oxidative metabolism of exogenous and endogenous compounds [8]. Several studies point to noteworthy quantitative differences concerning P450 isoenzymes following DIM treatment. Lake et.al., demonstrated in cultured precision-cut human liver slices that DIM induces CYP1A2 [9]. Using various CYP specific activity assays, Stressor et.al. found that DIM inhibits the human CYP1A1 and 1A2, and rat CYP2B1 [10]. DIM also inhibits in vitro CYP-mediated metabolism of the ubiquitous food contaminant and potent hepatocarcinogen-aflatoxin B1 [10], Together, these results point toward DIM’s ability to inhibit catalytic activities of a range of CYP isoforms may form the basis of its anti-carcinogenic mechanism of action. In contrast to these findings, Gross-Steinmeyer et.al., reported in cultured human primary hepatocytes that DIM exhibits a remarkably effective induction response to CYP1A1 (474-, 239- and 87-fold at 50, 25 and 10 μM, respectively) and CYP1A2 (113-, 70- and 31-fold at 50, 25 and 10 μM, respectively), While the results were semi-quantitatively reflected in mRNA level, no increase in GST-mRNA level were demonstrated [11;12]. Paradoxically, some isoforms within these CYP families are also responsible for the metabolism of estrogens. In agreement, it has been shown that DIM interferes with regulation of the estrogen-metabolizing CYP enzymes associated with cancer susceptibility. Evidence supporting such alliance has been put forth by Parkin and Malejka-Giganti [13;14] and Jellinck et.al., [15]. These investigators reported difference in hepatic P450-dependent metabolism of estrogen and tamoxifen in rats treated with DIM and its parent compound I3C wherein, treatment with DIM led to estrogen detoxification. Correspondingly, a significant new finding that emerged from one of these study relates to DIM–mediated inhibition of a CYP complement (e.g. CYP3A1/2 activity catalyzing 4-hydroxylation) that results in increased capacities of hepatic microsomes to metabolize 17β-estradiol (E2) and estrone (E1) to less estrogenic 2-catechols, estrogenic 4-catechols and the 6-α, 6-β and 16α-hydroxy (OH) derivatives [13]. Thus, inhibition in the formation of these metabolites following treatment with DIM provides an important mechanism for preventing the tumorigenic process in estrogen-responsive sites. Moreover, alterations in estrogen metabolism is important to suppress viral oncogene expression especially infections with types 16 and 18, two “high risk” HPV that cause (70%) cervical cancers. Other mechanisms based on estrogen receptor-β target genes by DIM are emerging [16] and remains to be elucidated.
Figure - 2.

Schematic representation of Phase I and II reactions and its modulation by DIM.
Microarray gene expression profiling published by our laboratory also attest anti-carcinogenic effect of DIM because DIM modulated CYP enzymes [17]. We found and reported that DIM up-regulates the expression of the phase-I enzyme CYP1A1 and the phase-II enzymes glutathione S transferase theta I and aldo-keto reductase in PC-3 prostate cancer cells. This evidence suggests DIM increases the capacity for detoxification and inhibition of carcinogen activation. Interestingly, the findings have been supported in animal models, specifically in the rat liver, where it has been reported that DIM elevates the activity of phase-II enzymes [18;19].
DIM and cell cycle
The hyper-proliferation of tumor cells progresses like normal cells through the four phases of cell cycle- G1, S G2 and M which are controlled by cyclin dependent kinase (CDK) and associated CDK inhibitors (CKI) (Fig-3). Accumulating evidence indicates that DIM causes a predominant G1 cytostatic cell cycle arrest in breast, ovarian, prostate, colon, thyroid cancer cells along with HUVEC cells [20-23]. Analysis of G1 acting cell cycle components indicate that the activity of cyclin-dependent kinase 2 (CDK2) is strongly reduced by DIM by a mechanism that involves upregulated expression of the CDK inhibitor- p21Cip1/Waf1 [24;25]. p21Cip1/Waf1 is a well-characterized Cip/Kip family CDK inhibitor, which induces G1 arrest and block entry into S phase by inactivating CDKs or by inhibiting the activity of proliferating cell nuclear antigen, PCNA [26]. Subsequent mechanistic investigations using deletion analysis of the p21Cip1/Waf1 promoter showed that DIM responsiveness was partially dependent on the binding of the Sp1 and Sp3 transcription factors to the GC-rich region of the proximal promoter [24;25]. Additional molecular insights in support of the role of Rb family of proteins in mediating the cell cycle progression are forthcoming [27]. In hypo-phosphorylated state- the Rb proteins associate with and inhibit the activity of E2F family transcription factors. Upon growth stimulus, the G1-specific cyclins/CDKs phosphorylate Rb, thereby causing the release of E2F factors and progression into S phase. In harmony with previous noted effect of DIM in inhibiting CDK2 and CDK4 protein expression, Vivar et.al., emohasized in LNCap prostate cancer cells showing that DIM inhibits phosphorylation of pRb by CDK4 and CDK2 and inhibits the kinase activity of cellular CDK2 [28]. Aditionally, Chinnakannu et.al., reported for the first time the cycle-dependent effects of B-DIM (a formulated DIM; supplied by BioResponse, Inc.) on synchronized LNCaP and C4-2B prostate cancer cells progressing from G1 to S phase [29]. Moreover the findings showed that B-DIM induces p27Kip1 - another CDK inhibitor in these cells. Further investigations revealed that beginning with G1 cell cycle arrest, down-regulation of AR and the inhibition of proteasome activity in S phase was imminent. This observed event in the S phase then lead to the inactivation of NF-κB signaling and finally conclude in the induction of apoptosis in these cells starting 4 hrs after release from isoleucine blockade. The molecular link between other key G1 acting cell cycle components being inhibited by DIM have been reported in human cancer cell lines from prostate and thyroid by Garipatty et.al. [30] and Rajoria S et.al [31]. Included among these components are CDK4, CDK6 and cyclin D. Data from other investigators surmised the effect of DIM in regulation of cell cycle in other cell lines such as colon, thyroid, breast and ovarian cancer cells. In HT-29 colon cancer cells 30μM DIM treatment caused cell cycle arrest mediated by markedly reduced activity of CDK2, phosphorylated Rb and E2F-1 resulting in an escalation of levels of CDK inhibitors, p21CIP1/WAF1 and p27KIPI [20]. It is also noteworthy that DIM diminishes the levels of cyclin A and D1 along with the activity of CDK4, CDC2, CDC25C phosphatase resulting in restriction of cell cycle progression [20]. Molecular analysis in HUVEC cells following 5μM DIM treatment revealed that these cells also utilize similar molecular target and signaling pathways including down-regulated expression of CDK2 and CDK6, and up-regulated expression of CDK inhibitor, p27 KIPI [23]. The anti-proliferative activity of DIM in follicular thyroid cancer (TCa) was also found to be mediated by G1 arrest followed by induction of apoptosis while in human breast cancer cells, DIM is able to stop the cell cycle progression independent of the estrogen dependence and p53 status [20;21;32]. In human ovarian cancer cells the growth inhibitory effect of DIM were mediated by cell cycle arrest in G2/M phase that was associated with DNA damage as indicated by phosphorylation of H(2)A.X at Ser139 and activation of checkpoint kinase 2 (Chk2) in all the three investigated ovarian cancer cell lines - SKOV-3, TOV-21G, and OVCAR-3 [33].
Figure - 3. Effect of DIM on some key regulators of the cell cycle.

DIM is able to block the cell cycle by activation or stimulation of cyclins, cyclin-dependent kinases (CDKs), inhibitor of CDKs. The places where DIM affects cell cycle are shown. The restriction point is the point at which cells progress through the cell cycle independent of external stimuli.
DIM and Oxidative Stress
Noticeable evidence indicates DIM could rapidly induce ROS production in breast and prostate cancer cells, hepatoma cells, leukemia cells, and primary endothelial cells. This initiates a cascade of pleiotropic effects including activation of DNA damage checkpoint signaling, hyperpolarization of mitochondrial inner membrane alongwith decreased cellular ATP level concluding in significant stimulation leading to generation of mitochondrial reactive oxygen species. The ROS production lead to the activation of stress activated pathways involving p38 and c-jun-NH2 terminal kinase alongwith increasingly inhibitory effect on cell proliferation. Antioxidants significantly attenuate DIM induced activation of p38 and JNK and induction of p21Cip1/Waf1 implying oxidative stress as a key trigger in mediating the cellular activity of this compound. Gong et al. reported that one hour after DIM treatment to breast cancer cells (MCF-7 and MDA-MB 231) hyper-polarization of mitochondrial membrane potential develops in a dose-responsive manner [24]. This effect has been deciphered ( or cracked ?) due to the noncompetitive inhibition of mitochondrial H+- ATPase by DIM which under normal conditions confer cells to synthesize and maintain cellular ATP levels [24]. Overall, the findings by Gong et al. suggest that DIM cause oxidative stress in cells through stimulation of ROS release from mitochondria. Of significance, a study involving ovarian cancer cells, Kandala and Srivastava reported DIM treatment cause ROS generation, which when blocked by N-acetyl cystine protected the cells from DIM mediated G2M arrest and apoptosis [33].
Intriguingly, a study reported by Fan et al. showed that low doses of DIM (1 μmol/L) could stimulate BRCA1 expression and protect normal or nontumor-derived HMECs cells against oxidative stress, in part, through BRCA1 [34]. This supports a previous report that BRCA1 protects against oxidative stress [35], which can now be mechanistically linked to the stimulation of antioxidant response via NRF2- a redox-sensitive transcription factor that stimulates the expression of phase II detoxifying enzymes and antioxidant genes through the ARE [36;37]. The study by Fan et al. illustrates the mechanism by which DIM protects against oxidative stress invoking the BRCA1-dependent stimulation of NRF2 activity [34;38]. DIM stimulates several NRF2-regulated promoters, including NQO1 (an oxidoreductase), GSTα1 (a glutathione S-transferase), and x-CT (cystine/glutamate transporter). NRF2 have been proposed earlier to be a target for some cancer preventive dietary agents [36] although this was not shown previously for DIM. These findings gain importance because normal cell types or pre-cancerous cells are presumably the main targets for cancer prevention. Because the protective effects of DIM occurs at physiologic concentrations, it is reasonable to speculate that DIM blocks carcinogenesis by enabling normal cells to mount a stringent antioxidant response. In contrast, tumor cells are often under oxidative duress due to impaired antioxidant homeostasis. Thus, from abovementioned account it is inferred that DIM executes a protective effect (via increased ability of normal cells to repair oxidative DNA damage by upregulating BRCA1) or mounts an oxidative stress response, based on cell type specificity. BRCA1 is known to contribute to various DNA repair processes [39].
DIM and ER Stress
There is growing evidence that selective pharmacologic active compounds generate stress within the endoplasmic reticulum (ER) of cells by unfolded protein response (UPR) activation coupled to apoptosis. Sun et al. perceived and investigated the induction of stress response genes GADD153, GADD34 and GADD45A, XBP-1, GRP78, GRP94, and asparagine synthase following exposure to DIM in cervical (C33A), breast (MCF-7) and prostate (DU145) cancer cell lines [40]. Their results concur with the activation of more than one stress response pathway in C33A cervical cancer cells exposed to 75 μM DIM which coincided with the onset of apoptosis [40]. Abdelrahim et al. hypothesized receptor-independent apoptosis by DIM and selected methylene-substituted DIMs (C-DIMs) leading to activation of ER stress in pancreatic cancer cells which was similar to that observed for thapsigargin (Tg), a prototypical inducer of ER stress [41]. These studies extend support to the hypothesis that cytotoxic concentrations of DIM can stimulate the cellular ER stress response pathways in vitro. Because DIM toxicity in vitro is enhanced in cells undergoing nutritional deprivation and ER stress, one may conceptualize within solid tumors an hierarchy of stressed cells may become increasingly sensitive to killing by DIM. A related study by Savino et al and Cheng et al. investigated perturbations in ER calcium ion homeostasis and concluded that alterations in Ca2+ homeostasis in the ER by DIM instigate apoptosis by both calcium dependent and independent mechanisms depending on cell types [42;43].
DIM and MAP kinase
Mitogen-activated protein kinases (MAPK) are ubiquitous in eukaryotic organisms that transmit signals from the cell membrane to the nucleus via a cascade of three sequentially acting kinases (Fig-4). The final MAPK in the cascade phosphorylates various transcription factors orchestrating activation of specific gene expression by regulating repertoire of required transcriptional machinery. There are three distinct but parallel MAPK cascades identified in mammalian cells such as extracellular signal-regulated kinase (ERK), c-Jun N terminal kinase (JNK) and p38 [44;45]. Although ERKs can be activated by mitogens and growth factors, JNK and p38 can be activated by many environmental stress stimuli such as UV and γ-irradiation as well as many anticancer drugs, e.g. cisplatin or etoposide [46;47]. Xue et al. showed that DIM can up-regulate the expression and stimulate secretion of interferon-gamma (IFNγ) in human MCF-7 breast cancer cell line which was mediated by activation of both JNK and p38 pathways [48]. This novel observation offer an important clue that explains the anticancer effects of DIM, because it is well known that IFNγ plays an important role in preventing the development of primary and transplanted tumors. DIM activates both JNK and p38 pathways, induces the phosphorylation of c-Jun and ATF-2, and increases binding of the homodimer or heterodimer of c-Jun/ATF-2 to the proximal AP-1/CREB-ATF-binding element [48]. A follow up on a previous cited study by Gong et al. revealed DIM treatment could lead to the activation of p38 and c-Jun NH(2)-terminal kinase which are typically stimulated by oxidative stress that cause the release of mitochondrial ROS and up-regulates p21 [24]. A study by Vivar et al. [28] characterized the effect of DIM on LNCaP (androgen receptor positive, p53 wild type) and DU145 (androgen receptor negative, p53 mutant) human prostate cancer cells. They used a dominant negative inhibitor of p38 MAPK that results in induction of p27Kip1 and subsequent G1 cell cycle arrest linked to activation of the p38 MAPK pathway. They further reconciled that DIM affects prostate cancer cells regardless of their androgen-dependence and p53 status. In oral squamous carcinoma cells, synthetic ring substituted DIM analogs- 5,5′-dibromo-DIM diminishes cell survival and inhibits the growth of oral cancer cells which is mechanistically mediated by the p38 mitogen-activated protein kinase pathway eventually leading to apoptosis [49]. An important therapeutic trait is the ability of DIM to disrupt MAPK signaling in vascular endothelial cells by functioning as a negative regulator of VEGF-stimulated MAPK activity that renders them insensitive to VEGF. This leads to impairment in vasculature including endothelial cell migration and differentiation affecting angiogenesis [22].
Figure - 4. Effect of DIM on the MAPK signaling mechanisms.

This pathway has been shown to be inhibited by DIM; these kinases transduce the signal by phosphorylating downstream protein kinases. Some of the phosphorylated signaling molecules translocate to the nucleus to activate transcription factors (such as AP-1) which regulate the expression of select set of genes.
The oncogene p75 (NTR) functions as a tumor suppressor in prostate epithelial cells, where its expression declines with progression to malignant cancer. In related context Khwaja et al. emphasized that DIM is capable of inducing p75 (NTR)-dependent apoptosis via the p38 MAPK pathway in prostate cancer cells [50]. They further mention that prostate (PC-3, DU-145) and bladder (T24) cancer cells were more sensitive than human breast cancer (MCF-7) and mouse fibroblast (3T3) cells to the effect of DIM mediated induction of p75(NTR) resulting in loss of viable tumor cells. Transfecting PC-3 cell line with a dominant-negative form of p75 (NTR) before DIM treatment abrogated the effect of DIM suggesting a cause and effect relationship between DIM mediated induction of p75 (NTR) levels and inhibition of cell survival. Based on aforementioned knowledge one may conclude that DIM activates both JNK and p38 MAPK kinase pathways in breast cancer cells, but p38 pathway is more prominent in prostate cancer cells. Activation of all three MAPK pathways in a single cell line by DIM has not been reported to date.
DIM and the inhibition of histone deacetylation
Histone deacetylase (HDAC) inhibitors are beginning to be recognized as novel, therapeutically efficacious anti-tumor agents with pleiotropic effects including stimulation of apoptosis. At molecular level HDAC’s regulate acetylation status of core nucleosomal histones impeding a variety of transcriptional activating factors accessibility to DNA, resulting in transcriptional repression of aberrant proliferation-linked gene expression. In 2006, FDA approved the HDAC inhibitor SAHA, which is at present tested in Phase I and II clinical trials for the treatment of various malignancies [51-53]. Another compound- butyrate is produced naturally in the colon by bacterial fermentation of dietary fibers. Butyrate has been extensively studied in colon cancer prevention and shown to inhibit tumor growth and induces apoptosis through the inhibition of histone deacetylase. Huang and Guo (2006) showed that mutations in the adenomatous polyposis coli (APC) gene confer resistance to HDAC inhibitor-induced apoptosis in colon cancers due to the failure of butyrate to down-regulate survivin in these cells [54]. Contextually, Bhatnagar et al. revealed that DIM inhibits survivin mRNA expression and promotes survivin protein degradation through inhibition of cdc2-cyclin B1-mediated survivin Thr(34) phosphorylation. Thus, conventional pretreatment of cells with DIM augmented butyrate-induced apoptosis in colon cancer cells expressing mutant APC [55]. It was found that DIM-butyrate combination treatment induces the expression of proapoptotic Bax and Bak proteins triggering Bax dimerization/activation that cause the release of cytochrome c and Smac proteins from mitochondria resulting in initiation of apoptosis [55]. Thus, DIM down-regulates survivin and enhances the effects of butyrate in apoptosis initiation and prevention in familial adenomatous polyposis in APC (min/+) mice. The findings are consistent, showing that the combination of DIM and butyrate could potentially be an effective strategy for the prevention of colon cancer. Other original findings originating from the same laboratory also indicate that DIM can selectively induce proteasome-mediated degradation of class I histone deacetylases (HDAC1, HDAC2, HDAC3, and HDAC8) without affecting the class II HDAC proteins in human colon cancer cells in vitro and in vivo in tumor xenograft [56]. As a corollary HDAC-mediated transcriptional inhibition of the cyclin-dependent kinase inhibitors p21WAF1 and p27KIP2 is abrogated leading to a significant increase in their expression causing cell cycle arrest in the G2 phase. Additionally, HDAC depletion is associated with induction of DNA damage that triggers apoptosis. Accordingly, the lead author of this investigation, Li et al. view ability of DIM to induce cell cycle arrest and apoptosis through a mechanism that involves degradation of class I HDACs in tumor cells [56].
DIM and inhibition of Autophagy
Autophagy is an evolutionary conserved intracellular pathway that sequesters and degrades cytosolic proteins and damaged organelles within autophagosomes to recycle substrates so as to maintain cellular energy homeostasis under conditions of limited nutrients or stress [34]. Paradoxically, depending on the circumstances, the process of autophagy can be used as a survival mechanism by tumor cells in nutrient limiting conditions [57]. The regulation of this pathway is complex and controlled by the coordinated actions of autophagy related genes. At present, it is difficult to appraise autophagy as a target for DIM-mediated chemoprevention although in vitro experiments using MDA-MB-231 human breast cancer cells treated with H2O2 and DIM for 24 hours lead to protein neutralization and degradation characterizing autophagy [34]. Using fluorescence confocal microscopy the findings provide evidence that autophagy could be one of the mechanisms of cell killing by DIM treatment as documented by multiple tests such as the presence of MDC (a dye that stains autophagic vacuoles) and conversion of LC3-I into LC3-II (marker of autophagy), and the knocked down or over-expression of the essential autophagy gene beclin 1[34]. Clearly further studies are needed to characterize DIM induced autophagy in other established models and cell lines.
DIM and Apoptosis
Based on our current state of knowledge between apoptosis and cancer therapy, one may exemplify apoptosis as a genetic and epigenetic controlled process therapeutically amenable by DIM leading to cytotoxic effects. One interesting observation regarding DIM is its apparent selective apoptosis inducing effects in cancerous cells but not on normal cells such as normal human keratinocytes [58]; CRL2221 human prostate epithelial cells [59]; human pancreatic ductal epithelial cells [60]. Studies from our laboratory as well as, other forthcoming reports overwhelmingly support anti-cancer effects of DIM in many different human cancers. DIM intervenes mechanistically by inducing disruption of mitochondrial membrane potential and cytochrome c release, followed by activation of caspase 9, 3 and poly(ADP-ribose) polymerase-all of which are mediated through the mitochondrial pathway (Fig-5). In addition, apoptosis is accompanied by down-regulation of anti-apoptotic Bcl-2 protein and an enhancement in the pro-apoptotic Bax expression following DIM treatment supporting the role of DIM as a potential anti-cancer agent and further underscoring the potential clinical importance of DIM as an adjunct to conventional therapies.
Figure - 5. A schematic summary of the molecular mechanism of action of DIM in induction of intrinsic apoptosis pathway.

In response to intrinsic death stimuli originating from the nucleus (chemotherapeutic drugs) or mitochondria (ROS) influence the release of cytochrome c controlled by the proapoptotic Bcl-2 family. DIM is able to stimulate ROS production in mitochondria and downregulates Bcl-2 and IAP protein expression causing caspase cascade activation leading to apoptosis.
The induction of apoptosis by DIM has been documented in human colon adenocarcinoma cells- S-174, Caco-2 [18], HT-29, HCT-116 [61], human esophageal adenocarcinoma cells (Seg-1 and Bic-1), breast carcinoma cells, including the highly invasive and metastatic breast cancer cell line, MDA-MB- 231 [62-64], melanoma cells [65], pancreatic carcinoma cells [60;66], hepatoma HepG2 cells [67], thyroid cancer cell lines representative of papillary (B-CPAP and 8505-C) and follicular carcinoma of the thyroid (CGTH-W-1 and ML-1) [21], numerous human leukemia cell lines [68] and lung cancer cells [69]. In human cervical cancer cells C33A, DIM was approximately four times more potent and faster acting than I3C in a mitochondrial function assay [58]. In androgen dependent human prostate carcinoma LNCaP cells, DIM-induced apoptosis was associated with stabilization of p53 and the down-regulation of NF-κB activity, resulting in a decreased expression of the anti-apoptotic protein Bcl-2 [58;70;71]. Nevertheless, in light of growing evidence Nachshon-Kedmi et al. emphasized in the androgen-independent PC-3 prostate cancer cell line DIM induces apoptosis mainly through the mitochondrial pathway and argued that the therapeutic response could be useful for the development of new strategies for the treatment of androgen-independent prostate cancer [71]. Interestingly, induction of apoptosis has been observed in synchronized androgen-sensitive (LNCaP) and androgen-insensitive (C4-2B) prostate cancer cells by B-DIM. It was noted that BDIM inhibits proteasome activity in the S phase of cell cycle, leading to the inactivation of NF-κB signaling as one of the molecular mechanisms of apoptosis induction by B-DIM. Based on their findings, the authors suggest that B-DIM could be a promising agent for the prevention and/or treatment of both hormone sensitive as well as hormone-refractory prostate cancer [72].
Supplementary studies have shown induction of apoptosis in breast cancer cells harboring either wild-type p53 (MCF-7), mutant p53 (T47-D), or cells deficient in p53 gene (Saos-2) and it was concluded that the induction of apoptosis by DIM treatment was independent of p53 status [73]. In addition, a study originating from our laboratory compared the effect of DIM treatment in breast cancer cell lines harboring different molecular signatures such as, over-expression of Her-2 and activated Akt [BT-20 and BT-474] and cells deficient in estrogen receptor [BT-20]; DIM treatment to these cells induced p27 KIPI transcript expression and induced nuclear localization of p27 KIPI in both cell lines independent of Her-2, Akt, or estrogen receptor status suggesting distinctive role of DIM against breast cancer [74]. Another study used estrogen receptor positive (MCF-7) and negative (MDA-MB-231) cells and demonstrated DIM cause inhibition of cell proliferation and persuade apoptosis in both the cell lines consistent with inhibition of Bcl-2, and induction of Bax proteins independent of ER status [75].
Moiseeva et al. [76] report treatment of breast cancer cells (MDA-MB-231) in long-term culture to low concentrations of DIM compromises sensitivity to radiation-induced DNA damage but not DNA repair capacity. Besides, their model revealed that apoptosis and not growth arrest is probable responsible for growth inhibition rationalizing additional new molecular target and activitiy of DIM. In colon adenocarcinoma cells- HCT116 and HT-29 cells, DIM increases the activation of caspase-3, -7, -8, and -9 and enhances poly (ADP-ribose) polymerase cleavage. In addition, DIM increased the translocation of cytochrome c and Smac/Diablo from the mitochondria to the cytoplasm during induction of apoptosis, which again was found to be a p53-independent process [77].
In addition to foregoing classical targets of apoptosis, other contemporary molecular targets have been identified and reported as being modulated by DIM culminating in apoptosis. These include p75 (NTR); decreased FLICE-like-inhibitory-protein (FLIP), activation and promotion of FasL-mediated apoptosis and Prostate apoptosis response-4 (Par-4). Par-4 is a unique pro-apoptotic protein that selectively induces apoptosis in prostate cancer cells. A study by Azmi et al. reported the influence of DIM in modulating the functional significance of Par-4 in a panel of pancreatic cancer cell lines showing apoptosis induction [78]. Interestingly, this was the first report that reveals DIM, and specifically its more bioavailable formulation B-DIM, at concentration as low as 20 micromol/L not only induced Par-4 in pancreatic cancer cells but also sensitized these cells to cytotoxic action of chemotherapeutic drug such as gemcitabine which was mediated through the up-regulation of Par-4 [78]. Another molecular entity-nonsteroidal anti-inflammatory drug-activated gene-1 (NAG-1) is a TGF-beta super family gene associated with pro-apoptotic and anti-tumorigenic activities. Studies report that DIM treatment increases luciferase activity of NAG-1 in HCT-116 cells transfected with NAG-1 promoter construct, This is suggestive of DIM repressing cell proliferation through up-regulation of NAG-1 and that activating transcription factor 3 (ATF3) plays a pivotal role in DIM-induced NAG-1 expression in human colorectal cancer cells [79]. These foregoing studies provide additional potential molecular basis for anti-tumorigenenic effects of DIM.
A recent study by Rahman et al. confirmed that anti-apoptotic protein, survivin is over-expressed in breast cancer cells and that DIM-mediated inhibition of cell growth and induction of apoptosis in MDA-MB-231 breast cancer cells was in part due to down-regulation of survivin [64]. Further, using elegantly designed experiment the authors showed that the down-regulation of survivin by siRNA prior to DIM treatment resulted in enhanced cell growth inhibition and apoptosis, whereas over-expression of survivin by cDNA transfection abrogates DIM-induced cell growth inhibition and apoptosis. Overall, the results support targeting survivin by DIM could be a valid approach for the prevention and/or treatment of breast cancer.
Related to apoptosis, a promising and unique molecular target involving inhibition of DNA topoisomerase (-I and -II alpha) by DIM affecting mitochondrial F0F1-ATP synthase (which in turn cause depletion of the mitochondrial ATP levels) has been cited [67;80]. Consequently, this drives a significant stimulation of mitochondrial reactive oxygen species (ROS) production followed by depolarization of mitochondrial membrane potential causing intracellular catastrophe and induction of programmed cell death. Based on these outcomes the authors suggest that one could exploit further development of DIM as a potential therapeutic agent.
Selective Molecular effects of DIM
DIM and Transcription factors: Nuclear transcription factor-κB
The Nuclear transcription factor-κB (NF-κB) signaling plays critical roles in the control of cell proliferation, survival, tumor invasion, metastasis, drug resistance and stress response. A large number of cancer cells, especially poorly differentiated cancer cells, show activated NF-κB in the nucleus, suggesting that activated NF-κB regulates its downstream genes to promote cancer cell growth. Therefore, NF-κB has emerged as a target for the prevention and/or treatment of cancer. Under non-stimulating conditions, NF-κB is sequestered in the cytoplasm through tight association with NF-κB inhibitory protein IκB. Following stimulation including cytokine binding to its receptor, activation of IκB kinase (IKK) complex occurs. This leads to phosphorylation and subsequent degradation of the inhibitory protein IκB, allowing NF-κB to translocate into the nucleus and binds to its target DNA to regulate the expression of NF-κB target genes (Fig-6). We found that DIM, or the formulated B-DIM treatment, could inactivate NF-κB DNA binding activity in prostate [81], breast [74;82;83], head and neck [84], and pancreatic cancer cells [60;66] resulting in inhibiting the transcription of NF-κB downstream genes. This process ultimately led to the inhibition of cell growth and induction of apoptotic cell death.
Figure - 6. Molecular targets and cell signaling pathways altered by DIM.

Multiple growth factor receptors are activated at the cell surface during cancer. Activation of these receptors activates several downstream signaling pathways. Among these pathways the Ras-MAPK, the P13K-Akt, mTOR and NF-κB pathways are of significance and targets of DIM. By dephosphorylating these molecules, DIM modulates downstream signaling pathways impinging on proliferation, angiogenesis and apoptosis. NF-kB pathway is inactive as a result of the binding of p50 and p65 to IκB. When IκB is phosphorylated by IKKs and degraded, p50 and p65 are set free and are translocated into the nucleus to activate a specific set of genes. This pathway has been shown to be inhibited both in vitro and in vivo by DIM. Activation of AP-1 is primarily mediated by MAPK, especially through the phosphorylation by c-Jun N-terminal kinase (JNK), which leads to translocation of AP-1 and subsequent transcriptional activation of target genes
DIM has also been found to potentiate the anti-tumor activity of chemotherapeutic agents through regulation of NF-κB. It has been reported that some chemotherapeutic agents such as cisplatin, gemcitabine and docetaxel could result in the activation of NF-κB in cancer cells, which may be responsible for drug resistance in cancer cells [2; 56; 57]. By in vitro and in vivo studies, we have found that pre-treatment of cancer cells with DIM followed by treatment with low concentrations of docetaxel, gemcitabine or oxaliplatin elicited significantly greater inhibition of cell growth and induction of apoptosis compared to either agent alone [41; 42; 56-58]. We also confirmed that NF-κB activity was significantly increased by docetaxel, gemcitabine or oxaliplatin treatment, and the NF-κB inducing activity of these agents was completely abrogated in cells pre-treated with DIM. The foregoing in vitro results were also recapitulated in our in vivo animal studies [60;82]. Collectively, these results clearly suggest that DIM pre-treatment, which inactivates NF-κB activity, together with other cellular effects of DIM, may contribute to increased cell growth inhibition and apoptosis with sub-optimal doses of docetaxel, oxaliplatin, or gemcitabine.
Aryl hydrocarbon receptor (AhR)
The AhR is a ligand-dependent transcription factor belonging to the basic helix-loop-helix/Per-ARNT-Sim family of proteins [85]. Prior to ligand binding, AhR exists in the cytoplasm in a complex with heat shock protein 90 [86], the cochaperone p23 [87], and the immunophilin homolog XAP2 [88]. Following ligand binding, AhR moves to the nucleus, dissociates from the chaperone complex, and forms a heterodimer with the basic helix-loop-helix/Per-ARNT-Sim protein ARNT. This heterodimer binds to xenobiotic response elements (XREs) in the promoter and enhancer regions of target genes including those genes involved in detoxification, inflammation, and cancer regulating their transcription (Fig-7) [89;90]. Induction of cytochrome P4501A1 (CYP1A1) expression has been studied extensively as a model of AhR action [91]. A variety of structurally diverse xenobiotic and bioactive food compounds, can bind in a ligand dependent manner as agonists and activate this transcription factor [92]. DIM antagonism of CYP1A1 induction appears to result from the poor recruitment of critical HATs as shown by Hestermann and Myles Brown [93].
Figure - 7. Model of the AhR ligand mediated activation of signaling pathways involved in activation of xenobiotic metabolism.

Upon ligand binding the AhR complex translocates to the nucleus and forms a functional AhR / Arnt complex. Binding of the AhR/Arnt complex to XRE cause initiation of the transcription of genes encoding phase- I and II xenobiotic metabolising enzymes. DIM can intervene at multiple sites in the process as described in the text, but not shown here for clarity.
Data from cultured cells and animal models indicate that AhR ligands can inhibit formation and proliferation of breast tumors and DIM has been shown to be a selective AhR modulator [16]. In estrogen-responsive breast cancer cells DIM binds to the AhR with lower affinity compared to AhR prototype agonists 2,3,7,8 tetrachlorodibenzo (p) dioxin (TCDD) [94], and thus, by analogy bear resemblance to selective estrogen receptor modulators (SERM) used in breast cancer treatment. In addition, it has been reported in MCF-7 breast cancer cells, DIM reverses the epigenetic activation of cyclooxygenase-2 (COX-2) expression induced by environmental (TCDD) agent, which is known to cause inflammation and tumorigenesis. In a study reported by Degner et al. [92], DIM (10 μmol/L) abrogates TCDD-induced recruitment of the AhR and AcH4 to the COX-2 promoter resulting in repression of COX-2 mRNA and protein. This suggests that naturally occurring modulators of the AhR such as DIM surmounts epigenetic activation of COX-2 expression by AhR agonists. Methyl-substituted DIM analogs with preclinical evidence against breast cancer also exhibit selective modulation of the AhR (SAhRMs) signaling in cancer cells [95].
Nuclear factor E2 p45-related factor (Nrf-2)
Nuclear factor E2 p45-related factor 2 (Nrf2), is a cap‘n’collar basic leucine zipper (Bzip) transcription factor, and after activation it translocates into the nucleus and binds to the “antioxidant response element” (ARE) in conjugation with small Maf proteins and plays a central role in the regulation (basal and/or inducible expression) of several Phase 2 genes (e.g., GST and NQO1) by binding to the ARE in their promoters [96] (Fig-8). DIM stimulates several Nrf2-regulated promoters, including NQO1 (an oxidoreductase), GSTα1(a glutathione S-transferase), and x-CT (cystine/glutamate transporter as stated earlier [34]. Our recent study indicate the pivotal role of this transcription factor in DIM stimulated signaling through the antioxidant response element in a BRCA1-dependent manner as described in previous paragraphs.
Figure - 8. Model of the signaling pathways involved in activation of Nrf2.

Nrf2 binds to Keap1 in the cytosol. Upon activation of upstream BRCA-1, proteins kinases (MAPKs, PI3K, PKC and PERK) and /or direct effect on Keap1, Nrf2 is released and translocate into the nucleus, where it binds to the ARE along with association of small Maf proteins. The binding results in the transcriptional activation of a battery of detoxification and antioxidant proteins.DIM induced BRCA1 may also directly cause the cleavage of disulfide bond between Nrf2 and Keap1.
Regulation of the Akt pathway
Most human cancers display reduced expression of the Akt inhibitor PTEN (the tumor suppressor phosphatase and tensin homologue deleted on chromosome 10). Hence, proteins regulating signaling through the phosphatidylinositol 3-kinase (PI3K)/Akt pathway are frequently activated in tumors because of the loss of PTEN. Activated Akt phosphorylates a plethora of downstream substrates involved in the regulation of cell survival, cell cycle progression, and cellular growth (Fig-6). Inhibition of phosphorylated Akt signaling sensitizes cancer cells to chemotherapy, suggesting the importance of the inhibition of phosphorylated Akt signaling as a vital therapeutic arsenal for cancer therapy. DIM has been found to inhibit cancer cell growth and induce apoptosis through the inhibition of the Akt pathway in breast, prostate and pancreatic cancer cells. This pathway does not operate in isolation, but instead studies have concluded that Akt is a central molecule that integrates and modulates other signaling including receptor tyrosine kinase signaling, NF-κB signaling, and extracellular regulated kinase (ERK) signaling cascade. Akt is essentially an inactive cytosolic protein which is recruited to the plasma membrane upon binding by growth factor and becomes fully active through phosphorylation at two key sites, threonine 308 and serine 473. Once activated, it dissociates from the plasma membrane and translocates into the cytosol and phosphorylate both cytoplasmic and nuclear target proteins notably, glycogen synthase kinase (GSK)-3β, p27Kip, mammalian target of rapamycin (mTOR) and forkhead transcription factor [97-101]; several of these are related to the regulation of proliferation, migration and invasion [65].
One significant finding reported by Bhuiyan et al. from our laboratory employed Akt gene transfection, reverse transcription-PCR, Western immunoblot, and EMSA, and concluded the existence of a potential crosstalk between Akt, NF-κB, and androgen receptor (AR). Furthermore, B-DIM significantly inhibited Akt activation, NF-κB DNA binding activity, AR phosphorylation and expressions of AR and prostate-specific antigen, suggesting that B-DIM could interrupt these cross-talks [102]. In hormone independent DU145 prostate cancer cells, Garikapatty et al. report that DIM induces the down-regulation of Akt, p-Akt, and PI3 kinase [30]. In breast cancer with constitutively active Akt, the ability of DIM to induce apoptosis and sensitization to taxotere chemotherapy implies the importance of the inhibition of phosphorylated Akt signaling as a clinically significant strategy [83]. Likewise, cholangiocarcinoma cells with increased constitutive phosphorylation of Akt, DIM inhibited phosphorylation of Akt and activation of FLICE-like-inhibitory-protein (FLIP) augmenting Fas-mediated apoptosis [103]. As mentioned earlier, findings from our group show DIM-mediated inhibition of the angiogenic features of human umbilical vein endothelial cells in vitro, which was correlated with the inactivation of Akt, the suppression of vascular endothelial growth factor (VEGF) secretion, and the down-regulation of VEGF receptor 2 protein levels [104]. In addition, Wang et al. from our laboratory reported that DIM inhibit the growth of breast cancer cell lines over-expressing Her-2 and activated Akt via the modulation of the PI3K/Akt pathway and p27Kip1 independent of estrogen receptor status [74], suggesting that DIM is a front runner pleiotropic agent, and thus could be useful for the prevention of tumor progression and/or treatment of cancers that have defects in multiple genes. In spot of clinical drug development Rajoria et al recently pointed to the inhibition of cell proliferation by DIM is exerted by the combined suppression of Akt and ERK pathways [31].
Mammalian target of rapamycin (mTOR) pathway and DIM
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase pathway and has emerged as an attractive cancer therapeutic target because it is a critical player for controlling many cellular processes e.g., cell growth, proliferation and cell division. The mTOR regulates translation rates and celln proliferation in part by phosphorylating two major targets, the eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and the ribosomal protein S6 kinases (S6K1 and S6K2) (Fig-6). Despite limited studies reported to date regarding DIM and the modulation of the mTOR pathway, one study by our group reported using a newly recognized-Platelet-derived growth factor-D (PDGF-D) over-expressing PC3 cells (PC3 cells stably transfected with PDGFD cDNA and referred to as PC3 PDGF-D) exhibits rapid growth rate and enhanced cell invasion associated with the activation of mammalian target of rapamycin (mTOR) and reduced Akt activity [105]. Rapamycin, an inhibitor that function by decreasing mTOR protein levels repress mTOR activity in these cells, but concomitantly resulted in the activation of Akt, nullifying the therapeutic effects of mTOR inhibitor-Rapamycin. Interestingly, B-DIM significantly inhibits both mTOR and Akt in PC3 PDGF-D cells. This correlates with decreased cell proliferation and invasion and also elicits other therapeutic effects by inactivation of both mTOR and Akt activity [105]. In this respect, B-DIM appears to be superior to Rapamycin and analogs that have otherwise shown clinical activity as a single agent in a limited number of tumor types [106].
Wnt signaling and DIM
The canonical Wnt signaling pathway operates by stabilizing β-catenin and has emerged as an important mechanism in the regulation of cell proliferation and survival. In the absence of Wnt/Wingless activation, β-Catenin is sequestered in the cytoplasm, undergoes phosphorylation and targeted for ubiquitination and proteolytic degradation. Activation of Wnt signaling by binding of Wnt ligands to a Frizzled receptor inhibits phosphorylation of β-catenin in the cytosol, which translocates to the nucleus and interacts with members of the T-cell factor/lymphoid enhancer binding factor (TCF/LEF) family of transcription factors causing transactivation of oncogenes including myc and cyclin D1 (Fig-9). A study reported from this laboratory unveiled that B-DIM significantly increased the phosphorylation of β-catenin, and inhibited β-catenin nuclear translocation, leading to the subsequent inhibition of cell growth and induction of apoptosis in hormone-sensitive LNCaP and hormone-insensitive C4-2B prostate cancer cells [107]. Further studies revealed alterations in the expression of AR and p27Kip1 signifying the effect of B-DIM in modulating the Akt/GSK-3β/β-catenin/AR signaling axis, which may be involved in the development of castrate resistant prostate cancer (CRPC). Thus, modulation of this signaling axis by B-DIM could be a promising non-toxic strategy for possible treatment of hormone-sensitive, and most importantly CRPC. Moreover, the results suggest the existence of a molecular cross-talk between Akt, Wnt and AR signaling pathways in prostate cancer cells and that GSK-3β is the key enzyme bridging these pathways [107].
Figure - 9. Model of the Canonical and non canonical Wnt signaling pathway.

Lef-1/ TCF proteins are the downstream DNA binding effectors of canonical Wnt signaling pathway. There is evidence that DIM can function downstream enhancing the phosphorylation of β-catenin and inhibit the nuclear translocation of β-catenin leading to inhibition of tumor cell proliferation and induction of apoptosis.
Androgen Receptor (AR)
As a member of the steroid receptor superfamily of transcription factors, AR and its cognate natural ligand - androgen, have been implicated in the normal prostate development as well as in the growth and maintenance of castrate resistant prostate cancer (CRPC) for which there is no curative therapy [108]. In prostate epithelial cells, ligand-free AR is sequestered in the cytoplasm in an inactive conformation by heat shock proteins. Binding of the ligand dissociates heat shock proteins. This leads to homodimerization and phosphorylation of the ligand AR complex and its translocation into the nucleus and binding to androgen response elements in the promoter regions of target genes including the androgen inducible prostate specific antigen (PSA) (Fig-10). Currently available treatment modalities for CRPC rely on therapies that target AR since most CPRC express AR and the androgen inducible PSA. Studies have shown that DIM function as a novel AR antagonist, represses AR function through competitive binding and utilize intracellular signaling pathways to alter AR nuclear accumulation and degradation by the ubiquitin proteasome pathway [72]. Li et.al., investigated and present evidence by which DIM impinges on other regulatory molecules that affects AR signaling pathways [107]. It has been known that FOXO3a, GSK-3β, and β-catenin are all AR coregulators which regulate the activity of AR. Accordingly Li et.al., from our laboratory investigated and reported the molecular effects of B-DIM (with higher bioavailability) on Akt/FOXO3a/GSK-3β/β-catenin/AR signaling in hormone-sensitive LNCaP and hormone-insensitive C4-2B prostate cancer cells. Utilizing innovative techniques such as electrophoretic mobility shift and chromatin immunoprecipitation (CHIP) assays, it was inferred that B-DIM inhibits FOXO3a binding to the promoter of AR and promotes FOXO3a binding to the p27KIP1 promoter, resulting in the alteration of AR and p27KIP1 expression; this leads to the inhibition of cell proliferation and the induction of apoptosis in both androgen-sensitive and insensitive prostate cancer cells. These results confirm that B-DIM-induced cell growth inhibition and apoptosis induction are partly mediated through the regulation of Akt/FOXO3a/GSK-3β/β-catenin/AR signaling, strengthening the notion that B-DIM could be a promising non-toxic agent for possible treatment of not only hormone-sensitive but also castrate resistant prostate cancer therapy (CRPC)..
Figure - 10. AR and ER signaling pathways and the effects of DIM on the pathways.

DIM induces BRCA1 expression- a key factor in recognizing and repairing DNA damage, mediating growth inhibition and cell cycle checkpoints and that both DIM and BRCA1 inhibit estrogen (E2)-stimulated estrogen receptor (ER-alpha) activity in human breast cancer cells.
DIM and cDNA Microarray data
cDNA microarray gene expression profiling to elucidate intracellular molecular mechanism(s) by which DIM exert its effect on cancer cells have been explored for prostate, breast and cervical cancer cells. Studies from our laboratory utilized the Affymetrix Human Genome U133A Array to interrogate the expression of 22,215 known genes following DIM treatment in PC3 prostate cancer cells. A total of 738 genes [677 genes were down-regulated and 61 were up-regulated as early as 6 h] showed a greater than two-fold change after 24 h of DIM treatment. Upon cluster analysis, it has been found, and reported that DIM up-regulates the expression of genes that are related to the Phase I and Phase II enzymes, suggesting their increased capacity for detoxification of carcinogens or chemicals. Additionally, it was noted that DIM down-regulates the expression of genes that are critically involved in the regulation of cell growth, cell cycle, apoptosis, signal transduction, Pol II transcription factor and oncogenesis. The findings corroborated with real-time reverse transcription-polymerase chain reaction analysis which was conducted to confirm the cDNA microarray data. In another related study, Rahman et al. found a total of 1,238 genes altered in DIM-treated breast cancer cells, among which 550 genes were down-regulated and 688 genes were seen up-regulated by DIM. Of greater significance were the findings from clustering analysis revealing significant down-regulation of survivin which is reportedly over-expressed in many human cancers including breast cancer. Thus down-regulation of survivin by DIM was explored from therapeutic point of view and the findings have been published [109]. An additional breast cancer related microarray profiling have been reported by Mulvey et al. in MCF7 breast cancer cells that were treated with E2 (1 nM) or DIM (25 μM) alone or in combination for 16 h. Analysis of their data revealed the interplay of E2 and DIM as reflected in the expression of a subset of genes (<90) in which the combination of E2 and DIM acted either additively or antagonistically altering gene expression [110].
Using cDNA microarrays, Carter et al. examined early changes in gene expression after treatment with 100 μmol/L DIM in cervical cancer cells [C33A and CaSki] and immortalized human epithelial cell line (HaCat), as well as in normal human foreskin keratinocytes (HFK) [111]. The authors conclude from their analysis that DIM consistently alters the expression of >100 genes at least two-fold, many of which stimulates genes encoding transcription factors and proteins involved in signaling, stress response and growth independent of integrated HPV sequences. Furthermore, it was noted that genes encoding bZip proteins were the ones most consistently and robustly stimulated including the stress-associated immediate early gene GADD153 (>50 fold in C33A) and nuclear factor-interleukin 6 (NF-IL6) and c/EBPbeta, (>5 fold in C33A), which have shown to reduce the expression of HPV oncogenes [111].
DIM and microRNA
microRNAs (miRNAs) are a class of endogenous small non-coding RNA molecules of 20-25 nucleotides in length cleaved from ~70- to 100 nucleotide hairpin premiRNAs precursors [112]. Increasing evidence reveals deregulation of miRNAs expression may promote tumorigenesis because they are key regulatory molecules in various biological and pathologic processes. There is also increasing evidence that over-expressed miRNAs (such mir-17-92), may function as oncogene and promote cancer development by negatively regulating tumor suppressor genes and /or genes that control cell differentiation or apoptosis [113]. Many studies have established this concept by discovering the up-regulation or down-regulation of specific miRNAs in various types of cancer and identifying some of their molecular targets [114-116]. Relatively few studies documented the effect of DIM in altering the expression of miRNAs profile. Li et al. from our laboratory assessed the expression profile of miRNAs between gemcitabine-sensitive and gemcitabine-resistant pancreatic cancer cells and reported the miRNA expression pattern was different between these two cell lines. By miRNA microarray and RT-PCR, they found that B-DIM treatments caused alterations in the expression of 28 miRNAs [117]. Further narrowing to miR200 and let-7 family revealed that miR-200b, miR-200c, let-7b, let-7c, let-7d, and let-7e were all increased by DIM in the gemcitabine-resistant cells concomitant with reversal of mesenchymal phenotype to epithelial phenotype (Fig-11).
Figure - 11.

Schematic representation of EMT and micro RNA and the effects of DIM on the pathways.
Biological effects of DIM
DIM and angiogenesis
Angiogenesis is the formation of new blood vessel from existing vasculature, an adaptive process to compensate tissue and cellular requirement for oxygen and nutrient. Accumulating evidence reveals VEGF to be a major mediator of angiogenesis by targeting endothelial cells. It has also been established that the α-subunit of potent tumorigenic factor, hypoxia inducible factor-1 (HIF-1α) becomes elevated in tumors due to hypoxia, and that HIF-1α transcriptional activity contributes to tumor angiogenesis, invasion and progression. Chang et.al., reported the effect of DIM on angiogenesis in an in vitro model of cultured human umbilical vein endothelial cells (HUVECs), as well as in an in vivo Matrigel plug angiogenesis assay [23]. According to these authors DIM produced a concentration-dependent decrease in proliferation, migration, invasion and capillary tube formation of cultured HUVECs and caused a G1 cell cycle arrest in actively proliferating HUVECs [23]. Additionally, within in vivo Matrigel plug angiogenesis assay, neovascularization was inhibited up to 76% compared with vehicle control following 5mg/kg DIM administration testifying not only anti-angiogenic activity but confirming that DIM is active in vivo. Further, probing into mechanism of DIM’s anti-angiogenic activity the authors conceded that DIM inhibits vascular endothelial growth factor (VEGF)-induced cell proliferation and DNA synthesis in HUVECs. Consistent with this inhibition, other downstream signaling events such as VEGF-induced extracellular signal-regulated kinase (ERK1/2) phosphorylation were found to be greatly reduced [23].
Other investigators disputed whether DIM exert a direct and specific effect on the tyrosine kinase activity of VEGF receptor; but instead DIM inhibits Ras signaling induced by VEGF and other growth factors which interferes with downstream biological effects necessary for angiogenesis [22]. In congruence, similar correlations have been recently reported by Kunimasa et al demonstrating that DIM has potential in the regulation of two principle survival signals ERK1/2 and Akt [118]. They report that pharmacological inhibition of these molecules was sufficient to inhibit tube formation and induce caspase-dependent apoptosis compromising tube forming capacity of HUVEC cells. As an extension to these preliminary investigations, studies carried out in the author’s laboratory sought to explore extensively investigating the molecular mechanism by which DIM inhibits angiogenesis and invasion by assessing the role of factors secreted by cancer cells which control all steps of angiogenesis. It was found that the formulated DIM (Bioresponse DIM) inhibits angiogenesis and invasion by reducing the bioavailability of vascular endothelial growth factor (VEGF) via repressing extracellular matrix-degrading proteases, such as matrix metalloproteinase (MMP)-9 and urokinase-type plasminogen activator (uPA) in human prostate cancer cells, resulting in reduced vascularity (angiogenesis) in vivo [104]. We also found that B-DIM treatment inhibited DNA binding activity of nuclear factor-κB (NF-κB), which is known to mediate the expression of many of its downstream target genes, including VEGF, IL-8, uPA, and MMP-9, all of which are involved in angiogenesis, invasion, and metastasis [104]. Based on our findings, we speculate that the inhibition of NF-κB DNA binding activity by B-DIM contribute to the regulated bioavailability of VEGF by MMP-9 and uPA which, sequentially, inhibits invasion and angiogenesis. Overall, these studies provide a rational and mechanistic viewpoint linking the anti-tumor activity of BDIM in a prostate cancer model.
Another report originating from our laboratory cited a significant increase in the tube forming capability of HUVEC cells following exposure to conditioned medium from platelet-derived growth factor-D over-expressing PC3 cells (PC3 PDGF-D cells) which turned out to be alleviated by B-DIM treatment [119]. These new findings suggest that B-DIM could serve as an efficient chemopreventive and/or therapeutic agent by inactivation of mTOR and Akt and by inhibiting angiogenic factors in PDGF-D over-expressing prostate cancer cells. Additional evidence in support of anti-angiogenic effect of DIM has been reported by Riby et.al., documenting that DIM reduces the level of hypoxia-inducible factor (HIF)-1alpha in hypoxic tumor cell lines, as well as HIF-1 transcriptional activity resulting in the reduced expression of key hypoxia responsive factors such as VEGF, furin, enolase-1, glucose transporter-1 and phosphofructokinase [120]. Collectively, these outcomes lend support showing that DIM can decrease the accumulation and activity of the key angiogenesis regulatory factor- HIF-1alpha, in hypoxic tumor cells.
DIM and Invasion and metastasis
Cancer metastasis represents the primary source of clinical morbidity and mortality in large majority of solid tumors attributable, at least, in part to entry of tumor cells into the systemic circulation (intravasation), survival in circulation, extravasation to distant organs, and finally growth of cancer cells resulting in secondary tumors. Gene expression analysis of human cancer specimens identified groups of genes, including cell migration genes, whose characteristic expression patterns can predict the risk of metastasis occurrence. Our knowledge related to the transcriptional factor that serves as a repressor in metastatic invasion remains obscure. However, closely related to this phenomenon, the expression and activation of uPA plays an important role in tumorigenicity, and high endogenous levels of uPA and uPAR has been found in advanced metastatic cancers. We therefore focused elucidating the role of uPA and uPAR in B-DIM-mediated inhibition of prostate and breast cancer cell growth and motility [62;121]. Accordingly, we noticed that uPA, as well as uPAR, induce the production of VEGF and MMP-9, and that the down-regulation of uPA/uPAR by siRNAs or B-DIM treatment resulted in the inhibition of VEGF and MMP-9 secretion thereby inhibiting cell migration. Interestingly, silencing of uPA/uPAR led to decreased sensitivity to BDIM. This indicates the importance of uPA/uPAR in B-DIM-mediated regulation of prostate and breast cancer cell growth and migration. These results provide additional mechanistic insights towards chemopreventive and/or therapeutic activity of B-DIM. We speculate this could partially be due to the down-regulation of uPA-uPAR leading to reduced production of VEGF/MMP-9 and ultimately leading to the inhibition of cell growth, migration and aggressiveness of cancer cells. A related study by Moiseeva et al. evaluated the modulation in the expression of a set of genes (cadherin-11, p21Cip1, urokinase-type plasminogen activator, and interleukin-6) in MDA-MB-231 breast cancer cells exposed for long-time in culture to a low concentrations of DIM. They inferred that DIM modified protein and/or RNA expression of the above mentioned genes, and decreased cadherin-11 [76].
Other molecules of relevance to metastasis are the proteolytic enzymes- MMP-2 and MMP-9. These are known to promote and enhance the metastatic phenotype of cancer cells and inhibited by DIM in thyroid cancer cells [122]. The effect of DIM in inhibiting growth and invasion of drug resistant human cancer cells from breast, glioma and non-small cell lung cancer expressing EGFR mutant have also been described. DIM is potentially effective in interrupting invasion phenotype and overcoming EGFR mutant associated drug resistance [123].
A common sequel to progression of breast, prostate and lung cancer is bone metastasis that leads to debilitating complications and intermittent pain. We simulated a model of experimental bone metastasis by injecting prostate and breast cancer cells into bone marrow using an experimental bone metastasis model whereby human fetal bone fragments were implanted in the subcutaneous site of SCID mice. Three to four weeks after injection of tumor cells into the marrow space, we reported evidence that B-DIM treatment results in the reduction of osteoblastic and osteoclastic reactions, and also had an effect in reducing tumor volume in mice with intact bone fragment. A closely related study by Dong et.al., investigated the influence of DIM on osteoclastogenesis in a mouse model of endotoxin-induced bone resorption (EIBR) and in vitro cultures of fibroblast-like cells and osteoblasts [124]. According to these authors, DIM reduces the expression of several inflammatory cytokines, and the expression of RANKL, leading to the obstruction of osteoclastogenesis. Thus, DIM could be useful in alleviating pain in bone metastasis and associated disorders. This requires further in-depth investigations.
DIM and Epithelial Mesenchymal transition (EMT)
For most epithelial tumors, progression toward malignancy is accompanied by a loss of epithelial differentiation and a shift toward mesenchymal phenotype defined as ‘Epithelial-to-Mesenchymal Transition’ (EMT) [125]. During the acquisition of EMT characteristics, cancer cells lose the expression of proteins that promote cell to cell contact such as E-cadherin and β-catenin, and gain the expression of mesenchymal markers such as vimentin, fibronectin, and N-cadherin (Fig-11). This leads to enhanced cancer cell migration and invasion [125]. Studies have also shown that cancer cells in the tumor center remain positive for the expression of E-cadherin and cytoplasmic β-catenin. However, cells in the periphery of tumor mass with penchant for invasion display loss of surface E-cadherin and up-regulation of vimentin, the typical characteristics of EMT phenotype. Therefore, it has been envisaged that inhibitors of EMT or agents that could either reverse the EMT phenotype or kill EMT-type cells would constitute a novel strategy for the treatment of most cancers. In this context, our laboratory investigated whether B-DIM could cause reversal of the EMT phenotype leading to cancer cell death in vitro. B-DIM treatment was found to increase the expression of E-cadherin and decreased the expression of transcription factors- ZEB1, slug, and mesenchymal marker- vimentin, as assessed by real-time RT-PCR and Western immunoblotting [126]. Furthermore, it has been reported that the morphology of MiaPaCa-2 cells changed from fibroblastoid to epithelial-like appearance after B-DIM treatment [126]. These limited studies are interesting because we believe that B-DIM could be useful to eradicate tumors by altering or killing the EMT-type cells, which are believed to be the root cause of tumor recurrence.
DIM and chemo-sensitization
The majority of patients diagnosed with cancers receive chemotherapy as a vital module of multimodality treatment. Unfortunately, findings from many clinical studies reveal increased chemo-resistance to certain chemotherapeutic agents. Reversal of such resistance is critical for the success of cancer treatment. Even though associations have been found between diet and cancer risk, DIM has not been established as a direct cause or cure for cancer. Overwhelming evidence from numerous studies indicates that DIM has more benefit in clinical therapy of cancer than chemotherapy alone rendering it as a highly attractive agent for therapeutic interventions. Agents so far tested with DIM include Taxotere, Cisplatin, Oxaliplatin, Gemcitabine, Erlotinib and TRAIL. Despite prevailing complexities in understanding the molecular mechanism of chemo-resistance, current knowledge and observational data relating to the underlying molecular mechanism of action of DIM conferring sensitivity is limited to altered apoptotic response mediated through the down-regulation of NF-κB. Clinical studies suggest that a common form of multidrug resistance in human cancers results from the expression of the MDR1 gene that encodes P-glycoprotein. A review of studies in pancreatic cancer reported recently by us showed that DIM pretreatment could enhance the apoptotic and therapeutic effectiveness of multiple chemotherapeutic agents (gemcitabine, oxaliplatin and cisplatin) in cell culture of pancreatic cell lines [60;127]. Interestingly, the effect exhibited by DIM was more robust compared to other chemopreventive compounds found in cruciferous vegetables. Furthermore, the ability of DIM to induce tumor cells to undergo apoptosis for therapeutic efficacy was recapitulated in a relevant preclinical orthotopic mouse model of pancreatic cancer wherein, the response between only oxaliplatin treatment vs. DIM and oxaliplatin was significant greater compared to either agent alone [60]. DIM also sensitizes pancreatic tumor cells to targeted therapeutic agents such as EGFR inhibitor- Erlotinib, potentiating apoptotic effect through altered regulation of EGFR signaling. This indeed is the logical explanation for the observed effect. Furthermore, in preclinical mouse xenograft model and in vitro studies conducted by us revealed that gemcitabine and erlotinib in combination with DIM could be promising in future clinical setting [128]. In HER2/Neu human breast cancer cells, DIM in combination with paclitaxel inhibited cell growth by 74 % compared with 42 % and 62 by DIM and paclitaxel alone, respectively [129]
Other successful combination studies using DIM have been reported for breast and prostate cancer [82;83;130]. The outcome shows an enhancement of cytotoxicity especially at lower concentration of the cytoxic agent – Taxotere and further advocate that NF-κB/ FOX-M1 over-expression leads to chemo resistance. Another study reported in literature bolster synergistic anticancer properties of DIM following co-treatment with activation of death receptor pathways in vitro suggesting its potential evaluation in cancer therapy to overcome TRAIL resistance [131]. Aberrant high expression level of c-FLIP found in many cancer cells renders a strong TRAIL resistance. Mechanistically, DIM sensitizes TRAIL-resistant cancer cells to TRAIL-induced apoptosis via enhancement of c-FLIP ubiquitination and proteasome -dependent degradation.
Other allied studies elucidating synergism between isothiocyanate (ITCs) compounds found in cruciferous vegetables such as sulforaphane (SFN) and DIM in human colon cancer cell lines have been put forth by Pappa, Gerlinde et al. citing cytostatic mechanisms [132]. Based on combination index (CI) method of Chou and Talalay [133], a total drug concentration of 2.5 μM of SFN and DIM was seen as antagonistic; whereas, with increasing cytotoxic concentrations, the antagonistic effect gradually graded into a synergistic interaction. Furthermore, SFN (10μM) in combination with DIM (10μM) resulted in strong G2/M cell-cycle arrest, which was not observed with either compound alone [132]. These results strongly suggest that cytotoxic concentrations of combined bioactive broccoli compounds- SFN and DIM combinations at physiologically relevant concentrations symbolize antagonistic interactions in terms of cell growth inhibition.
DIM and Bioavailability
Low oral bioavailability of crystalline DIM is expected from its low aqueous and lipid solubility [134]. Thus, in an effort to improve relative bioavailability, most pharmacokinetic studies regarding DIM have been reported using BR-DIM, a patented, oral formulation containing D-α-tocopheryl acid succinate, phosphatidylcholine, and silica microencapsulated in starch which exhibits 50% higher bioavailability than does crystalline DIM. Consequentially, higher concentrations of DIM have been observed in the blood of humans who received the same dose of BR-DIM as the crystalline form along with increased net exposure to DIM [135].
Reed et al. looked at the pharmacokinetics, clinical safety, and the tolerability of single ascending doses (50,100,150,200,and 300 mg) of Bioresponse-DIM (BR-DIM) in drug- free, non-smoking healthy men and women [136]. Their pharmacokinetics data revealed a linear dose exposure relationship over the range of 50-300mg. It was concluded that BR-DIM is well tolerated at single doses up to 200mg, and that increasing the dose to 300mg did not increase Cmax. The single 200 mg dose produced a mean Cmax of 104 ng/ml, and a mean AUC of 553 h* ng/ml [136].
Another study reported use of a physiological pharmacokinetic (PBPK) model. This model is reportedly extremely useful in studying the pharmacokinetics of compounds and extrapolating dose response relationship between species, routes of administration, and various dosing regimens. Employing this PBPK model Anderton et al. compared pharmacokinetic properties and bio-distribution of pure crystalline DIM and that of the formulated BR-DIM after oral administration to mice [38;137]. Based on their findings which included, the relative drug exposure and absorption parameters, it was inferred that BR-DIM exhibited approximately 50% higher bioavailability than the crystalline formulation of DIM [38].
DIM has been quantified in the urine of patients who received I3C and also in the urine of a patient who received DIM by the oral route [138]. According to Chung et.al., it has been estimated that consumption of 200 gms of broccoli provides ~12 mg of DIM; with maximum absorption the blood concentration of DIM would be expected to reach ~10μM similar to the effective levels of DIM in cultured cells [22].
DIM and preclinical studies
Preclinical rodent models in support of DIM per se are limited to xenograft and orthotopic models but studies supporting chemopreventive effects are limited to its precursor molecule- I3C. Nevertheless, with few exceptions, most preclinical studies reported thus far which utilized DIM are essentially chemosensitization related or of a kind involving combination therapy aimed at achieving a superior chemotherapeutic efficacy relative to monotherapy. Chen et.al., reported DIM given as 5mg/kg body weight, every other day inhibited DMBA induced rat mammary tumor growth and volume that was not accompanied by induction of hepatic CYP1A1-dependent activity [139]. From a therapeutic viewpoint, DIM has been evaluated as a single agent in xenograft model established with TRAMP-C2- a mouse prostate cancer cell line. Intraperitoneal injections of DIM (2.5, 5, or 10 mg per kg body weight) 3-times a week, for 3 weeks, caused a significant inhibitory effect as reflected in diminished tumor growth accompanied by apoptosis and reduced cell proliferation relative to vehicle controls [140]. Of interest, DIM didn’t affected body weights or kidney and liver functioning, strengthening assurance of its non-toxic systemic effect. Reinforcing their original findings, Cho et. al., looked at and deduced DIM feeding inhibited prostate carcinogenesis in the transgenic adenocarcinoma mouse prostate (TRAMP) model [141]. Compared with mice who did not receive DIM treatment, the number of cells expressing the SV40 large tumor antigen and proliferating cell nuclear antigen were diminished, and the number of terminal dUTP nick-end labeling-positive cells in the dorsolateral lobes of the prostate were increased. Additionally, DIM feeding reduced the expression of cyclin A, cyclin-dependent kinase (CDK)2, CDK4, and Bcl-xL, and increased p27 and Bax expression. Another study that addressed this within context of therapeutic response of DIM to metastasis is the study by Kim et al appraising oral efficacy of DIM in inhibiting lung metastasis of 4T1 murine mammary carcinoma cells in syngenic BALB/c mice [61]. Thirteen days of treatment with oral DIM resulted in marked reduction in the number of pulmonary tumor nodules along with reduction in the levels of matrix metalloproteinase (MMP-2 and 9),Tissue Inhibitor of Matrix Metalloproteinase-1 (TIMP-1), and the serum concentration of IL-β, IL-6 and TNF-α [61]. This merits serious consideration since in principle these parameters are relevant to human cancer pathogenesis and prevention. The efficacy of DIM in K14-HPV16 transgenic mouse model of cervical cancer has also been reported [142]. Of relevance an important consideration within their study objective was to inhibit cervical lesions alter estrogen metabolism in favor of C-2 hydroxylation, and to enhance immune response. Wild-type and transgenic mice were given either AIN76A diet alone or one with 2,000 ppm DIM for 12 weeks. On sacrifice, histopathological examination showed a marked decrease in cervical dysplasia in both wild-type and transgenic mice, signifying that DIM delays or inhibits the progression from cervical dysplasia to cervical cancer. Additionally, the authors revealed that DIM inhibits the development of E6/E7 oncogene-induced cervical lesions and increased serum INF-γ indicating increased immune response in the DIM-fed animals. Furthermore, to define the extent and the most effective minimum dose of DIM administration to augment the efficacy of preventive and therapeutic HPV vaccines, these investigators utilizing the K14-HPV16 transgenic mouse model concluded 1000ppm of DIM as the minimum effective and viable dose for future human studies [143]. The urinary DIM concentrations following 1000ppm DIM dose compare favorably to DIM measured in women taking either 200 or 400 mg of DIM twice daily for 4 weeks as reported earlier [138].
DIM Analogs
In search for additional effective compounds with strong anti-tumorigenic potency than DIM, several analogs of DIM have been synthesized and their activity examined against known biological targets including superior therapeutic potency and pharmacological profile (i.e., improvement in their selectivity, bioavailability and stability) from a curative viewpoint. In this direction of development, Cho et al. published 5,5′-dibromoDIM among a series of synthetic symmetrical ring-substituted DIM analogues, as being more potent than DIM in inhibiting the growth of colon and oral cancer cells [1;49;144;145]. In the investigated colon cancer cells (HT-29 and RKO cells], 5,5′-dibromoDIM decreased cell proliferation and inhibited G0 - G1- to S-phase progression, accompanied by induction of the cyclin-dependent kinase inhibitor p21 which was mechanistically found dependent on Kruppel-like factor 4 (KLF4) family of transcription factors. Furthermore, a dose of 30 mg/kg/day of 5,5′-DibromoDIM was able to inhibit tumor growth and induce p21 in athymic nude mice bearing RKO cells as xenografts [144;145]. Thus, ring-substituted DIM such as 5,5′-dibromoDIM have been envisaged by the authors as a viable class of mechanism-based drugs for clinical treatment of colon cancer. Upcoming information from same investigator laboratory also report that 5,5′-dibromoDIM induces caspase dependent apoptosis through the decrease in Bcl-2 protein expression mediated by MAPK (specifically p38 MAPK and not through ERK1/2 and JNK pathway) [49]. In a different study originating from the Dr Safe’s lab, DIM was used as a starting material to synthesize a series of 1,1-bis(3′indolyl)-1-(substituted aromatic) methanes (i.e.C-DIMs). Included among these were compounds containing p-CF3 (DIM-C-pPhCF3), p-t-butyl (DIM-C-pPhtBu), and p-phenyl (DIM-C-pPhC6H5). These were also found to be cytotoxic to cancer cells and tumors through a mechanism involving modulation of the nuclear receptor superfamily of transcription factors [144;146;147]. Further structure-activity studies revealed that the aromatic ring retention results in the generation of unique PPAR gamma receptor agonists, which activates the pro-apoptotic orphan nuclear receptor- nerve growth factor-induced-Bα (NGFI-Bα, Nur77). This underscores the development of novel DIM analogs harboring promising potential in cancer therapeutics [144]. Further evaluation of these compounds in pancreatic cancer cells [Panc-1 cells] revealed that C-DIMs (triarylmethanes), such as the p-t-butyl derivative (DIM-C-pPhtBu), activate endoplasmic reticulum (ER) stress and release calcium, accompanied by increased expression of glucose related protein 78 and C/EBP homologous transcription factor (CHOP/GADD153) proteins synchronizing apoptosis through induction of death receptor DR5 and subsequent cleavage of caspase 8, caspase 3, Bid and PARP. Similar results were noted after treatment with a prototypical inducer of ER stress- thapsigargin. These provocative findings including activation of both receptor-dependent and receptor-independent (ER stress) pathways utilizing DIM and DIM-C-pPhtBu in pancreatic cancer cells have been speculated to enhance efficacy of treatment and emphasizes the potential clinical importance of these compounds for cancer chemotherapeutic applications [41;148]. As further corollary to above mentioned analogs, Ichite et al., reported the anti tumorigenic activity of combination of docetaxel and the analog- DIM-C- pPhC6H5 against orthotopic lung tumors in mice [69]. DIM-C-pPhC6H5 alone showed anti tumorigenic activity against orthotopic lung tumors in mice (22% reduction in lung weight), while the combination of DIM-C-pPhC6H5 + docetaxel caused a significant (57%; p < 0.01) decrease in lung tumor weights compared with controls or either agent alone. These effects were found associated with enhanced apoptosis in the orthotopic tumors [69].
A few other investigators also made explorative studies to obtain information on efficacy of DIM analogs and thus have addressed on this issue. Among them McDougal et al. documented the synthesis of six methyl-substituted DIMs: 1,1′-dimethyl-, 2,2′-dimethyl-, 5,5′-dimethyl-, 6,6′-dimethyl-, and 7,7′-dimethylDIM and 1,1′,2,2′-tetramethyl DIM that were later investigated for aryl hydrocarbon receptor (AhR) agonist activity and for their potential to inhibit AhR-estrogen receptor crosstalk [95]. Of interest, the methyl-substituted DIMs inhibited estrogen-induced T47D human breast cancer cell growth and the four most active compounds (1,1′-, 2,2′-, 5,5′-dimethylDIM and 1,1′,2,2′-tetramethylDIM) inhibited estrogen-induced responses in 21-day-old female B6C3F1 mice at a dose of 100 mg/kg/day (x3). Furthermore, the authors evaluated the anti-tumorigenic activity of these compounds in 7,12-dimethylbenz[a]anthracene-induced rat mammary tumor model in which the DIM analogs were orally administered (by gavage in corn oil) at a dose of 1 mg/kg/day (x10). 1,1′- dimethyl DIM, 5,5′-dimethylDIM and 1,1′,2,2′-tetramethyl DIM resulted in a significant inhibition of mammary tumor growth, mimicking selective AhR modulators (SAhRMs), suggesting their potential role for clinical treatment of breast cancer. Other structural analogs of DIM have been reported and examined in H295R human adrenocortical carcinoma cells by Sanderson et.al.,[149] exploring for their effects on three cytochrome P450 (CYP) enzymes involved in estrogen synthesis and/or metabolism- CYP1A1, CYP1B1, and CYP19 (aromatase). Based on their findings, it was concluded that their analogs at 3 μM concentrations induced aromatase and EROD activity (as a measure of CYP1A1 and possibly CYP1B1 activity), alongwith the mRNA levels of CYP1A1 and CYP1B1. Macijewska et al. synthesized a series of DIM derivatives bearing fluoro, bromo, iodo, and nitro substituents in indole or benzene rings and tested for cytotoxicity against human melanoma cell lines after characterizing their structures. These derivatives were found to cause 50% inhibition of the viability of ME18 and ME18/R cell lines at concentrations ranging between 9.7-17.3 μM [65]. This finding clearly suggests that synthetic analog of DIM have potential role in cancer therapy in the near future once their systemic toxicity studies have been determined.
Clinical trials for cancer therapies by DIM
Based on information available at http//www.clinicaltrials.gov, several clinical trials are under way which mirrors the clinical usefulness of DIM in cancer patients (Table 1). Most of these clinical trials focus on investigating the effects of DIM in the treatment of prostate cancer or cervical dysplasia. It has been anticipated that in the future DIM could be useful as a potent agent in combination with conventional therapeutics for the treatment of human malignancies. As mentioned by us earlier, Phase I data support the non-toxic nature of DIM in healthy volunteers [136]. This is complemented by studies from our laboratory showing that formulated DIM (B-DIM) is biologically active in vitro and in pre-clinical animal model in vivo [102]. The findings provoked us to conduct a Phase- I clinical trials in prostate cancer patients. We found that B-DIM was well tolerated, and the pharmacokinetic studies revealed that drug exposure was dose proportional with measurable levels of B-DIM in the plasma [150]. Serum PSA stabilization and partial response were seen at the recommended Phase II dose of B-DIM (225 mg twice daily), suggesting the beneficial effects of B-DIM in prostate cancer treatment. This is being tested by a phase II study in our institution at the present time.
Table 1.
Clinical trials elucidating the value of DIM in cancer clinic (* Clinicaltrials,gov)
| NCT ID | Status | Site /organ | Title of study and End points |
|---|---|---|---|
| NCT00888654 | Recruiting | Prostate | DIM in treating patients with Stage I or Stage II prostate cancer undergoing radical prostatectomy |
End Points:
|
|||
| NCT00450229 | Completed | Prostate | Patients undergoing surgery for stage I or stage II prostate cancer |
End Points:
|
|||
| NCT00305747 | Active, not recruiting |
Prostate | DIM in treating patients with nonmetastatic prostate cancer has not responded to previous hormone therapy |
End Points:
|
|||
| NCT00462813 | Unknown | Cervix | DIM in treating patients with abnormal cervical cells. |
End Points:
|
|||
| NCT00212381 | Unknown | Cervix | DIM for the treatment of cervical dysplasia |
End Points:
|
|||
| NCT00784394 | Completed | Healthy | DIM in healthy nonsmokers |
End Points:
|
|||
| NCT01022333 | Recruiting | Breast | The potential for oral DIM supplementation to increase the production of the BRCA1 protein in BRCA1 mutation carriers. |
End Points:
|
|||
| NCT00591305 | Recruiting | Papilloma | New therapy of laryngeal papilloma In children |
End Points:
|
As an alternative to surgical treatment, a pilot study on patients with cervical intraepithelial neoplasia (CIN) grade 2 or 3 lesions has been reported recently [151]. In this study, which was independent of level of dysplasia, age, race, HPV status, tobacco use or contraceptive usage, the authors found that subjects given DIM at dose of 2mg/kg/day for 12 weeks had improved CIN in 47% cases with either a decrease by 1-2 grades or reversion to normal stage. Furthermore, oral DIM at the above mentioned dose was well tolerated with no significant toxicity and clinically significant improvement was confirmed in patients with grade 2 or 3 CIN in this randomized clinical trial [151]. Other preferred uses of DIM in a clinical setting include its inclusion as an adjunct to conventional therapy for recurrent respiratory papillomatosis [152].
Conclusions and perspectives
In conclusion, this review article provides a global overview suggesting that DIM could have potential benefit for chemoprevention. In addition, DIM could be useful as an adjunct to conventional therapeutics for the prevention of tumor progression and/or treatment of human malignancies in the future. The foregoing narrative are “proof-of-principle” that must be tested in controlled clinical trials especially because diet derived molecules such as DIM have pleiotropic molecular mechanism with different molecular targets including, but not limited to, its action on cell cycle, cell apoptotic processes, angiogenesis, invasion, and metastasis. Moreover, with emerging technologies including the use of genetically modified mouse models of human diseases and computational analysis, further molecular insights into basic, translational and clinical research will aid in the advancement of our knowledge to prove or disprove whether DIM could fulfill its promise as a chemopreventive and/or therapeutic agent against human cancers. However, the data available in the literature to-date provide strong support in favor of the use of DIM as a cancer preventive and therapeutic agent for human malignancies, and suggest the need for definitive clinical trial results.
Acknowledgement
Grant support from National Cancer Institute/NIH grants 5R01CA083695, 5R01CA108535, 5R01CA101870 is gratefully acknowledged. The authors also express their sincere appreciation to Ms Jaclyne Schaffert and Ms. Amenda Schmer for her editorial assistance.
Funding Source All sources of funding should also be acknowledged and you should declare any involvement of study sponsors in the study design; collection, analysis and interpretation of data; the writing of the manuscript; the decision to submit the manuscript for publication. If the study sponsors had no such involvement, this should be stated.
Please state any sources of funding for your research National Cancer Institute/NIH grants 5R01 CA083695, 5R01CA108535, 5R01CA101870
Footnotes
Declarations Mutation Research-Reviews in Mutation Research requires that all authors sign a declaration of conflicting interests. If you have nothing to declare in any of these categories then this should be stated.
Conflict of Interest A conflicting interest exists when professional judgement concerning a primary interest (such as patient’s welfare or the validity of research) may be influenced by a secondary interest (such as financial gain or personal rivalry). It may arise for the authors when they have financial interest that may influence their interpretation of their results or those of others. Examples of potential conflicts of interest include employment, consultancies, stock ownership, honoraria, paid expert testimony, patent applications/registrations, and grants or other funding.
Please state any competing interests Not Applicable
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- [1].Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J.Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- [2].Reed GA, Arneson DW, Putnam WC, Smith HJ, Gray JC, Sullivan DK, Mayo MS, Crowell JA, Hurwitz A. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3,3′-diindolylmethane. Cancer Epidemiol.Biomarkers Prev. 2006;15:2477–2481. doi: 10.1158/1055-9965.EPI-06-0396. [DOI] [PubMed] [Google Scholar]
- [3].Reed GA, Sunega JM, Sullivan DK, Gray JC, Mayo MS, Crowell JA, Hurwitz A. Single-dose pharmacokinetics and tolerability of absorption-enhanced 3,3′-diindolylmethane in healthy subjects. Cancer Epidemiol.Biomarkers Prev. 2008;17:2619–2624. doi: 10.1158/1055-9965.EPI-08-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reed GA, Arneson DW, Putnam WC, Smith HJ, Gray JC, Sullivan DK, Mayo MS, Crowell JA, Hurwitz A. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3,3′-diindolylmethane. Cancer Epidemiol.Biomarkers Prev. 2006;15:2477–2481. doi: 10.1158/1055-9965.EPI-06-0396. [DOI] [PubMed] [Google Scholar]
- [5].Crowell JA, Page JG, Levine BS, Tomlinson MJ, Hebert CD. Indole-3-carbinol, but not its major digestive product 3,3′-diindolylmethane, induces reversible hepatocyte hypertrophy and cytochromes P450. Toxicol.Appl.Pharmacol. 2006;211:115–123. doi: 10.1016/j.taap.2005.06.011. [DOI] [PubMed] [Google Scholar]
- [6].Morse MA, Stoner GD. Cancer chemoprevention: principles and prospects. Carcinogenesis. 1993;14:1737–1746. doi: 10.1093/carcin/14.9.1737. [DOI] [PubMed] [Google Scholar]
- [7].Wattenberg LW. Chemoprevention of cancer. Cancer Res. 1985;45:1–8. [PubMed] [Google Scholar]
- [8].Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227:115–124. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- [9].Lake BG, Tredger JM, Renwick AB, Barton PT, Price RJ. 3,3′-Diindolylmethane induces CYP1A2 in cultured precision-cut human liver slices. Xenobiotica. 1998;28:803–811. doi: 10.1080/004982598239227. [DOI] [PubMed] [Google Scholar]
- [10].Stresser DM, Bjeldanes LF, Bailey GS, Williams DE. The anticarcinogen 3,3′-diindolylmethane is an inhibitor of cytochrome P-450. J.Biochem.Toxicol. 1995;10:191–201. doi: 10.1002/jbt.2570100403. [DOI] [PubMed] [Google Scholar]
- [11].Gross-Steinmeyer K, Stapleton PL, Liu F, Tracy JH, Bammler TK, Quigley SD, Farin FM, Buhler DR, Safe SH, Strom SC, Eaton DL. Phytochemical-induced changes in gene expression of carcinogen-metabolizing enzymes in cultured human primary hepatocytes. Xenobiotica. 2004;34:619–632. doi: 10.1080/00498250412331285481. [DOI] [PubMed] [Google Scholar]
- [12].Gross-Steinmeyer K, Stapleton PL, Tracy JH, Bammler TK, Strom SC, Buhler DR, Eaton DL. Modulation of aflatoxin B1-mediated genotoxicity in primary cultures of human hepatocytes by diindolylmethane, curcumin, and xanthohumols. Toxicol.Sci. 2009;112:303–310. doi: 10.1093/toxsci/kfp206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Parkin DR, Malejka-Giganti D. Differences in the hepatic P450-dependent metabolism of estrogen and tamoxifen in response to treatment of rats with 3,3′-diindolylmethane and its parent compound indole-3-carbinol. Cancer Detect.Prev. 2004;28:72–79. doi: 10.1016/j.cdp.2003.11.006. [DOI] [PubMed] [Google Scholar]
- [14].Parkin DR, Lu Y, Bliss RL, Malejka-Giganti D. Inhibitory effects of a dietary phytochemical 3,3′-diindolylmethane on the phenobarbital-induced hepatic CYP mRNA expression and CYP-catalyzed reactions in female rats. Food Chem.Toxicol. 2008;46:2451–2458. doi: 10.1016/j.fct.2008.03.029. [DOI] [PubMed] [Google Scholar]
- [15].Jellinck PH, Makin HL, Sepkovic DW, Bradlow HL. Influence of indole carbinols and growth hormone on the metabolism of 4-androstenedione by rat liver microsomes. J.Steroid Biochem.Mol.Biol. 1993;46:791–798. doi: 10.1016/0960-0760(93)90320-v. [DOI] [PubMed] [Google Scholar]
- [16].Vivar OI, Saunier EF, Leitman DC, Firestone GL, Bjeldanes LF. Selective Activation of Estrogen Receptor-{beta} Target Genes by 3,3′-Diindolylmethane. Endocrinology. 2010 doi: 10.1210/en.2009-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li Y, Li X, Sarkar FH. Gene expression profiles of I3C- and DIM-treated PC3 human prostate cancer cells determined by cDNA microarray analysis. J.Nutr. 2003;133:1011–1019. doi: 10.1093/jn/133.4.1011. [DOI] [PubMed] [Google Scholar]
- [18].Bonnesen C, Eggleston IM, Hayes JD. Dietary indoles and isothiocyanates that are generated from cruciferous vegetables can both stimulate apoptosis and confer protection against DNA damage in human colon cell lines. Cancer Res. 2001;61:6120–6130. [PubMed] [Google Scholar]
- [19].Wortelboer HM, de Kruif CA, van Iersel AA, Falke HE, Noordhoek J, Blaauboer BJ. Acid reaction products of indole-3-carbinol and their effects on cytochrome P450 and phase II enzymes in rat and monkey hepatocytes. Biochem.Pharmacol. 1992;43:1439–1447. doi: 10.1016/0006-2952(92)90200-3. [DOI] [PubMed] [Google Scholar]
- [20].Choi HJ, Lim d.Y., Park JH. Induction of G1 and G2/M cell cycle arrests by the dietary compound 3,3′-diindolylmethane in HT-29 human colon cancer cells. BMC.Gastroenterol. 2009;9:39. doi: 10.1186/1471-230X-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tadi K, Chang Y, Ashok BT, Chen Y, Moscatello A, Schaefer SD, Schantz SP, Policastro AJ, Geliebter J, Tiwari RK. 3,3′-Diindolylmethane, a cruciferous vegetable derived synthetic anti-proliferative compound in thyroid disease. Biochem.Biophys.Res.Commun. 2005;337:1019–1025. doi: 10.1016/j.bbrc.2005.09.143. [DOI] [PubMed] [Google Scholar]
- [22].Chang X, Firestone GL, Bjeldanes LF. Inhibition of growth factor-induced Ras signaling in vascular endothelial cells and angiogenesis by 3,3′-diindolylmethane. Carcinogenesis. 2006;27:541–550. doi: 10.1093/carcin/bgi230. [DOI] [PubMed] [Google Scholar]
- [23].Chang X, Tou JC, Hong C, Kim HA, Riby JE, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane inhibits angiogenesis and the growth of transplantable human breast carcinoma in athymic mice. Carcinogenesis. 2005;26:771–778. doi: 10.1093/carcin/bgi018. [DOI] [PubMed] [Google Scholar]
- [24].Gong Y, Sohn H, Xue L, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane is a novel mitochondrial H(+)-ATP synthase inhibitor that can induce p21(Cip1/Waf1) expression by induction of oxidative stress in human breast cancer cells. Cancer Res. 2006;66:4880–4887. doi: 10.1158/0008-5472.CAN-05-4162. [DOI] [PubMed] [Google Scholar]
- [25].Hong C, Kim HA, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane (DIM) induces a G(1) cell cycle arrest in human breast cancer cells that is accompanied by Sp1-mediated activation of p21(WAF1/CIP1) expression. Carcinogenesis. 2002;23:1297–1305. doi: 10.1093/carcin/23.8.1297. [DOI] [PubMed] [Google Scholar]
- [26].Gartel AL, Serfas MS, Tyner AL. p21--negative regulator of the cell cycle. Proc.Soc.Exp.Biol.Med. 1996;213:138–149. doi: 10.3181/00379727-213-44046. [DOI] [PubMed] [Google Scholar]
- [27].Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- [28].Vivar OI, Lin CL, Firestone GL, Bjeldanes LF. 3,3′-Diindolylmethane induces a G(1) arrest in human prostate cancer cells irrespective of androgen receptor and p53 status. Biochem.Pharmacol. 2009;78:469–476. doi: 10.1016/j.bcp.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chinnakannu K, Chen D, Li Y, Wang Z, Dou QP, Reddy GP, Sarkar FH. Cell cycle-dependent effects of 3,3′-diindolylmethane on proliferation and apoptosis of prostate cancer cells. J.Cell Physiol. 2009;219:94–99. doi: 10.1002/jcp.21650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Garikapaty VP, Ashok BT, Tadi K, Mittelman A, Tiwari RK. 3,3′-Diindolylmethane downregulates pro-survival pathway in hormone independent prostate cancer. Biochem.Biophys.Res.Commun. 2006;340:718–725. doi: 10.1016/j.bbrc.2005.12.059. [DOI] [PubMed] [Google Scholar]
- [31].Rajoria S, Suriano R, Wilson YL, Schantz SP, Moscatello A, Geliebter J, Tiwari RK. 3,3′-diindolylmethane inhibits migration and invasion of human cancer cells through combined suppression of ERK and AKT pathways. Oncol.Rep. 2011;25:491–497. doi: 10.3892/or.2010.1076. [DOI] [PubMed] [Google Scholar]
- [32].Jin Y, Zou X, Feng X. 3,3′-Diindolylmethane negatively regulates Cdc25A and induces a G2/M arrest by modulation of microRNA 21 in human breast cancer cells. Anticancer Drugs. 2010;21:814–822. doi: 10.1097/CAD.0b013e32833e53ea. [DOI] [PubMed] [Google Scholar]
- [33].Kandala PK, Srivastava SK. Activation of checkpoint kinase 2 by 3,3′-diindolylmethane is required for causing G2/M cell cycle arrest in human ovarian cancer cells. Mol.Pharmacol. 2010;78:297–309. doi: 10.1124/mol.110.063750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fan S, Meng Q, Saha T, Sarkar FH, Rosen EM. Low concentrations of diindolylmethane, a metabolite of indole-3-carbinol, protect against oxidative stress in a BRCA1-dependent manner. Cancer Res. 2009;69:6083–6091. doi: 10.1158/0008-5472.CAN-08-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bae I, Fan S, Meng Q, Rih JK, Kim HJ, Kang HJ, Xu J, Goldberg ID, Jaiswal AK, Rosen EM. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004;64:7893–7909. doi: 10.1158/0008-5472.CAN-04-1119. [DOI] [PubMed] [Google Scholar]
- [36].Gopalakrishnan A, Tony Kong AN. Anticarcinogenesis by dietary phytochemicals: cytoprotection by Nrf2 in normal cells and cytotoxicity by modulation of transcription factors NF-kappa B and AP-1 in abnormal cancer cells. Food Chem.Toxicol. 2008;46:1257–1270. doi: 10.1016/j.fct.2007.09.082. [DOI] [PubMed] [Google Scholar]
- [37].Kelner MJ, Bagnell RD, Montoya MA, Estes LA, Forsberg L, Morgenstern R. Structural organization of the microsomal glutathione S-transferase gene (MGST1) on chromosome 12p13.1-13.2. Identification of the correct promoter region and demonstration of transcriptional regulation in response to oxidative stress. J.Biol.Chem. 2000;275:13000–13006. doi: 10.1074/jbc.275.17.13000. [DOI] [PubMed] [Google Scholar]
- [38].Anderton MJ, Manson MM, Verschoyle R, Gescher A, Steward WP, Williams ML, Mager DE. Physiological modeling of formulated and crystalline 3,3′-diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab Dispos. 2004;32:632–638. doi: 10.1124/dmd.32.6.632. [DOI] [PubMed] [Google Scholar]
- [39].Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–5874. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- [40].Sun S, Han J, Ralph WM, Jr., Chandrasekaran A, Liu K, Auborn KJ, Carter TH. Endoplasmic reticulum stress as a correlate of cytotoxicity in human tumor cells exposed to diindolylmethane in vitro. Cell Stress.Chaperones. 2004;9:76–87. doi: 10.1379/CSC-2R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S. 3,3′-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis. 2006;27:717–728. doi: 10.1093/carcin/bgi270. [DOI] [PubMed] [Google Scholar]
- [42].Savino JA, III, Evans JF, Rabinowitz D, Auborn KJ, Carter TH. Multiple, disparate roles for calcium signaling in apoptosis of human prostate and cervical cancer cells exposed to diindolylmethane. Mol.Cancer Ther. 2006;5:556–563. doi: 10.1158/1535-7163.MCT-05-0355. [DOI] [PubMed] [Google Scholar]
- [43].Cheng JS, Shu SS, Kuo CC, Chou CT, Tsai WL, Fang YC, Kuo LN, Yeh JH, Chen WC, Chien JM, Lu T, Pan CC, Cheng HH, Chai KL, Jan CR. Effect of diindolylmethane on Ca(2+) movement and viability in HA59T human hepatoma cells. Arch.Toxicol. 2011 doi: 10.1007/s00204-011-0670-9. [DOI] [PubMed] [Google Scholar]
- [44].Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem.Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- [45].Juge N, Mithen RF, Traka M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol.Life Sci. 2007;64:1105–1127. doi: 10.1007/s00018-007-6484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J.Biol.Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- [47].Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- [48].Xue L, Firestone GL, Bjeldanes LF. DIM stimulates IFNgamma gene expression in human breast cancer cells via the specific activation of JNK and p38 pathways. Oncogene. 2005;24:2343–2353. doi: 10.1038/sj.onc.1208434. [DOI] [PubMed] [Google Scholar]
- [49].Choi KH, Kim HK, Kim JH, Choi ES, Shin JA, Lee SO, Chintharlapalli S, Safe S, Abdelrahim M, Kong G, Choi HS, Jung JY, Cho HT, Cho NP, Cho SD. The p38 MAPK pathway is critical for 5,5′-dibromodiindolylmethane-induced apoptosis to prevent oral squamous carcinoma cells. Eur.J.Cancer Prev. 2010;19:153–159. doi: 10.1097/CEJ.0b013e328333d088. [DOI] [PubMed] [Google Scholar]
- [50].Khwaja FS, Wynne S, Posey I, Djakiew D. 3,3′-diindolylmethane induction of p75NTR-dependent cell death via the p38 mitogen-activated protein kinase pathway in prostate cancer cells. Cancer Prev.Res.(Phila Pa) 2009;2:566–571. doi: 10.1158/1940-6207.CAPR-08-0202. [DOI] [PubMed] [Google Scholar]
- [51].Kelly WK, O’Connor OA, Marks PA. Histone deacetylase inhibitors: from target to clinical trials. Expert.Opin.Investig.Drugs. 2002;11:1695–1713. doi: 10.1517/13543784.11.12.1695. [DOI] [PubMed] [Google Scholar]
- [52].Kelly WK, Richon VM, O’Connor O, Curley T, Gregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin.Cancer Res. 2003;9:3578–3588. [PubMed] [Google Scholar]
- [53].Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, Gregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J.Clin.Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Huang X, Guo B. Adenomatous polyposis coli determines sensitivity to histone deacetylase inhibitor-induced apoptosis in colon cancer cells. Cancer Res. 2006;66:9245–9251. doi: 10.1158/0008-5472.CAN-06-0887. [DOI] [PubMed] [Google Scholar]
- [55].Bhatnagar N, Li X, Chen Y, Zhou X, Garrett SH, Guo B. 3,3′-diindolylmethane enhances the efficacy of butyrate in colon cancer prevention through down-regulation of survivin. Cancer Prev.Res.(Phila Pa) 2009;2:581–589. doi: 10.1158/1940-6207.CAPR-08-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Li Y, Li X, Guo B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70:646–654. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009;14:376–391. doi: 10.1007/s10495-008-0307-5. [DOI] [PubMed] [Google Scholar]
- [58].Chen DZ, Qi M, Auborn KJ, Carter TH. Indole-3-carbinol and diindolylmethane induce apoptosis of human cervical cancer cells and in murine HPV16-transgenic preneoplastic cervical epithelium. J.Nutr. 2001;131:3294–3302. doi: 10.1093/jn/131.12.3294. [DOI] [PubMed] [Google Scholar]
- [59].Li Y, Chinni SR, Sarkar FH. Selective growth regulatory and pro-apoptotic effects of DIM is mediated by AKT and NF-kappaB pathways in prostate cancer cells. Front Biosci. 2005;10:236–243. doi: 10.2741/1523. [DOI] [PubMed] [Google Scholar]
- [60].Banerjee S, Wang Z, Kong D, Sarkar FH. 3,3′-Diindolylmethane enhances chemosensitivity of multiple chemotherapeutic agents in pancreatic cancer. Cancer Res. 2009;69:5592–5600. doi: 10.1158/0008-5472.CAN-09-0838. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [61].Kim EJ, Shin M, Park H, Hong JE, Shin HK, Kim J, Kwon DY, Park JH. Oral administration of 3,3′-diindolylmethane inhibits lung metastasis of 4T1 murine mammary carcinoma cells in BALB/c mice. J.Nutr. 2009;139:2373–2379. doi: 10.3945/jn.109.111864. [DOI] [PubMed] [Google Scholar]
- [62].Ahmad A, Kong D, Wang Z, Sarkar SH, Banerjee S, Sarkar FH. Down-regulation of uPA and uPAR by 3,3′-diindolylmethane contributes to the inhibition of cell growth and migration of breast cancer cells. J.Cell Biochem. 2009;108:916–925. doi: 10.1002/jcb.22323. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [63].Rahman KW, Sarkar FH. Inhibition of nuclear translocation of nuclear factor-{kappa}B contributes to 3,3′-diindolylmethane-induced apoptosis in breast cancer cells. Cancer Res. 2005;65:364–371. [PubMed] [Google Scholar]
- [64].Rahman KW, Li Y, Wang Z, Sarkar SH, Sarkar FH. Gene expression profiling revealed survivin as a target of 3,3′-diindolylmethane-induced cell growth inhibition and apoptosis in breast cancer cells. Cancer Res. 2006;66:4952–4960. doi: 10.1158/0008-5472.CAN-05-3918. [DOI] [PubMed] [Google Scholar]
- [65].Maciejewska D, Rasztawicka M, Wolska I, Anuszewska E, Gruber B. Novel 3,3′-diindolylmethane derivatives: synthesis and cytotoxicity, structural characterization in solid state. Eur.J.Med.Chem. 2009;44:4136–4147. doi: 10.1016/j.ejmech.2009.05.011. [DOI] [PubMed] [Google Scholar]
- [66].Ali S, Banerjee S, Ahmad A, El-Rayes BF, Philip PA, Sarkar FH. Apoptosis-inducing effect of erlotinib is potentiated by 3,3′-diindolylmethane in vitro and in vivo using an orthotopic model of pancreatic cancer. Mol.Cancer Ther. 2008;7:1708–1719. doi: 10.1158/1535-7163.MCT-08-0354. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [67].Gong Y, Firestone GL, Bjeldanes LF. 3,3′-diindolylmethane is a novel topoisomerase IIalpha catalytic inhibitor that induces S-phase retardation and mitotic delay in human hepatoma HepG2 cells. Mol.Pharmacol. 2006;69:1320–1327. doi: 10.1124/mol.105.018978. [DOI] [PubMed] [Google Scholar]
- [68].Contractor R, Samudio IJ, Estrov Z, Harris D, McCubrey JA, Safe SH, Andreeff M, Konopleva M. A novel ring-substituted diindolylmethane,1,1-bis[3′-(5-methoxyindolyl)]-1-(p-t-butylphenyl) methane, inhibits extracellular signal-regulated kinase activation and induces apoptosis in acute myelogenous leukemia. Cancer Res. 2005;65:2890–2898. doi: 10.1158/0008-5472.CAN-04-3781. [DOI] [PubMed] [Google Scholar]
- [69].Ichite N, Chougule MB, Jackson T, Fulzele SV, Safe S, Singh M. Enhancement of docetaxel anticancer activity by a novel diindolylmethane compound in human non-small cell lung cancer. Clin.Cancer Res. 2009;15:543–552. doi: 10.1158/1078-0432.CCR-08-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nachshon-Kedmi M, Yannai S, Haj A, Fares FA. Indole-3-carbinol and 3,3′-diindolylmethane induce apoptosis in human prostate cancer cells. Food Chem.Toxicol. 2003;41:745–752. doi: 10.1016/s0278-6915(03)00004-8. [DOI] [PubMed] [Google Scholar]
- [71].Nachshon-Kedmi M, Yannai S, Fares FA. Induction of apoptosis in human prostate cancer cell line, PC3, by 3,3′-diindolylmethane through the mitochondrial pathway. Br.J.Cancer. 2004;91:1358–1363. doi: 10.1038/sj.bjc.6602145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chinnakannu K, Chen D, Li Y, Wang Z, Dou QP, Reddy GP, Sarkar FH. Cell cycle-dependent effects of 3,3′-diindolylmethane on proliferation and apoptosis of prostate cancer cells. J.Cell Physiol. 2009;219:94–99. doi: 10.1002/jcp.21650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ge X, Yannai S, Rennert G, Gruener N, Fares FA. 3,3′-Diindolylmethane induces apoptosis in human cancer cells. Biochem.Biophys.Res.Commun. 1996;228:153–158. doi: 10.1006/bbrc.1996.1631. [DOI] [PubMed] [Google Scholar]
- [74].Wang Z, Yu BW, Rahman KM, Ahmad F, Sarkar FH. Induction of growth arrest and apoptosis in human breast cancer cells by 3,3-diindolylmethane is associated with induction and nuclear localization of p27kip. Mol.Cancer Ther. 2008;7:341–349. doi: 10.1158/1535-7163.MCT-07-0476. [DOI] [PubMed] [Google Scholar]
- [75].Hong C, Firestone GL, Bjeldanes LF. Bcl-2 family-mediated apoptotic effects of 3,3′-diindolylmethane (DIM) in human breast cancer cells. Biochem.Pharmacol. 2002;63:1085–1097. doi: 10.1016/s0006-2952(02)00856-0. [DOI] [PubMed] [Google Scholar]
- [76].Moiseeva EP, Almeida GM, Jones GD, Manson MM. Extended treatment with physiologic concentrations of dietary phytochemicals results in altered gene expression, reduced growth, and apoptosis of cancer cells. Mol.Cancer Ther. 2007;6:3071–3079. doi: 10.1158/1535-7163.MCT-07-0117. [DOI] [PubMed] [Google Scholar]
- [77].Kim EJ, Park SY, Shin HK, Kwon DY, Surh YJ, Park JH. Activation of caspase-8 contributes to 3,3′-Diindolylmethane-induced apoptosis in colon cancer cells. J.Nutr. 2007;137:31–36. doi: 10.1093/jn/137.1.31. [DOI] [PubMed] [Google Scholar]
- [78].Azmi AS, Ahmad A, Banerjee S, Rangnekar VM, Mohammad RM, Sarkar FH. Chemoprevention of pancreatic cancer: characterization of Par-4 and its modulation by 3,3′ diindolylmethane (DIM) Pharm.Res. 2008;25:2117–2124. doi: 10.1007/s11095-008-9581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lee SH, Kim JS, Yamaguchi K, Eling TE, Baek SJ. Indole-3-carbinol and 3,3′-diindolylmethane induce expression of NAG-1 in a p53-independent manner. Biochem.Biophys.Res.Commun. 2005;328:63–69. doi: 10.1016/j.bbrc.2004.12.138. [DOI] [PubMed] [Google Scholar]
- [80].Roy A, Ganguly A, BoseDasgupta S, Das BB, Pal C, Jaisankar P, Majumder HK. Mitochondria-dependent reactive oxygen species-mediated programmed cell death induced by 3,3′-diindolylmethane through inhibition of F0F1-ATP synthase in unicellular protozoan parasite Leishmania donovani. Mol.Pharmacol. 2008;74:1292–1307. doi: 10.1124/mol.108.050161. [DOI] [PubMed] [Google Scholar]
- [81].Li Y, Chinni SR, Sarkar FH. Selective growth regulatory and pro-apoptotic effects of DIM is mediated by AKT and NF-kappaB pathways in prostate cancer cells. Front Biosci. 2005;10:236–243. doi: 10.2741/1523. [DOI] [PubMed] [Google Scholar]
- [82].Rahman KM, Banerjee S, Ali S, Ahmad A, Wang Z, Kong D, Sakr WA. 3,3′-Diindolylmethane enhances taxotere-induced apoptosis in hormone-refractory prostate cancer cells through survivin down-regulation. Cancer Res. 2009;69:4468–4475. doi: 10.1158/0008-5472.CAN-08-4423. [DOI] [PubMed] [Google Scholar]
- [83].Rahman KM, Ali S, Aboukameel A, Sarkar SH, Wang Z, Philip PA, Sakr WA, Raz A. Inactivation of NF-kappaB by 3,3′-diindolylmethane contributes to increased apoptosis induced by chemotherapeutic agent in breast cancer cells. Mol.Cancer Ther. 2007;6:2757–2765. doi: 10.1158/1535-7163.MCT-07-0336. [DOI] [PubMed] [Google Scholar]
- [84].Ali S, Varghese L, Pereira L, Tulunay-Ugur OE, Kucuk O, Carey TE, Wolf GT, Sarkar FH. Sensitization of squamous cell carcinoma to cisplatin induced killing by natural agents. Cancer Lett. 2009;278:201–209. doi: 10.1016/j.canlet.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [85].Hankinson O. The aryl hydrocarbon receptor complex. Annu.Rev.Pharmacol.Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- [86].Perdew GH. Association of the Ah receptor with the 90-kDa heat shock protein. J.Biol.Chem. 1988;263:13802–13805. [PubMed] [Google Scholar]
- [87].Kazlauskas A, Sundstrom S, Poellinger L, Pongratz I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol.Cell Biol. 2001;21:2594–2607. doi: 10.1128/MCB.21.7.2594-2607.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Carver LA, Bradfield CA. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J.Biol.Chem. 1997;272:11452–11456. doi: 10.1074/jbc.272.17.11452. [DOI] [PubMed] [Google Scholar]
- [89].Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu.Rev.Pharmacol.Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- [90].Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem.Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- [91].Whitlock JP., Jr. Induction of cytochrome P4501A1. Annu.Rev.Pharmacol.Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- [92].Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3′-diindolylmethane in breast cancer cells. J.Nutr. 2009;139:26–32. doi: 10.3945/jn.108.099259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Hestermann EV, Brown M. Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Mol.Cell Biol. 2003;23:7920–7925. doi: 10.1128/MCB.23.21.7920-7925.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Jellinck PH, Forkert PG, Riddick DS, Okey AB, Michnovicz JJ, Bradlow HL. Ah receptor binding properties of indole carbinols and induction of hepatic estradiol hydroxylation. Biochem.Pharmacol. 1993;45:1129–1136. doi: 10.1016/0006-2952(93)90258-x. [DOI] [PubMed] [Google Scholar]
- [95].McDougal A, Gupta MS, Morrow D, Ramamoorthy K, Lee JE, Safe SH. Methyl-substituted diindolylmethanes as inhibitors of estrogen-induced growth of T47D cells and mammary tumors in rats. Breast Cancer Res.Treat. 2001;66:147–157. doi: 10.1023/a:1010608000074. [DOI] [PubMed] [Google Scholar]
- [96].Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- [97].Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- [98].Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat.Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- [99].Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC., Jr. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc.Natl.Acad.Sci.U.S.A. 1998;95:7772–7777. doi: 10.1073/pnas.95.13.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- [101].Kucab JE, Lee C, Chen CS, Zhu J, Gilks CB, Cheang M, Huntsman D, Yorida E, Emerman J, Pollak M, Dunn SE. Celecoxib analogues disrupt Akt signaling, which is commonly activated in primary breast tumours. Breast Cancer Res. 2005;7:R796–R807. doi: 10.1186/bcr1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Bhuiyan MM, Li Y, Banerjee S, Ahmed F, Wang Z, Ali S, Sarkar FH. Down-regulation of androgen receptor by 3,3′-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in both hormone-sensitive LNCaP and insensitive C4-2B prostate cancer cells. Cancer Res. 2006;66:10064–10072. doi: 10.1158/0008-5472.CAN-06-2011. [DOI] [PubMed] [Google Scholar]
- [103].Chen Y, Xu J, Jhala N, Pawar P, Zhu ZB, Ma L, Byon CH, McDonald JM. Fas-mediated apoptosis in cholangiocarcinoma cells is enhanced by 3,3′-diindolylmethane through inhibition of AKT signaling and FLICE-like inhibitory protein. Am.J.Pathol. 2006;169:1833–1842. doi: 10.2353/ajpath.2006.060234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kong D, Li Y, Wang Z, Banerjee S, Sarkar FH. Inhibition of angiogenesis and invasion by 3,3′-diindolylmethane is mediated by the nuclear factor-kappaB downstream target genes MMP-9 and uPA that regulated bioavailability of vascular endothelial growth factor in prostate cancer. Cancer Res. 2007;67:3310–3319. doi: 10.1158/0008-5472.CAN-06-4277. [DOI] [PubMed] [Google Scholar]
- [105].Kong D, Banerjee S, Huang W, Li Y, Wang Z, Kim HR, Sarkar FH. Mammalian target of rapamycin repression by 3,3′-diindolylmethane inhibits invasion and angiogenesis in platelet-derived growth factor-D-overexpressing PC3 cells. Cancer Res. 2008;68:1927–1934. doi: 10.1158/0008-5472.CAN-07-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat.Rev.Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- [107].Li Y, Wang Z, Kong D, Murthy S, Dou QP, Sheng S, Reddy GP, Sarkar FH. Regulation of FOXO3a/beta-catenin/GSK-3beta signaling by 3,3′-diindolylmethane contributes to inhibition of cell proliferation and induction of apoptosis in prostate cancer cells. J.Biol.Chem. 2007;282:21542–21550. doi: 10.1074/jbc.M701978200. [DOI] [PubMed] [Google Scholar]
- [108].Narayanan R, Yepuru M, Szafran AT, Szwarc M, Bohl CE, Young NL, Miller DD, Mancini MA, Dalton JT. Discovery and mechanistic characterization of a novel selective nuclear androgen receptor exporter for the treatment of prostate cancer. Cancer Res. 2010;70:842–851. doi: 10.1158/0008-5472.CAN-09-3206. [DOI] [PubMed] [Google Scholar]
- [109].Li Y, Chinni SR, Sarkar FH. Selective growth regulatory and pro-apoptotic effects of DIM is mediated by AKT and NF-kappaB pathways in prostate cancer cells. Front Biosci. 2005;10:236–243. doi: 10.2741/1523. [DOI] [PubMed] [Google Scholar]
- [110].Mulvey L, Chandrasekaran A, Liu K, Lombardi S, Wang XP, Auborn KJ, Goodwin L. Interplay of genes regulated by estrogen and diindolylmethane in breast cancer cell lines. Mol.Med. 2007;13:69–78. doi: 10.2119/2006-00038.Mulvey. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Carter TH, Liu K, Ralph W, Jr., Chen D, Qi M, Fan S, Yuan F, Rosen EM, Auborn KJ. Diindolylmethane alters gene expression in human keratinocytes in vitro. J.Nutr. 2002;132:3314–3324. doi: 10.1093/jn/132.11.3314. [DOI] [PubMed] [Google Scholar]
- [112].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev.Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- [114].Casalini P, Iorio MV. MicroRNAs and future therapeutic applications in cancer. J.BUON. 2009;14(Suppl 1):S17–S22. [PubMed] [Google Scholar]
- [115].Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J.Clin.Oncol. 2009;27:5848–5856. doi: 10.1200/JCO.2009.24.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- [117].Li Y, VandenBoom TG, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Kunimasa K, Kobayashi T, Kaji K, Ohta T. Antiangiogenic effects of indole-3-carbinol and 3,3′-diindolylmethane are associated with their differential regulation of ERK1/2 and Akt in tube-forming HUVEC. J.Nutr. 2010;140:1–6. doi: 10.3945/jn.109.112359. [DOI] [PubMed] [Google Scholar]
- [119].Kong D, Banerjee S, Huang W, Li Y, Wang Z, Kim HR, Sarkar FH. Mammalian target of rapamycin repression by 3,3′-diindolylmethane inhibits invasion and angiogenesis in platelet-derived growth factor-D-overexpressing PC3 cells. Cancer Res. 2008;68:1927–1934. doi: 10.1158/0008-5472.CAN-07-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Riby JE, Firestone GL, Bjeldanes LF. 3,3′-diindolylmethane reduces levels of HIF-1alpha and HIF-1 activity in hypoxic cultured human cancer cells. Biochem.Pharmacol. 2008;75:1858–1867. doi: 10.1016/j.bcp.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ahmad A, Kong D, Sarkar SH, Wang Z, Banerjee S, Sarkar FH. Inactivation of uPA and its receptor uPAR by 3,3′-diindolylmethane (DIM) leads to the inhibition of prostate cancer cell growth and migration. J.Cell Biochem. 2009;107:516–527. doi: 10.1002/jcb.22152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Rajoria S, Suriano R, George A, Shanmugam A, Schantz SP, Geliebter J, Tiwari RK. Estrogen induced metastatic modulators MMP-2 and MMP-9 are targets of 3,3′-diindolylmethane in thyroid cancer. PLoS.One. 2011;6:e15879. doi: 10.1371/journal.pone.0015879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Rahimi M, Huang KL, Tang CK. 3,3′-Diindolylmethane (DIM) inhibits the growth and invasion of drug-resistant human cancer cells expressing EGFR mutants. Cancer Lett. 2010;295:59–68. doi: 10.1016/j.canlet.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Dong L, Xia S, Gao F, Zhang D, Chen J, Zhang J. 3,3′-Diindolylmethane attenuates experimental arthritis and osteoclastogenesis. Biochem.Pharmacol. 2010;79:715–721. doi: 10.1016/j.bcp.2009.10.010. [DOI] [PubMed] [Google Scholar]
- [125].Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- [126].Li Y, VandenBoom TG, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Ali S, Banerjee S, Ahmad A, El-Rayes BF, Philip PA, Sarkar FH. Apoptosis-inducing effect of erlotinib is potentiated by 3,3′-diindolylmethane in vitro and in vivo using an orthotopic model of pancreatic cancer. Mol.Cancer Ther. 2008;7:1708–1719. doi: 10.1158/1535-7163.MCT-08-0354. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [128].Ali S, Banerjee S, Schaffert JM, El-Rayes BF, Philip PA, Sarkar FH. Concurrent inhibition of NF-kappaB, cyclooxygenase-2, and epidermal growth factor receptor leads to greater anti-tumor activity in pancreatic cancer. J.Cell Biochem. 2010 doi: 10.1002/jcb.22523. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [129].McGuire KP, Ngoubilly N, Neavyn M, Lanza-Jacoby S. 3,3′-diindolylmethane and paclitaxel act synergistically to promote apoptosis in HER2/Neu human breast cancer cells. J.Surg.Res. 2006;132:208–213. doi: 10.1016/j.jss.2006.02.008. [DOI] [PubMed] [Google Scholar]
- [130].Ahmad A, Ali S, Wang Z, Ali AS, Sethi S, Sakr WA, Raz A, Rahman KW. 3, 3′-diindolylmethane enhances taxotere-induced growth inhibition of breast cancer cells through down-regulation of FoxM1. Int.J.Cancer. 2010 doi: 10.1002/ijc.25839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Zhang S, Shen HM, Ong CN. Down-regulation of c-FLIP contributes to the sensitization effect of 3,3′-diindolylmethane on TRAIL-induced apoptosis in cancer cells. Mol.Cancer Ther. 2005;4:1972–1981. doi: 10.1158/1535-7163.MCT-05-0249. [DOI] [PubMed] [Google Scholar]
- [132].Pappa G, Strathmann J, Lowinger M, Bartsch H, Gerhauser C. Quantitative combination effects between sulforaphane and 3,3′-diindolylmethane on proliferation of human colon cancer cells in vitro. Carcinogenesis. 2007;28:1471–1477. doi: 10.1093/carcin/bgm044. [DOI] [PubMed] [Google Scholar]
- [133].Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv.Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- [134].Jacobs IC, Zeligs MA. Facilitated absorption of a hydrophobic dietary ingredient. Proc Int Symp Control Rel Bioact Mater. 1999;26:958–959. [Google Scholar]
- [135].Jacobs IC, Zeligs MA. New formulation strategies for bioavailability enhancement of two poorly absorbed phytonutrient supplements: diindolylmethane and yohimbine bark extract. Proc Int Symp Control Rel Bioact Mater. 2000;27:1324–1325. [Google Scholar]
- [136].Reed GA, Sunega JM, Sullivan DK, Gray JC, Mayo MS, Crowell JA, Hurwitz A. Single-dose pharmacokinetics and tolerability of absorption-enhanced 3,3′-diindolylmethane in healthy subjects. Cancer Epidemiol.Biomarkers Prev. 2008;17:2619–2624. doi: 10.1158/1055-9965.EPI-08-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Nestorov I. Whole body pharmacokinetic models. Clin.Pharmacokinet. 2003;42:883–908. doi: 10.2165/00003088-200342100-00002. [DOI] [PubMed] [Google Scholar]
- [138].Sepkovic DW, Bradlow HL, Bell M. Quantitative determination of 3,3′-diindolylmethane in urine of individuals receiving indole-3-carbinol. Nutr.Cancer. 2001;41:57–63. doi: 10.1080/01635581.2001.9680612. [DOI] [PubMed] [Google Scholar]
- [139].Chen I, McDougal A, Wang F, Safe S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis. 1998;19:1631–1639. doi: 10.1093/carcin/19.9.1631. [DOI] [PubMed] [Google Scholar]
- [140].Nachshon-Kedmi M, Fares FA, Yannai S. Therapeutic activity of 3,3′-diindolylmethane on prostate cancer in an in vivo model. Prostate. 2004;61:153–160. doi: 10.1002/pros.20092. [DOI] [PubMed] [Google Scholar]
- [141].Cho HJ, Park SY, Kim EJ, Kim JK, Park JH. 3,3′-Diindolylmethane inhibits prostate cancer development in the transgenic adenocarcinoma mouse prostate model. Mol.Carcinog. 2011;50:100–112. doi: 10.1002/mc.20698. [DOI] [PubMed] [Google Scholar]
- [142].Sepkovic DW, Stein J, Carlisle AD, Ksieski HB, Auborn K, Bradlow HL. Diindolylmethane inhibits cervical dysplasia, alters estrogen metabolism, and enhances immune response in the K14-HPV16 transgenic mouse model. Cancer Epidemiol.Biomarkers Prev. 2009;18:2957–2964. doi: 10.1158/1055-9965.EPI-09-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Sepkovic DW, Stein J, Carlisle AD, Ksieski HB, Auborn K, Raucci L, Nyirenda T, Bradlow HL. Results from a dose response study using 3, 3′-diindolylmethane in the K14-HPV16 transgenic mouse model: Cervical histology. Cancer Prev.Res.(Phila) 2011 doi: 10.1158/1940-6207.CAPR-10-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Cho SD, Lei P, Abdelrahim M, Yoon K, Liu S, Guo J, Papineni S, Chintharlapalli S, Safe S. 1,1-bis(3′-indolyl)-1-(p-methoxyphenyl)methane activates Nur77-independent proapoptotic responses in colon cancer cells. Mol.Carcinog. 2008;47:252–263. doi: 10.1002/mc.20378. [DOI] [PubMed] [Google Scholar]
- [145].Cho SD, Chintharlapalli S, Abdelrahim M, Papineni S, Liu S, Guo J, Lei P, Abudayyeh A, Safe S. 5,5′-Dibromo-bis(3′-indolyl)methane induces Kruppel-like factor 4 and p21 in colon cancer cells. Mol.Cancer Ther. 2008;7:2109–2120. doi: 10.1158/1535-7163.MCT-07-2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Lee SO, Li X, Khan S, Safe S. Targeting NR4A1 (TR3) in cancer cells and tumors. Expert.Opin.Ther.Targets. 2011;15:195–206. doi: 10.1517/14728222.2011.547481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Yoon K, Lee SO, Cho SD, Kim K, Khan S, Safe S. Activation of Nuclear TR3 (Nr4a1) by A Diindolylmethane Analog Induces Apoptosis and Proapoptotic Genes in Pancreatic Cancer Cells and Tumors. Carcinogenesis. 2011 doi: 10.1093/carcin/bgr040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Dalessandri KM, Firestone GL, Fitch MD, Bradlow HL, Bjeldanes LF. Pilot study: effect of 3,3′-diindolylmethane supplements on urinary hormone metabolites in postmenopausal women with a history of early-stage breast cancer. Nutr.Cancer. 2004;50:161–167. doi: 10.1207/s15327914nc5002_5. [DOI] [PubMed] [Google Scholar]
- [149].Sanderson JT, Slobbe L, Lansbergen GW, Safe S, van den BM. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and diindolylmethanes differentially induce cytochrome P450 1A1, 1B1, and 19 in H295R human adrenocortical carcinoma cells. Toxicol.Sci. 2001;61:40–48. doi: 10.1093/toxsci/61.1.40. [DOI] [PubMed] [Google Scholar]
- [150].Heath EI, Heilbrun lk, Vaishampayan UN, Li J, Harper F, Zeligs M, Pemberton P, Sarkar FH. A phase-I dose escalation study of oral BR-DIM (BioResponse 3,3′-Diindolylmethane) in castrate-resistant,non-metastatic,PSA relapse prostate cancer patients. ASCO Meeting Genitourinary Cancer Symposium; 2009. [PMC free article] [PubMed] [Google Scholar]
- [151].Del PG, Gudipudi DK, Montemarano N, Restivo AM, Malanowska-Stega J, Arslan AA. Oral diindolylmethane (DIM): Pilot evaluation of a nonsurgical treatment for cervical dysplasia. Gynecol.Oncol. 2010;116:464–467. doi: 10.1016/j.ygyno.2009.10.060. [DOI] [PubMed] [Google Scholar]
- [152].Auborn KJ. Therapy for recurrent respiratory papillomatosis. Antivir.Ther. 2002;7:1–9. [PubMed] [Google Scholar]