

Abstract
Pre-existing serum antibodies have long been associated with graft loss in transplant candidates. While most studies have focused on HLA-specific antibodies, the contribution of non-HLA-reactive antibodies has been largely overlooked. We have recently characterized monoclonal antibodies secreted by B cell clones derived from kidney allograft recipients with rejection that selectively bind to apoptotic cells. Here, we assessed the presence of such antibodies in pre-transplant serum from 300 kidney transplant recipients and examined their contribution to the graft outcomes. Kaplan-Meier survival analysis revealed that patients with high pre-transplant IgG reactivity to apoptotic cells had a significantly increased rate of late graft loss. The effect was only apparent after approximately 1 year post-transplant. Moreover, the association between pre-transplant IgG reactivity to apoptotic cells and graft loss was still significant after excluding patients with high reactivity to HLA. This reactivity was almost exclusively mediated by IgG1 and IgG3 with complement fixing and activating properties. Overall, our findings support the view that IgG reactivity to apoptotic cells contribute to pre-sensitization. Taking these antibodies into consideration alongside anti-HLA antibodies during candidate evaluation would likely improve the transplant risk assessment.
Keywords: Anti-apoptotic cell antibodies, sensitization, apoptotic cells, kidney transplantation, graft loss, complement
Introduction
Pre-sensitization has been a major limitation to solid organ transplantation for decades. Candidate recipients with pre-existing antibodies to potential donor grafts have a higher risk of rejection and eventually graft loss (1–6). It is commonly accepted that these antibodies are either naturally pre-formed or had developed after exposure to allogeneic antigens occurring during pregnancy, blood transfusion or previous allografts. In ABO compatible donor-recipient pairs, sensitizing antibodies are primarily IgG reactive to human leukocyte antigen (HLA). However, a number of observations suggest that non-HLA reactive antibodies also contribute to pre-sensitization and may influence the overall graft outcome (7–9). Cases of early humoral rejection in the absence of detectable donor-specific antibodies (DSA) have also been reported (10, 11). In a landmark collaborative transplant study, Opelz and colleagues revealed the association between high panel reactive antibodies (PRA) before transplantation and late graft loss in recipients of kidney transplants from HLA identical siblings (12). Since the donors and recipients shared both HLA loci, the PRA impact on graft survival could not be attributed to donor specific HLA antibodies. Additional studies support a contribution of non-HLA antibodies to pre-sensitization (7, 13). More specifically, serum IgG reactivity to autoantigens such as cardiac myosin, vimentin, collagen, oxidized lipids and LG3 has been associated with increased rejection rates and reduced graft survival (14–23).
Natural antibodies are distinct antibodies that develop without any evidence of immunization (24). An important characteristic of natural antibodies is their capacity to react to apoptotic cells (25–28). These antibodies are primarily IgM, although IgG have also been detected in various pathological conditions, indicating class switch recombination (CSR) of the producing B cells. Despite their essential role in health and diseases, anti-apoptotic cell antibodies have seldom been examined in the context of human transplantation.
In previous studies, we isolated a number of B cell clones secreting antibodies reactive to apoptotic cells from a kidney transplant recipient with antibody mediated rejection (AMR) (29). More generally, we also observed elevated IgG reactivity to apoptotic cells in kidney transplant recipients experiencing AMR compared to patients with stable graft function (30). Collectively, these findings alluded to a contribution of anti-apoptotic cell IgG to the pathophysiology of graft rejection. In this study, we examined the contribution of serum IgG reacting to apoptotic cells to pre-sensitization and graft outcome on a large cohort of patients who received a kidney transplant at Massachusetts General Hospital (MGH) between 1999 and 2007.
Materials and Methods
Patient characteristics and sample collection
The collection of all specimens used in this study was approved by MGH internal review board. The patient group consisted of 300 non-consecutive kidney transplant recipients who received a kidney transplant at MGH between May 1999 and July 2007 and whose pre-transplant serum specimens were available. Patients with pre-transplant DSA were excluded in this study. All serum specimens were collected prior to transplantation as part of the patients’ standard clinical care. Serum samples collected from 20 healthy subjects were used as control in this study. The baseline characteristics of all patients included in this study are summarized as Table 1. Fourty six of the 300 patients included in our study lost their grafts and returned to dialysis. For 42 of these patients, the cause of graft loss was based on pathological changes seen in biopsy specimens. For the remaining 4 patients, the cause of graft loss was determined by clinical criteria and serological tests.
Table 1.
Patient characteristics
| Parameters | Values |
|---|---|
| Age in year (mean±SD) | 49.0±14.4 |
| Sampling time pre-tx, month (mean±SD) | 3.7±2.5 |
| Follow-up, month (mean±SD) | 81.2 ±35.3 |
| Gender, n (%) | |
| Male | 192 (64.0%) |
| Female | 108 (36.0%) |
| Race, n (%) | |
| Caucasian | 239 (79.7%) |
| African | 29 (9.6%) |
| Asian | 14 (4.7%) |
| Hispanic | 18 (6.0%) |
| No. of transplant, n (%) | |
| First | 259 (86.3%) |
| Second | 35 (11.7 %) |
| Third | 6 (2.0%) |
| Donation, n (%) | |
| Deceased | 147 (49.0%) |
| Living related | 84 (28.0%) |
| Living unrelated | 69 (23.0%) |
| Transfusion pre-Tx, (n %) | |
| Yes | 94 (31.3%) |
| No | 206 (68.7%) |
| Induction therapy, (n %) | |
| Yes | 84 (27.9%) |
| No | 216 (72.1%) |
| Cause of end-stage renal disease, n (%) | |
| Polycystic kidney | 36 (12.0%) |
| Focal Segmental Glomerulosclerosis (Primary) | 26 (8.7%) |
| IgA nephropathy | 39 (13.0%) |
| Type I diabetes mellitus | 28 (9.3%) |
| Hypertension | 21 (7.0%) |
| Obstructive/reflux uropathy/anatomical issues | 22 (7.3%) |
| Systemic lupus erythematosus | 6 (2.0%) |
| Immune complex disease | 29 (9.7%) |
| Congenital/hereditary disease | 18 (6.0%) |
| Interstitial nephritis | 23 (7.7%) |
| Focal Segmental Glomerulosclerosis (Secondary) | 12 (4.0%) |
| Type II diabetes mellitus | 25 (8.3%) |
| Others | 4 (1.3%) |
| Unknow | 11 (3.7%) |
Serum IgG purification
Serum IgG were purified from patients specimens using the Melon Gel IgG Purification Kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions. Briefly, serum samples were diluted 1:10 in a dilution buffer and incubated with a resin that retains non-IgG immunoglobulin as well as other abundant serum proteins.
Assessment of reactivity to apoptotic cells
We used flow cytometry to assess the reactivity of serum IgM, IgG and purified IgG to apoptotic Jurkat cells in samples collected pre-transplant from 300 patients who received a kidney transplant at MGH between 1999 and 2007 as well as 20 control healthy subjects. In these experiments, human Jurkat T cell leukemia cells were cultured overnight with 200ng/ml of anti-FAS antibody (clone CH11, Millipore, Billerica, MA) or exposed to UV light (240×10−3 J) to induce apoptosis using a UV stratalinker 2400 (Stratagene, Santa Clara, CA). Then 1×106 apoptotic Jurkat T cells were incubated for 30 minutes at 37°C with 100μl serum IgM and IgG samples diluted 1:5 in PBS or 60μl purified IgG samples diluted 1:2 in PBS. After two washes in 3ml PBS at 4°C, samples were incubated with FITC-conjugated anti-IgM or anti-IgG F(ab′)2 secondary antibodies, respectively (Invitrogen, Camarillo, CA) for 30 minutes at 4°C. After two additional washes in 3ml PBS at 4°C, cells were acquired using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) after gating on apoptotic cells. To avoid inter-experiment variations, all samples were assessed at the same time in the same experiment using the same instrument settings. Results are reported as log2 values of the mean fluorescence intensity (MFI) of positive cells. A titration experiments carried out with purified IgG samples from 4 patients with highest IgG reactivity to apoptotic cells is reported in Figure S1.
Generation of HLA class I negative Jurkat cells
Supernatants containing packaged pLKO.1 lentiviral vector containing a shRNA specific for the human β2-microglobulin was obtained from Dr. Roberto Bellucci (Dana-Farber Cancer Institute). Preparation of the lentiviral vector is described elsewhere (31). Jurkat cells were transduced with virus supernatants and polybrene at 8μg/ml (Millipore) two times and selected with puromycin for 24 hours after the second transduction. HLA class I negative cells were then purified from transduced cells by sorting. This operation was repeated three times until homogeneous HLA class I negative was obtained.
Quantification of IgM and IgG concentration
Serum IgM and IgG in patients and healthy subjects were quantified using a Cytometric Bead Array kit (BD Biosciences, San Jose, CA). Briefly, capture beads were incubated with 50μl samples diluted at 1:2,500 for IgM, 1:100,000 for IgG and 1:10,000 for purified IgG with dilution buffer for 1h at room temperature. After 1 wash in 1ml wash buffer, 50μl mixed PE detection reagent was added in each sample and incubated for 2h at room temperature. After 1 wash in 1 ml wash buffer, beads were resuspended in 300μl wash buffer and acquired using an Accuri C6 flow cytometer (BD Biosciences). Immunoglobulin concentrations were calculated using FCAP Array v3.0 software (BD Biosciences). IgG subclasses (IgG1 ~ IgG4) in patients before and after IgG purification were quantified by ELISA using Human IgG Subclass Profile Kit (Invitrogen, Camarillo, CA) according to the manufacturer’s instruction. Briefly, 50μl serum samples diluted at 1:2500 were incubated for 30 minutes at room temperature with anti-human IgG1~ IgG4 subclasses specific antibodies, respectively. Plates were washed 4 times in wash buffer, peroxidase anti-human IgG solution was added and incubated for 30 minutes at room temperature, after 4 washes, developed using 3, 3′, 5, 5′-tetramethylbenzidine. Optical density was measured at 450 nm.
Detection of reactivity to HLA and MICA molecules
The reactivity of patients’ serum to HLA Class I, HLA Class II, or MICA was assessed using beads coated with mixed HLA molecules (LABScreen Mixed, One Lambda, Los Angeles, CA). Antibodies reactive to beads were detected with an anti-IgG (One Lambda) PE-conjugated secondary antibody on a Luminex 200 apparatus (Luminex, Austin, TX). A MFI of 1,000 was arbitrarily used as a cutoff value.
Serum IgG subclass reactivity to apoptotic cells
Apoptotic Jurkat (1×106 cells) cells were incubated for 30 minutes at 37°C with 60μl IgG purified from the 50 most reactive patient serum specimens diluted 1:2. After two washes in 3ml PBS at 4°C, samples were incubated with PE-conjugated anti-IgG1, IgG2, IgG3, IgG4 secondary antibodies for each patient, respectively (Clone 4E3, HP6002, HP6050, HP6025, Southern Biotech, Birmingham, AL) at 4°C for 30 minutes. After two washes, cells were acquired using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) after gating on apoptotic cells.
C4d binding assay
Apoptotic Jurkat cells (0.5×106 cells) were incubated for 30 minutes at 37ºC with 60μl purified serum IgG (see above) diluted 1:2. Human serum from a healthy subject diluted 1:100 in HBSS was then added as a source of complement and incubated for 15 minutes at 37ºC. After 2 washes in PBS, cells were incubated for 30 minutes at 4ºC with an anti-C4d antibody (Quidel, San Diego, CA), washed twice in PBS, and then incubated for 30 minutes at 4ºC with a FITC-conjugated anti-mouse IgG secondary antibody (BD Biosciences). After 2 final washes at 4ºC, C4d binding was measured after gating on apoptotic cells on an Accuri C6 flow cytometer (BD Biosciences).
Statistical analysis
Mann-Whitney tests were performed to compare IgM or IgG reactivity to apoptotic cells between all subgroups of patients and healthy subjects. We used Spearman correlation tests to determine the association between IgG1 and IgG3 reactivity to apoptotic cells and C4d binding on target cells, between IgG concentration before and after purification, between IgG reactivity to apoptotic cells and patient age and between IgG reactivity to wild type Jurkat cells and reactivity to class I negative Jurkat cells. Kidney graft loss post transplant was reported as death censored graft loss. Graft-survival rates were computed using the Kaplan-Meier method and groups were compared by the log-rank test. Cox models were used to adjust the effects of IgG reactivity to apoptotic cells for potentially confounding factors including reactivity to HLA antibodies, sex, age, race, calculated panel reactive antibody (cPRA), delayed graft function (DGF), HLA mismatch, cold ischemia time, induction therapy and living donor transplants. Hazard ratio and 95% confidence interval are provided as measures of strength of association and precision, respectively. All tests were two-sided and a P-value of < 0.05 was considered to be statistically significant. General data analysis was conducted using SAS Analytics Software (SAS Institute Inc, Cary, NC) and GraphPad Prism (GraphPad Software 4.0, San Diego, CA).
Results
Serum reactivity to apoptotic cells before transplantation
In order to evaluate the contribution of anti-apoptotic cell antibodies to pre-sensitization, we assessed pre-transplant serum IgG reactivity to apoptotic cells in 300 kidney transplant recipients as well as 20 healthy controls. We had previously observed that the binding of IgG to apoptotic cells can be masked by serum proteins (30). We thus purified serum IgG before assessing their reactivity to apoptotic cells. As shown in Figure 1, purified IgG reactivity to apoptotic cells was significantly higher in pre-transplant serum compared to healthy subjects (P = 0.011). Comparable results were obtained when assessing non purified serum IgG reactivity to apoptotic cells (Figure S2). In contrast, no significant difference in IgM reactivity to apoptotic cells was observed between pre-transplant patients and healthy subjects (P = 0.922, Figure S2).
Figure 1. Pre-transplant purified IgG reactivity to apoptotic cells.

The purified IgG reactivity to apoptotic Jurkat cells was measured by flow cytometry in samples collected pre-transplant from kidney transplant recipients as well as healthy subjects. Log2 values of MFI are reported (y-axis). The numbers of samples tested in each group are shown below the box plot. The horizontal bar represents the median value; the bottom and top of each box represent the 25th and 75th percentiles; the lower and upper bars of each box represent the minimum and maximum values.
We next verified that the difference observed in reactivity to apoptotic cells between the two groups was not solely due to serum immunoglobulin levels. As illustrated in Figure S3A, concentrations of serum IgM, IgG as well as purified IgG were not significantly different between pre-transplant patients and healthy subjects (P = 0.900, P = 0.665, P = 0.420, respectively). Additionally, concentrations of purified IgG were comparable to that of unpurified serum IgG concentrations in all 300 pre-transplant patients (P < 0.001, Figure S3B).
We then examined whether pre-transplant IgG reactivity to apoptotic cells was associated with discrete patient characteristics. We observed a positive correlation between age and purified IgG reactivity to apoptotic cells (P = 0.024, not shown), However, reactivity of purified IgG to apoptotic cells did not appear to significantly correlate with sex, race, donor, etiology of kidney failure, previous transplants or history of blood transfusions. In addition, we did not observe any significant difference between patients with autoimmune diseases (including primary focal segmental glomerulosclerosis, IgA nephropathy, type I diabetes, systemic lupus erythematosus and immune complex diseases) and patients with non-autoimmune diseases (Figure S4).
Serum reactivity to HLA class I negative Jurkat cells and viable Jurkat cells
To ensure that reactivity to apoptotic Jurkat cells was not due to the recognition of HLA class I, we generated class I negative Jurkat cells through β-2 microglobulin knockdown using shRNA transfection to use as target (Figure S5A). As shown in Figure S5B, the binding of purified IgG to apoptotic class I negative cells is comparable to that of wild type Jurkat cells in the 39 patients with high IgG reactivity to HLA class I (MFI > 1000), (r = 0.874, P < 0.001), indicating that these antibodies recognize other antigenic structures than HLA on apoptotic cells. Lastly, as illustrated in Figure S6A for 3 representative pre-transplant samples, no reactivity was detected on viable Jurkat cells.
Pre-transplant purified IgG reactivity to apoptotic cells and kidney graft survival
The mean duration of follow-up for all patients included in this retrospective study was 81.2 ± 35.3 months. Forty-six patients lost their grafts and returned to dialysis due to various complications. The causes of graft loss are reported in Table 2. As depicted in Figure 2A, these patients had significantly higher purified IgG reactivity to apoptotic cells before transplantation compared to those with functioning graft (P < 0.001). In contrast, pre-transplant IgG reactivity to viable cells was not significantly different between patients with functioning graft and patients who experienced graft loss (P = 0.634, Figure S6B). Remarkably, among the 46 patients who lost their grafts, pre-transplant purified IgG reactivity to apoptotic cells was significantly increased in those whose graft loss was attributed to AMR compared to patients with other causes of graft loss (P = 0.033, Figure 2B).
Table 2.
Cause of graft loss (N=46)
| Cause of graft loss | Number |
|---|---|
| Antibody mediated rejection | 17 |
| Antibody mediated rejection & Cellular rejection | 6 |
| Cellular rejection | 4 |
| BK nephropathy | 5 |
| Recurrent IgA nephropathy | 3 |
| Recurrent FSGS | 4 |
| Transplant glomerulopathy | 7 |
Figure 2. Pre-transplant purified IgG reactivity to apoptotic cells and graft loss.

(A) Pre-transplant IgG reactivity to apoptotic cells was compared between patients with functioning graft and patients who experienced graft loss and (B) between patients whose graft loss was attributed to AMR and patients with other causes of graft loss. The numbers of samples analyzed in each group are shown below the box plot. The horizontal bar represents the median value; the bottom and top of each box represent the 25th and 75th percentiles; the lower and upper bars of each box represent the minimum and maximum values.
Figure 3A reports the death with functioning graft censored Kaplan-Meier survival outcome for patients with pre-transplant purified IgG reactivity to apoptotic cells above or below the median value (P = 0.002). We next separated the patients into 4 groups according to their reactivity to apoptotic cells: below the 1st quartile, between the 1st quartile and the 2nd quartile, between the 2nd quartile and the 3rd quartile or above the 3rd quartile value. As shown in this figure, the graft survival rate was significantly different between these groups (P < 0.001, Figure 3B). Patients with pre-transplant purified IgG reactivity to apoptotic cells above the 3rd quartile experienced the worst outcome while patients whose purified IgG reactivity to apoptotic cells was below the 1st quartile value had the highest graft survival rate. The influence of IgG reactivity to apoptotic cells on graft survival was only noticeable after approximately 1 year post-transplantation as apparent in the graft survival curve.
Figure 3. Pre-transplant purified IgG reactivity to apoptotic cells and graft outcome.
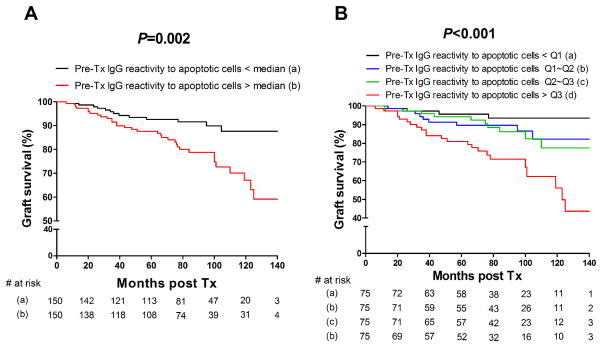
Kaplan-Meier cumulative, death censored, graft survival plot analyzing the effect of pre-transplant purified IgG reactivity to apoptotic Jurkat cells (A) below (black line) or above (red line) the median value. (B) below the 1st quartile (black line), between the 1st quartile and the 2nd quartile (blue line), between the 2nd and 3rd quartile (green line) or above the 3rd quartile value (red line). The number of patients at risk is shown below each time point. P value among groups was computed using log-rank (Mantel-Cox) test.
None of the 300 patients included in this study had detectable DSA at time of transplant. However, we assessed the overall pre-transplant serum reactivity to mixed HLA class I, II and MICA by Luminex in order to evaluate its effect on the graft outcome. Results showed that pre-existing reactivity to HLA class II and MICA but not HLA class I above a cutoff value of 1000 MFI was associated with decreased graft survival (P = 0.039, P = 0.006, P = 0.539, respectively; Figure 4A). We next investigated whether reactivity to apoptotic cells still correlated with graft loss when patients with reactivity to HLA class I, class II or MICA above 1000 MFI were excluded. As shown in Figure 4B, pre-transplant IgG reactivity to apoptotic cells still affected graft outcome in patients with low reactivity to HLA class I, class II and MICA (P = 0.003, P = 0.003, P = 0.023, respectively). Conversely, we examined whether reactivity to HLA class I, class II and MICA influenced the graft outcome in patients with IgG reactivity to apoptotic cells below the median value. As depicted in Figure 4C, pre-existing HLA class I, class II and MICA reactive antibodies did not correlate with graft loss in patients with IgG reactivity to apoptotic cells below the median value (P = 0.794, P = 0.091, P = 0.665, respectively). Of note, the number of patients included in this latter analysis was limited. To rule out the possibility that the risk-elevation for graft loss with increased levels of pre-transplant IgG reactivity to apoptotic cells was an artifact due to confounding with other factors including other IgG reactivities, sex, age, race, cPRA, DGF, HLA mismatch, cold ischemia time, induction therapy and living donor transplants, we carried out a Cox proportional hazards analysis including these covariates. The results confirmed that IgG reactivity to apoptotic cells as an explanatory factor remained significantly associated to graft loss even when the reactivity to HLA class I, class II and MICA as well as other variables were included and adjusted for in the statistical model (Table 3). In additional Cox models not shown, the hazard ratio for IgG reactivity to apoptotic cells remained constant throughout post-transplant follow-up time.
Figure 4. Kaplan-Meier cumulative graft survival.

(A) Kaplan-Meier cumulative, death censored, graft survival plot analyzing the effect of pre-transplant IgG reactivity to HLA class I, HLA class II or MICA above (dot line) or below (solid line) a cutoff MFI value of 1000. (B) Kaplan-Meier cumulative, death censored, graft survival plot analyzing the effect of pre-transplant purified IgG reactivity to apoptotic Jurkat cells above (dotted line) or below (solid line) the median value after exclusion of patients with high reactivity to HLA class I, HLA class II or MICA (MFI > 1000). (C) Kaplan-Meier cumulative, death censored, graft survival plot analyzing the effect of pre-transplant IgG reactivity to HLA class I, HLA class II or MICA above (dot line) or below (solid line) a cutoff MFI value of 1000 after exclusion of patients with reactivity to apoptotic cells above the median value. The number of patients at risk is shown below each time point. P value between two groups was computed using log-rank (Mantel-Cox) test.
Table 3.
Cox proportional hazards model. Hazard ratio (HR) with 95% confidence interval (95% CI) for graft survival in accordance with different variables
| Variable | Hazard ratio | 95% confidence interval | p-value |
|---|---|---|---|
| IgG reactivity to apoptotic cellsa | 2.271 | 1.530~3.369 | <0.001 |
| IgG reactivity to HLA class Ia | 0.909 | 0.739~1.118 | 0.366 |
| IgG reactivity to HLA class IIa | 1.212 | 0.979~1.501 | 0.077 |
| IgG reactivity to MICAa | 1.015 | 0.863~1.195 | 0.857 |
| Sex (male vs female) | 0.964 | 0.499~1.865 | 0.915 |
| African indicator | 1.209 | 0.481~3.040 | 0.687 |
| Asians indicator | 0.600 | 0.079~4.539 | 0.621 |
| Hispanic indicator | 2.062 | 0.683~6.220 | 0.199 |
| Age | 0.996 | 0.972~1.019 | 0.715 |
| cPRA | 0.993 | 0.970~1.017 | 0.574 |
| DGF | 1.981 | 0.851~4.611 | 0.113 |
| HLA mismatch | 1.227 | 0.977~1.541 | 0.079 |
| Cold ischemia time | 1.000 | 0.947~1.057 | 0.994 |
| Induction therapy | 0.692 | 0.326~1.470 | 0.338 |
| Living related donor | 0.659 | 0.205~2.122 | 0.484 |
| Living unrelated donor | 1.029 | 0.372~2.846 | 0.956 |
per unit increase in log2(1+IgG reactivity)
Subclasses of serum IgG reactive to apoptotic cells
The activation of the complement pathway is a primary effector function of antibodies implicated in graft failure (32–34). We investigated whether IgG reactive to apoptotic cells could also activate complement. We first used distinct secondary antibodies in flow cytometry experiments to determine the subclasses of IgG reactive to apoptotic cells. These experiments were conducted using IgG purified from the 50 most reactive serum specimens. For all specimens the purified serum IgG reactivity to apoptotic cells was almost exclusively mediated by complement fixing IgG1 or IgG3 subclasses or a combination of the two (Figure 5). Since only few samples showed IgG4 or IgG2 reactivity to apoptotic cells, we verified that our purification procedure did not eliminate these two IgG subclasses. As shown in Figure S7, the concentrations of all four IgG subclasses were comparable before and after IgG purification. We also tested the reactivity of all anti-IgG subclass secondary antibodies used in these assays on peripheral blood B cells from 2 healthy subjects. A distinct positive subset was detectable for each anti-IgG subclass as depicted in Figure S8.
Figure 5. Subclasses of serum IgG reactive to apoptotic cells.

(A) Example of subclass analysis by flow cytometry of IgG reactive to apoptotic Jurkat cells purified from 4 representative pre-transplant serum specimens, using secondary antibodies specific to IgG1 ~ IgG4. Filled gray histograms show background reactivity obtained with secondary antibodies alone. IgG1 ~ IgG4 reactivity to apoptotic Jurkat cells is depicted as solid line histograms. (B) Heat-map representation of IgG1 ~ IgG4 reactivity to apoptotic cells for the 50 most reactive serum samples. The shade of gray corresponds to the level of reactivity (MFI) as reported on the scale.
IgG1 and IgG3 reactive to apoptotic cells activate complement
The implication of the complement pathway in graft dysfunction is commonly revealed by the deposition of C4d into the allograft tissue (35–38). We used an in vitro assay to assess whether IgG reactive to apoptotic cells purified from pre-transplant serum specimens have the capacity to activate complement, resulting in the deposition of C4d on target cells (Figure S9). As expected, purified IgG comprising primarily complement fixing IgG1 and IgG3 had the capacity to activate complement. Moreover, we observed a strong association between IgG1 and IgG3 reactivity to apoptotic cells and C4d deposition on target cells (P < 0.001 and P = 0.005, respectively; Figure 6).
Figure 6. Correlation between complement activation and IgG subclasses reactivity to apoptotic cells.
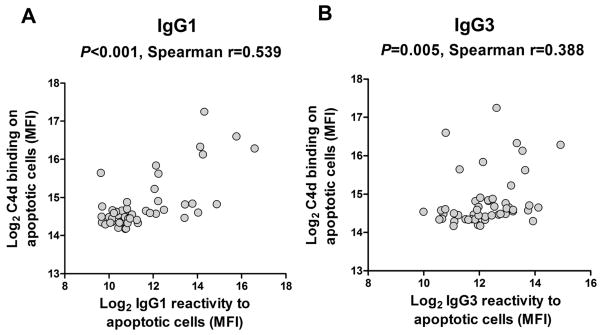
The deposition of C4d on the surface of apoptotic Jurkat cells was detected by flow cytometry after complement activation in vitro with IgG purified from the 50 most reactive pre-transplant serum samples. C4d deposition is reported together with IgG1 (A) or IgG3 (B) reactivity to apoptotic cells measured in the same samples.
Discussion
Pre-sensitization has been a long-standing limitation to solid organ transplantation. Until now, pre-sensitization has been explained by the presence of allospecific antibodies reactive to donor HLA or ABO blood group antigens. Our retrospective studies conducted on a relatively large series of patients are the first to implicate IgG reactive to apoptotic cells in pre-sensitization. The observation that pre-transplant anti-apoptotic cell IgG levels correlate with long-term graft loss even after exclusion of patients with pre-existing reactivity to HLA (MFI>1000), highlights the important value of these antibodies in kidney transplantation. Further, the association between serum reactivity to apoptotic cells and AMR even suggests an active role in this form of rejection. It is remarkable that the effect on graft survival was only visible after approximately 1 year post-transplantation as noticeable in the graft survival curve. This observation seems to match that made by Opelz and colleagues who examined the predictive value of pre-transplant PRA on graft survival in recipients of kidney transplants from HLA identical siblings (12). In this study, the effect of high PRA measurements on graft survival was only apparent after one year post-transplant. Our results suggest that anti-apoptotic cell IgG may have contributed to sensitization in these patients. The delayed effect is difficult to interpret. Immune events occurring early post-transplant and resulting from pre-existing anti-apoptotic cell antibodies may polarize the global host response to the graft and significantly influence the late outcome. Another hypothesis is that patients with elevated serum reactivity to apoptotic cells are immunologically more “sensitive” than those with lower pre-transplant serum reactivity. “Sensitive” patients would have an increased probability of complications, resulting in a higher rate of graft loss. Under this hypothesis, the pre-transplant serum reactivity would be a surrogate marker of “immune sensitivity” without any direct relation of cause and effect between pre-transplant antibodies and graft loss.
The generation of de novo DSA and non donor-specific antibodies (NDSA) post-transplant would also provide valuable information regarding the development of immunity to the organ graft in these patients. Unfortunately, our collection of post-transplant serum specimens was limited and we could not establish whether elevated pre-transplant IgG reactivity to apoptotic cells correlated with the development of de novo anti-HLA antibodies especially DSA for these patients. This lack of longitudinal follow-up is a significant limitation to our study. Additional studies will be required to address this point.
The origin of IgG reactive to apoptotic cells in patients with end-stage kidney disease is unclear. While “natural” IgM have been described in animals and humans for decades, class switched IgG bearing the same reactivity have only been reported in the context of certain autoimmune diseases such as systemic lupus erythematosus and more recently AMR (39, 40). Since they share the same distinctive reactivity, both natural IgM and anti-apoptotic cell IgG are likely produced by the same “innate” B cells that underwent class switch recombination (CSR). The exact cause for innate B cell CSR in kidney transplant candidates is unknown. Long-term hemodialysis treatments which most of the transplant candidates in our study underwent are often accompanied by an increase in inflammatory markers (41, 42). Likewise, several chronic kidney diseases have been associated with heightened levels of systemic inflammation that may have contributed to the generation of anti-apoptotic cell IgG (43).
It is still unclear as to what antigens are recognized by these antibodies at the surface of apoptotic cells. In a previous study, we reported the characterization of monoclonal antibodies reactive to apoptotic cells established from a patient with AMR (30). These monoclonal antibodies were all polyreactive, i.e. they reacted to multiple antigens as diverse as DNA or lipopolysaccharide. We also found that 2 of 6 polyreactive antibodies bound equally to phosphatidylserine and lysophosphatidylcholine. Both antigens are known immunogenic structures exposed at the surface of apoptotic cells. These results indicate that polyreactive antibodies can recognize distinct antigens on apoptotic cells.
The fact that anti-apoptotic cell IgG in our series are almost exclusively composed of complement fixing IgG1 and IgG3 is novel and intriguing. The reason for such a restriction is unclear. The preferential switch to IgG1 and IgG3 may be driven by the type of stimulation received by B cells reactive to apoptotic cells. It is possible that apoptotic cells deliver concomitant signals through the B cell receptor (BCR) as well as through co-stimulatory molecules and Toll-like receptor (TLR) directing CSR towards these 2 IgG subclasses. It is noteworthy however that both IgG1 and IgG3 have complement activating properties. Late allograft failures often involve complement activation, as revealed by the deposition of C4d on the graft tissue. IgG1 and IgG3 appear to play a dominant role in this process (44–47). Along these lines, our findings support the view that IgG1 and IgG3 reactive to apoptotic cells participate in the overall inflammatory response to the allograft and eventually contribute to graft loss.
Results presented in this report shed a new light on anti-apoptotic cell IgG as important contributors to pre-sensitization along with allospecific antibodies. The association between elevated levels of these antibodies and the transplant outcome even suggests a direct physiological role in graft destruction. Our findings have significant implications for the assessment of transplant candidates. Taking these IgG into consideration in addition to anti-HLA antibodies during pre-transplant evaluation would likely improve the risk assessment of the graft outcome. Moreover, patients with high reactivity to apoptotic cells pre-transplant could also possibly benefit from desensitization treatments. Prospective studies are now warranted to extend our observations to additional transplant recipients and further corroborate the involvement of anti-apoptotic cell IgG in the host response to kidney allografts.
Supplementary Material
Acknowledgments
We thank Dr. Roberto Bellucci for the packaged lentiviral vector containing a β2-microglobulin shRNA.
This work was supported by the Fahd and Nadia Alireza’s Research Fund, the Roche Organ Transplantation Research Foundation (ROTRF) and the National Institute of Health, National Institute of Diabetes, Digestive and Kidney Diseases Grant DK083352.
Abbreviations
- HLA
human leukocyte antigen
- DSA
donor-specific antibodies
- PRA
panel reactive antibodies
- CSR
class switch recombination
- AMR
antibody mediated rejection
- MFI
mean fluorescence intensity
- cPRA
calculated panel reactive antibody
- DGF
delayed graft function
- NDSA
nondonor-specific antibodies
- BCR
B cell receptor
- TLR
Toll-like receptor
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
References
- 1.Terasaki PI. Humoral theory of transplantation. Am J Transplant. 2003;3(6):665–673. doi: 10.1034/j.1600-6143.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 2.Jordan SC, Pescovitz MD. Presensitization: the problem and its management. Clin J Am Soc Nephrol. 2006;1(3):421–432. doi: 10.2215/CJN.01651105. [DOI] [PubMed] [Google Scholar]
- 3.Susal C, Opelz G. Kidney graft failure and presensitization against HLA class I and class II antigens. Transplantation. 2002;73(8):1269–1273. doi: 10.1097/00007890-200204270-00014. [DOI] [PubMed] [Google Scholar]
- 4.Susal C, Dohler B, Opelz G. Presensitized kidney graft recipients with HLA class I and II antibodies are at increased risk for graft failure: a Collaborative Transplant Study report. Hum Immunol. 2009;70(8):569–573. doi: 10.1016/j.humimm.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Susal C, Dohler B, Sadeghi M, Ovens J, Opelz G. HLA antibodies and the occurrence of early adverse events in the modern era of transplantation: a collaborative transplant study report. Transplantation. 2009;87(9):1367–1371. doi: 10.1097/TP.0b013e3181a24073. [DOI] [PubMed] [Google Scholar]
- 6.Amico P, Honger G, Mayr M, Steiger J, Hopfer H, Schaub S. Clinical relevance of pretransplant donor-specific HLA antibodies detected by single-antigen flow-beads. Transplantation. 2009;87(11):1681–1688. doi: 10.1097/TP.0b013e3181a5e034. [DOI] [PubMed] [Google Scholar]
- 7.Zou Y, Stastny P, Susal C, Dohler B, Opelz G. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med. 2007;357(13):1293–1300. doi: 10.1056/NEJMoa067160. [DOI] [PubMed] [Google Scholar]
- 8.Dragun D. Humoral responses directed against non-human leukocyte antigens in solid-organ transplantation. Transplantation. 2008;86(8):1019–1025. doi: 10.1097/TP.0b013e3181889748. [DOI] [PubMed] [Google Scholar]
- 9.Sumitran-Holgersson S. Relevance of MICA and other non-HLA antibodies in clinical transplantation. Curr Opin Immunol. 2008;20(5):607–613. doi: 10.1016/j.coi.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Jackson AM, Kuperman MB, Montgomery RA. Multiple hyperacute rejections in the absence of detectable complement activation in a patient with endothelial cell reactive antibody. Am J Transplant. 2012;12(6):1643–1649. doi: 10.1111/j.1600-6143.2011.03955.x. [DOI] [PubMed] [Google Scholar]
- 11.Crespo M, Pascual M, Tolkoff-Rubin N, Mauiyyedi S, Collins AB, Fitzpatrick D, et al. Acute humoral rejection in renal allograft recipients: I. Incidence, serology and clinical characteristics. Transplantation. 2001;71(5):652–658. doi: 10.1097/00007890-200103150-00013. [DOI] [PubMed] [Google Scholar]
- 12.Opelz G. Non-HLA transplantation immunity revealed by lymphocytotoxic antibodies. Lancet. 2005;365(9470):1570–1576. doi: 10.1016/S0140-6736(05)66458-6. [DOI] [PubMed] [Google Scholar]
- 13.Solgi G, Furst D, Mytilineos J, Pourmand G, Amirzargar AA. Clinical relevance of pre and post-transplant immune markers in kidney allograft recipients: anti-HLA and MICA antibodies and serum levels of sCD30 and sMICA. Transpl Immunol. 2012;26(2–3):81–87. doi: 10.1016/j.trim.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Dragun D, Muller DN, Brasen JH, Fritsche L, Nieminen-Kelha M, Dechend R, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med. 2005;352(6):558–569. doi: 10.1056/NEJMoa035717. [DOI] [PubMed] [Google Scholar]
- 15.Kalache S, Dinavahi R, Pinney S, Mehrotra A, Cunningham MW, Heeger PS. Anticardiac myosin immunity and chronic allograft vasculopathy in heart transplant recipients. J Immunol. 2011;187(2):1023–1030. doi: 10.4049/jimmunol.1004195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joosten SA, Sijpkens YW, van Ham V, Trouw LA, van der Vlag J, van den Heuvel B, et al. Antibody response against the glomerular basement membrane protein agrin in patients with transplant glomerulopathy. Am J Transplant. 2005;5(2):383–393. doi: 10.1111/j.1600-6143.2005.00690.x. [DOI] [PubMed] [Google Scholar]
- 17.Rose ML. Role of anti-vimentin antibodies in allograft rejection. Hum Immunol. 2013;74(11):1459–1462. doi: 10.1016/j.humimm.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linke AT, Marchant B, Marsh P, Frampton G, Murphy J, Rose ML. Screening of a HUVEC cDNA library with transplant-associated coronary artery disease sera identifies RPL7 as a candidate autoantigen associated with this disease. Clin Exp Immunol. 2001;126(1):173–179. doi: 10.1046/j.1365-2249.2001.01654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porcheray F, DeVito J, Yeap BY, Xue L, Dargon I, Paine R, et al. Chronic humoral rejection of human kidney allografts associates with broad autoantibody responses. Transplantation. 2010;89(10):1239–1246. doi: 10.1097/TP.0b013e3181d72091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warraich RS, Pomerance A, Stanley A, Banner NR, Dunn MJ, Yacoub MH. Cardiac myosin autoantibodies and acute rejection after heart transplantation in patients with dilated cardiomyopathy. Transplantation. 2000;69(8):1609–1617. doi: 10.1097/00007890-200004270-00015. [DOI] [PubMed] [Google Scholar]
- 21.Wheeler CH, Collins A, Dunn MJ, Crisp SJ, Yacoub MH, Rose ML. Characterization of endothelial antigens associated with transplant-associated coronary artery disease. J Heart Lung Transplant. 1995;14(6 Pt 2):S188–197. [PubMed] [Google Scholar]
- 22.Cristol JP, Vela C, Maggi MF, Descomps B, Mourad G. Oxidative stress and lipid abnormalities in renal transplant recipients with or without chronic rejection. Transplantation. 1998;65(10):1322–1328. doi: 10.1097/00007890-199805270-00007. [DOI] [PubMed] [Google Scholar]
- 23.Cardinal H, Dieude M, Brassard N, Qi S, Patey N, Soulez M, et al. Antiperlecan antibodies are novel accelerators of immune-mediated vascular injury. Am J Transplant. 2013;13(4):861–874. doi: 10.1111/ajt.12168. [DOI] [PubMed] [Google Scholar]
- 24.Avrameas S. Natural autoantibodies: from ‘horror autotoxicus’ to ‘gnothi seauton’. Immunol Today. 1991;12(5):154–159. doi: 10.1016/S0167-5699(05)80045-3. [DOI] [PubMed] [Google Scholar]
- 25.Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. 2010;10(11):778–786. doi: 10.1038/nri2849. [DOI] [PubMed] [Google Scholar]
- 26.Fu M, Fan PS, Li W, Li CX, Xing Y, An JG, et al. Identification of poly-reactive natural IgM antibody that recognizes late apoptotic cells and promotes phagocytosis of the cells. Apoptosis : an international journal on programmed cell death. 2007;12(2):355–362. doi: 10.1007/s10495-006-0581-z. [DOI] [PubMed] [Google Scholar]
- 27.Silverman GJ, Gronwall C, Vas J, Chen Y. Natural autoantibodies to apoptotic cell membranes regulate fundamental innate immune functions and suppress inflammation. Discov Med. 2009;8(42):151–156. [PubMed] [Google Scholar]
- 28.Peng Y, Kowalewski R, Kim S, Elkon KB. The role of IgM antibodies in the recognition and clearance of apoptotic cells. Mol Immunol. 2005;42(7):781–787. doi: 10.1016/j.molimm.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 29.Porcheray F, DeVito J, Helou Y, Dargon I, Fraser JW, Nobecourt P, et al. Expansion of polyreactive B cells cross-reactive to HLA and self in the blood of a patient with kidney graft rejection. Am J Transplant. 2012;12(8):2088–2097. doi: 10.1111/j.1600-6143.2012.04053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porcheray F, Fraser JW, Gao B, McColl A, Devito J, Dargon I, et al. Polyreactive antibodies developing amidst humoral rejection of human kidney grafts bind apoptotic cells and activate complement. Am J Transplant. 2013;13(10):2590–2600. doi: 10.1111/ajt.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellucci R, Nguyen HN, Martin A, Heinrichs S, Schinzel AC, Hahn WC, et al. Tyrosine kinase pathways modulate tumor susceptibility to natural killer cells. J Clin Invest. 2012;122(7):2369–2383. doi: 10.1172/JCI58457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colvin RB. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. J Am Soc Nephrol. 2007;18(4):1046–1056. doi: 10.1681/ASN.2007010073. [DOI] [PubMed] [Google Scholar]
- 33.Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat Rev Nephrol. 2012;8(11):670–678. doi: 10.1038/nrneph.2012.212. [DOI] [PubMed] [Google Scholar]
- 34.Fukuda M. Evaluation and clinical significance of circulating immune complexes after renal transplantation. Transplantation. 1983;36(2):155–160. doi: 10.1097/00007890-198308000-00009. [DOI] [PubMed] [Google Scholar]
- 35.Bohmig GA, Exner M, Habicht A, Schillinger M, Lang U, Kletzmayr J, et al. Capillary C4d deposition in kidney allografts: a specific marker of alloantibody-dependent graft injury. J Am Soc Nephrol. 2002;13(4):1091–1099. doi: 10.1681/ASN.V1341091. [DOI] [PubMed] [Google Scholar]
- 36.Magil AB, Tinckam K. Monocytes and peritubular capillary C4d deposition in acute renal allograft rejection. Kidney Int. 2003;63(5):1888–1893. doi: 10.1046/j.1523-1755.2003.00921.x. [DOI] [PubMed] [Google Scholar]
- 37.Regele H, Bohmig GA, Habicht A, Gollowitzer D, Schillinger M, Rockenschaub S, et al. Capillary deposition of complement split product C4d in renal allografts is associated with basement membrane injury in peritubular and glomerular capillaries: a contribution of humoral immunity to chronic allograft rejection. J Am Soc Nephrol. 2002;13(9):2371–2380. doi: 10.1097/01.asn.0000025780.03790.0f. [DOI] [PubMed] [Google Scholar]
- 38.Lorenz M, Regele H, Schillinger M, Exner M, Rasoul-Rockenschaub S, Wahrmann M, et al. Risk factors for capillary C4d deposition in kidney allografts: evaluation of a large study cohort. Transplantation. 2004;78(3):447–452. doi: 10.1097/01.tp.0000128344.94808.03. [DOI] [PubMed] [Google Scholar]
- 39.Liu S, Cerutti A, Casali P, Crow MK. Ongoing immunoglobulin class switch DNA recombination in lupus B cells: analysis of switch regulatory regions. Autoimmunity. 2004;37(6–7):431–443. doi: 10.1080/08916930400010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci U S A. 2008;105(28):9727–9732. doi: 10.1073/pnas.0803644105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jofre R, Rodriguez-Benitez P, Lopez-Gomez JM, Perez-Garcia R. Inflammatory syndrome in patients on hemodialysis. J Am Soc Nephrol. 2006;17(12 Suppl 3):S274–280. doi: 10.1681/ASN.2006080926. [DOI] [PubMed] [Google Scholar]
- 42.Carrero JJ, Qureshi AR, Axelsson J, Avesani CM, Suliman ME, Kato S, et al. Comparison of nutritional and inflammatory markers in dialysis patients with reduced appetite. Am J Clin Nutr. 2007;85(3):695–701. doi: 10.1093/ajcn/85.3.695. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Jacobi AM, Wang T, Berlin R, Volpe BT, Diamond B. Polyreactive autoantibodies in systemic lupus erythematosus have pathogenic potential. J Autoimmun. 2009;33(3–4):270–274. doi: 10.1016/j.jaut.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colvin RB, Smith RN. Antibody-mediated organ-allograft rejection. Nat Rev Immunol. 2005;5(10):807–817. doi: 10.1038/nri1702. [DOI] [PubMed] [Google Scholar]
- 45.Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. 1987;166(5):1351–1361. doi: 10.1084/jem.166.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Michaelsen TE, Garred P, Aase A. Human IgG subclass pattern of inducing complement-mediated cytolysis depends on antigen concentration and to a lesser extent on epitope patchiness, antibody affinity and complement concentration. Eur J Immunol. 1991;21(1):11–16. doi: 10.1002/eji.1830210103. [DOI] [PubMed] [Google Scholar]
- 47.Lucisano Valim YM, Lachmann PJ. The effect of antibody isotype and antigenic epitope density on the complement-fixing activity of immune complexes: a systematic study using chimaeric anti-NIP antibodies with human Fc regions. Clin Exp Immunol. 1991;84(1):1–8. doi: 10.1111/j.1365-2249.1991.tb08115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.