

Abstract
In cytochrome c oxidase (CcO), a redox-driven proton pump, protons are transported by the Grotthuss shuttling via hydrogen-bonded water molecules and protonatable residues. Proton transport through the D-pathway is a complicated process that is highly sensitive to alterations in the amino acids or the solvation structure in the channel, both of which can inhibit proton pumping and enzymatic activity. Simulations of proton transport in the hydrophobic cavity showed a clear redox state dependence. To study the mechanism of proton pumping in CcO, multi-state empirical valence bond (MS-EVB) simulations have been conducted, focusing on the proton transport through the D-pathway and the hydrophobic cavity next to the binuclear center. The hydration structures, transport pathways, effects of residues, and free energy surfaces of proton transport were revealed in these MS-EVB simulations. The mechanistic insight gained from them is herein reviewed and placed in context for future studies.
Keywords: cytochrome c oxidase, proton pump, proton transport, computer simulation, multi-state empirical valence bond (MS-EVB)
1. Introduction
Biological proton pumps are integral membrane proteins that drive protons across membranes against their electrochemical gradient. These pumping mechanisms have attracted great interest for several decades because they play a critical role in controlling energy conversion in biology. Cytochrome c oxidase (CcO), the fourth complex in the mitochondrial electron transport chain, is one of three types of proton pumps that function in cellular respiration [1-9]. It catalyzes the reduction of O2 into H2O and uses the released energy to pump protons from the negative inside (N-side) of the membrane to the positive outside (P-side). CcO transports electrons from cytochrome c to O2 at its active site through multiple metal centers: the copper cluster (CuA), heme a, and the binuclear center (BNC), which consists of heme a3 and another copper site (CuB). Different redox states of the metal centers formed by electron transfer provide the driving force for proton pumping. In each catalytic cycle, four “chemical” protons are transported from the N-side of the membrane to the active site (near CuB) to form H2O, and another four “pumped” protons are transferred from the N-side to the P-side of the membrane (Fig 1). Two questions have been extensively addressed and studied in the CcO catalytic cycle. One is how proton pumping is coupled to redox state transitions, while the other is how the two proton transport processes are regulated. Understanding the energetics, dynamics, and resulting kinetics of the proton transport in CcO is the key to answering these questions.
Figure 1.
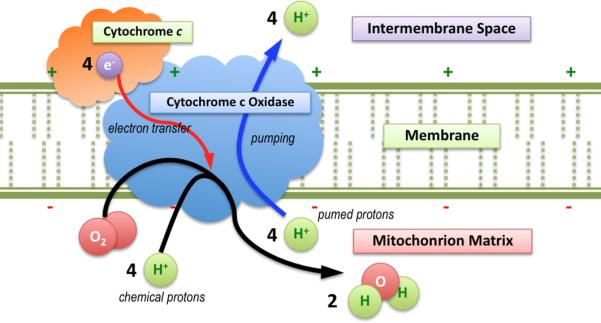
The mechanism of the redox-driven proton pumping by CcO in mitochondria, including two types of proton substrates, is shown.
The excess proton (H+) possesses unique structural and dynamical characteristics in aqueous systems. For a long period, it was believed that in aqueous environments the proton exists as a hydronium cation (H3O+), transported via both the vehicular motion of H3O+ and a hopping mechanism entitled “Grotthuss shuttling” [10,11] via the rearrangement of covalent bonds (Fig. 2A). However, this conception has been largely extended and modified by theoretical studies. Supported by different types of computer simulations, it was found that the classical hydronium structure, in which the positive charge is localized, is rarely observed. Instead, the electronic charge defect associated with the excess proton is more delocalized and shared by the surrounding water molecules, forming more complicated structures, namely (H3O)nH+ [12-15]. The dominant structures are usually referred to as the asymmetric Eigen cation (H9O4+), in which the positive charge is delocalized on four water molecules, and the more symmetric Zundel cation (H5O2+), in which the positive charge is delocalized on two water molecules. Nevertheless, there is no clear boundary between these two types of structures. They can interchange frequently in aqueous environments, and a series of continuously transitioning structures (Eigen-like or Zundel-like) can be observed. Therefore, proton transport is significantly dependent upon the rearrangement of the electron structure, due to the charge defect, and is strongly correlated to the dynamics of its hydration structure [14-16].
Figure 2.
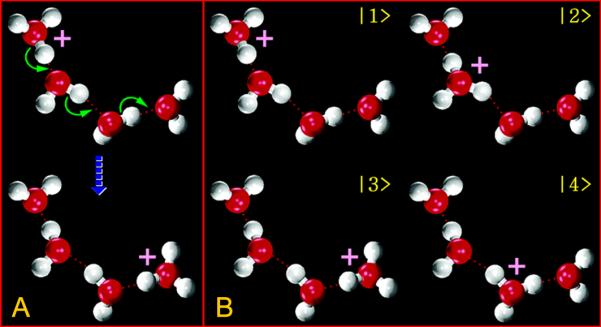
A. The transport of an excess proton in a chain of four water molecules via Grotthuss shuttling is shown. B. Diabatic states are used (of the system in A) in MS-EVB simulations to describe this process and the excess proton charge defect delocalization.
Researchers have often suggested that, in proteins, proton transport also undergoes the Grotthuss shuttling through the hydrogen bonded chains (HBC) formed by water molecules [17, 18]. However, this process differs from the proton diffusion in homogeneous systems for at least two reasons. One difference is that the water configurations and dynamics are restrained by the spatial confinement and complicated electrostatic effects on the surface, or in the interior regions of the proteins [19-21]. Another difference is that titratable residues and other charged groups are likely to be directly involved in proton transport and charge defect delocalization, thus potentially behaving as proton donors/acceptors.
Over the years, X-ray crystal structures of CcO from different sources (i.e., mitochondria from bovine heart cells [22] and bacteria such as P. denitrificans [23] and R. sphaeroides [24]) have been reported. In this article, the structure and residue indexing of CcO from R. sphaeroides is used. X-ray crystal structures of CcO and other enzymes from the heme-copper binuclear center oxidase superfamily show similar structures of subunits and metal sites. Furthermore, catalytic kinetics, mutagenesis, and computer simulation studies on these enzymes yield similar results [2]. By combining structural analysis and site-directed mutagenesis, three proton pathways in CcO, which are named as D, K (Fig. 3), and H, have been located and treated as the main targets in experimental and theoretical studies. The D-pathway starts from Asp132 of subunit I near the N-side membrane surface, and ends at Glu286, which is located below (closer to the N-side, Fig. 3) a hydrophobic cavity near the heme a/a3 molecules (Fig. 3A). It is a proton uptake channel conducting both pumped protons and chemical protons [25, 26]. The K-pathway is another proton uptake channel, from Lys362 of subunit II to the active site, but it is only used for chemical protons and has a much lower conductance than the D-pathway [25]. Finally, the H-pathway, which is from the N-side membrane surface to the P-side and conducts only pumped protons, was proposed based on the structures from bovine heart oxidases [27-31]. The location of the proton loading site (PLS), where the pumped protons reside above (closer to the P-side, Fig. 3) the hydrophobic cavity [32], is still uncertain. Two possible candidates are the D-propionate of heme a3 (PRDa3, Fig. 3B) [33, 34] and one of the three CuB-bound histidines (Fig. 3C) [35-37]. Of the three proton channels, the D-pathway has received the most attention, possibly due to its high proton conductance and influence on both enzyme activity and pumping. This pathway appears to be a highly solvated water channel with crystal structures revealing approximately ten water molecules, and simulations report 11~12 waters that are rather dynamic [38, 39].
Figure 3.

Structure of R. sphaeroides CcO (PDB1M56) with colored subunits (I-blue, II-red, III-green, and IV-orange), cellular membrane (gray), two proton transport pathways (D and K), and important metal sites and amino-acid residues is shown. Inset A: The pore water structure in D-pathway, which is from Asp132 to Glu286 in subunit I, and two critical residue clusters, Ser200/Ser201 and Asn125/Asn139, are shown. Inset B: The salt-bridge formed by Arg481 and PRDa3 is shown. Inset C: The catalytic center, which consists of heme a3 and a copper atom (CuB) coordinated by three histidine residues, is shown.
Structural characterizations from experiments have provided valuable starting points for theoretical studies and computational modeling. Extensive computational studies, such as electronic-structure calculations and molecular dynamics (MD) simulations, have been conducted on CcO and have generally extended our understanding of its mechanisms [38-61]. MD simulation is a useful technique in biophysical and biochemical studies, since it allows researchers to track and analyze the dynamics of biomolecular systems at the atomic level. Nevertheless, traditional MD simulations are not able to study directly the Grotthuss shuttling of proton transport dynamics, because the standard molecular mechanics (MM) potentials used in MD do not allow changes in the covalent bonding topology. Instead of simulating proton transport directly, standard MD simulations have been used to reveal the hydration structure and dynamic properties of water molecules along the proton transport pathways. Another useful tool, the quantum-mechanics/molecular-mechanics (QM/MM) hybrid approach, is able to treat the chemical bond rearrangement involved in the proton transport process from electronic structure calculations, revealing certain bonding and solvation structures of the excess proton [62-71]. However, due to its high computational cost in the QM region, it is still very expensive to run QM/MM simulations for proton transport over large scales of length (i.e., tens of nanometers) and time (i.e., tens to thousands of nanoseconds) at present.
Instead of directly simulating the proton transport process, the alternative is to simply study the energetics of certain key titratable residues, i.e., to calculate its pKa value or pKa shift from a bulk water environment [54, 55, 61, 70-73], then attempt to piece together a proposed proton mechanism based on the resulting data set. Some commonly used approaches for pKa calculations include the Poisson-Boltzmann (PB) model [74], the generalized Born (GB) model [75], the multi-conformation continuum electrostatic (MCCE) model [76], the free energy perturbation (FEP) methods [77], and the thermodynamic integration (TI) methods [78]. These can be used to estimate the change in free energies of important residues between their protonated and deprotonated states. These approaches cannot, however, reveal the complete potential of mean force (PMF) for the excess proton migration, and thus lacks certain key kinetic information along the vectorial proton transport pathways.
The Multi-State Empirical Valence Bond (MS-EVB) method, which is a reactive MD methodology, has proven to be a successful and much more computationally efficient alternative to electronic structure methods for the computer simulation of excess protons in aqueous environments and biomolecules [12, 79-84]. During the past several years, a series of MS-EVB studies on the proton transport behavior of CcO have been conducted, focusing on the proton uptake in the D-pathway and redox-coupled proton pumping in the hydrophobic cavity. These studies have contributed to the understanding the proton pumping in CcO by directly providing a view of the explicit proton transport dynamics [39, 46, 48, 50, 53, 85]. The rest of this article is organized as follows: an introduction of the MS-EVB methodology and its advantages are introduced in section 2; the results from the MS-EVB simulations are discussed in section 3; and conclusions are summarized in section 4.
2. Materials and Methods
MS-EVB is an efficient and validated reactive MD approach to simulating excess protons in condensed phase systems. It is a dynamic multi-state generalization of the simple EVB approach extensively developed by Warshel and co-workers [86, 87]. In MS-EVB the molecular system ΨEVB is described as a linear combination of a series of diabatic states, {ψi}, each of which indicates a different bonding topology, as represented by molecular mechanics (Fig. 2B) [12, 79-84],
| (1) |
As in linear variational quantum theory, the Hamiltonian operator is built as a matrix,
| (2) |
in which the diagonal elements, hii, are the empirical potentials of the diabatic states and the off-diagonal elements, hij, are analytic functions of all nuclear coordinates, , selected to enable the transitions between states. The eigenvectors of this matrix are obtained for each nuclear configuration via diagonalization, and the one with the lowest eigenvalue represents the ground state of the system, and by definition, the actual potential for the dynamics of the excess proton. The parameters in the MS-EVB were obtained by carefully fitting the potential energy surface calculated on ab-initio level, i.e., MP2/aug-cc-pVDZ. Also it was showed that the MS-EVB parameters are able to give the binding energies and structures of small protonated water clusters in a very good agreement with with ab-initio calculations. The multiple diabatic states and their amplitudes {ci} are actual system variables that represent the effects of an evolving electronic structure empirically, although variationally. Therefore, MS-EVB achieves i) a smooth and reversible transition of bonding topologies for a chemical reaction, ii) a complicated and accurate potential energy surface constructed along the reaction coordinate for the excess proton Grotthuss shuttling, and iii) charge defect delocalization and its thermal fluctuations.
One of the keys to conducting a meaningful MS-EVB simulation is the algorithm that dynamically selects diabatic states. Initially, the excess proton is considered as a classical hydronium (H3O+), which is the most favorable diabatic state (it possesses the maximum weight in the ground-state eigenvector). Then, a breadth-first search (BFS) is adopted on solvation shells, one at a time (for details please refer to Ref. 84, Sec. 2.2). More importantly, states in an outer shell are searched by their hydrogen bond networks, which are the essential pathways for proton transport [80]. At each MD step, the state-search algorithm is conducted, which tracks the movement of the excess proton. Therefore, the critical area containing those atoms involved in the bond breaking/forming process, referred to as the “EVB-complex”, is dynamically created for the system during the MD simulation. Thus, the candidate diabatic states are intelligently selected based on the hydrogen bond topology, thus reducing the computational cost while still ensuring the key proton transfer pathways are collected.
Another difficulty that exists in the simulations of excess protons is the sampling of the proton transport pathway through the water HBC. This is because the traditional free energy, or PMF sampling techniques, such as umbrella-sampling, use nuclear coordinates (i.e., atomic position, centers of mass) to define the reaction coordinate. Proton transport, on the other hand, is actually a “charge translocation”, defined significantly by the ever-changing electronic structure. MS-EVB simulations solve this problem by introducing the center-of-excess-charge (CEC) as the weighted averaged charge centers of all diabatic states,
| (3) |
where denotes the center of charges (COC) of a classical hydronium (or equivalent amino acid structure),
| (4) |
Since the excess proton is seldom found as a localized hydronium cation in the system [15], the CEC is an optimal choice for monitoring the “position” of the protonic charge defect, and can be used to calculate structural and dynamical properties that can be compared to experimental observables. Furthermore, the CEC has been successfully used as the reaction coordinate to study the proton transport pathways in biomolecules, such as proton channels and enzymes [88-92]. Utilizing the CEC, combined with umbrella-sampling [93, 94], the potential of mean force of certain proton transport segments in CcO has thus been explicitly obtained and analyzed in MS-EVB simulations [46, 48, 50, 53, 85].
3. Results and Discussion
3.1 Glu286 at subunit I: the branching site in proton transport and its pKa
Glu286, which lies at the end of the D-pathway [95, 96] and is near to the hydrophobic cavity in subunit I, is highly conserved in different forms of CcO. Mutations of this residue also significantly inhibit, if not block completely, enzymatic activity [4, 26, 41, 97]. Thus, it is considered that Glu286 is an essential residue for proton pumping in CcO. The first MS-EVB simulations focused on proton transport through the D-pathway to Glu286 [46]. It was shown that an excess proton placed in the D-pathway with ~15Å away from Glu286 travels to Glu286 very quickly (less than 30 ps) when it is deprotonated (negatively charged). This result suggested that Glu286 has a very strong proton affinity, which is in agreement with the pKa estimations from previous experimental and computational studies [54, 61, 70, 72, 98]. The experimental pKa of Glu286 in the wild-type CcO is determined to be 9.4 from the kinetics of forming the F state [98]. Ghosh and co-workers conducted a computational TI study combined with the SCC-DFTB/MM-GSBP-based model. They computed a series of pKa values ranging from 8.1 to 21.3, based on different sets of the systems. Their results showed that the pKa of Glu286 is very sensitive to the electrostatic effect from the protein environment (e.g., the ionization states of surrounding residues), but is less affected by the number of pore water molecules in the hydrophobic cavity [70]. A recent study conducted by Warshel and co-workers reported the calculation of pKa based on their EVB/FEB approach, combined with the PDLD/S-LRA semimacroscopic model. In this work, the authors argued that the apparent pKa of Glu286 is contributed by the values of two different configurations, which were revealed from the X-ray crystal structures of the wild-type and the Asn139Asp mutant from P. denitrificans. The computed apparent pKa from the two-configuration-based model is in a range from 9.1 to 10.3, which is in good agreement with experimental results [61].
Glu286 has been thought to transport the proton from the D-pathway to the hydrophobic cavity by a dihedral angle rotation of its side chain [52, 95]. From the PMF for this rotation, the reorientation of Glu286 from the “down” state (~75°) to the “up” state (~295°) crosses a 6 kcal/mol barrier. The “down” state is more stable because it is about 2 kcal/mol lower in free energy than the “up” state [39]. While in almost all crystal structures the Glu286 is also observed in the “down” state, it is reported in some simulations that it instead prefers to stay in the “up” state with the lower free energy; this is explained by the hydration (four or more water molecules) in the hydrophobic cavity [52, 99]. In these simulations it was found that Glu286 favors the “down” state only when the simulation is conducted without water molecules in this cavity [52], or without the membrane and full solvation environment [99]. Since the free energy difference and barrier between the “up” and “down” states are very small, the orientation of Glu286 will clearly be sensitive to its hydration and electrostatic environment. This implies that in computational studies on CcO, one should be very careful when constructing the hydration structures in its hydrophobic cavity and D-pathways.
3.2 Proton uptake in the D-pathway: from Asp132 to Glu286
An MS-EVB study on the D-pathway proton transport was conducted by tracking the CEC position time-evolution when Glu286 was deprotonated. The excess proton was first placed and equilibrated at the bottom of the D-pathway in the hydronium form, then was relaxed to move to Glu286. The simulation showed a two-step mechanism, in which the proton resided for about 10 ps near a position about 7Å lower than Glu286, which was postulated as a sort of “proton trap” [46]. However, it is not a true thermodynamic trap for the proton, based on the PMF computed with umbrella sampling. The free energy profile indicates a straight downhill trend along the proton translocation to deprotonated Glu286, and implies that the proton uptake in the D-pathway is not a rate-limiting step. An explanation for the “trapping” can be based on the two serine residues (Ser200 and Ser201, Fig. 3A) in that region of the D-channel. Their polar side chains can form hydrogen bonds with nearby water molecules, restricting the water dynamics. Another explanation is based on the pore radius analysis along the D-pathway. A maximum radius of about 2.5Å is located at this region, which means the area can contain enough water molecules to form the most favorable structure of hydrated protons in bulk water, the Eigen cation (H9O4+) [46]. Another simulation was performed with the Glu286 in the neutral/protonated state [39]. In this case, the excess proton can only reach the region near Ser200/Ser201, and the PMF around Ser200/Ser201 becomes the minimum along the proton pathway. This implies that this region may be a sort of proton pool, where a hydrated excess proton can reside and wait until the deprotonation of Glu286. After deprotonation, Glu286 is immediately re-protonated from this region. This concept was supported to the same degree by another MS-EVB simulation that was compared to a higher-resolution PDB entry [50]. The comparison suggests that the positive charge defect of the excess proton is delocalized by four water molecules to form an Eigen-like species, which is stabilized by a hydrogen bond network formed by the surrounding residues Phe108/Ser197/Ser200/Ser201 and a fifth water molecule [50]. However, it should be noted that this structure is a static crystal structure and thus may only represent a snap shot of the real functioning membrane-bound CcO.
To study further the proton uptake in the D-pathway, a comprehensive investigation was conducted recently, combining both experiments and simulations [39]. The site-directed mutants in the D-pathway, which replace Ser200 and Ser201 with more hydrophobic residues, including Ser200Val/Ser201Val (VV) and Ser200Val/Ser201Tyr (VY), were investigated and compared alongside wild-type (WT) enzymes. Experiments showed that both VV and VY mutants lower the steady-state activity of CcO to 37% and 11% of the WT value, respectively, the proton pumping may be up to 54% less efficient in the VV mutant, and the pumping is totally absent in the VY mutant [39]. One possible explanation is that the mutants introduce a hydrophobic region into the D-pathway, thus creating an energy barrier for proton uptake; this effect was not seen in the simulation. When Glu286 is deprotonated, the PMF of proton uptake in the VY mutant showed a downhill trend similar to WT, suggesting that proton transport in the D-pathway is never the rate-limiting step for the redox transitions in wild-type and mutated CcO. This is possible because the negatively charged Glu286 has strong electrostatic attractive forces acting on the excess protons. Experiments and electrostatic potential calculations both showed that the mutants elevated the apparent pKa of Glu286. This effect cannot be easily explained as being due to broken hydrogen bonds after the introduction of the hydrophobic residues. In fact, in the MD simulations the mutants cause the D-pathway to be more hydrated when Glu286 is protonated. The role of water molecules in the D-pathway is therefore intricate and the mutants evidently shift the pKa by increasing the solvation for protonated Glu286, decreasing the solvation for the deprotonated form. To support this conclusion, simulations were carried out for structures having the D-pathway filled with varying numbers of water molecules and the results were found to be similar. From this study [39], it was also noted that the hydrogen bond structures in the D-pathway are strongly affected by the presence of the excess proton. This is due to the response of the water configurations to the excess proton and implies that using water structures alone, in the absence of excess protons, to predict the proton transport mechanism must be interpreted with care.
Another important cluster of residues in the D-pathway is Asn121/Asn139, located about 4Å above the entrance Asp132 (Fig. 3A). An inter-residue hydrogen bond (-N-H---O=) crossing the D-pathway can be observed in the crystal structure and is maintained in MD simulations with a lengtH3Of about 1.8Å [22-24]. This hydrogen bond breaks the water HBC in the D-pathway, thereby eliminating the water density along the pathway and introducing a 5 kcal/mol energy barrier in proton uptake from Asp132 to the Ser200/Ser201 structure when Glu286 is in its protonated form [48]. In fact, there is an important class of mutants for the D-pathway called “uncoupled” mutants, which can totally block the proton pumping, yet have no effect on or even increase the oxidase activity [98, 100-102]. Asn139Asp is one member in this class. The MS-EVB simulations of the D-pathway with the mutant Asn139Asp were thus conducted to study the molecular mechanism of this process. In contrast to the downhill trend of the PMF in the WT enzyme, Asn139Asp significantly changes the proton transport free energy landscape of the D-pathway, such that an energy barrier of about 10 kcal/mol is present [48]. Proton transport from the Glu286 to the PLS in CcO is believed to require a very precise time window (as will be discussed in section 3.3), so the mutants that affect the apparent pKa of Glu286 will affect the proton pumping significantly [55, 103]. Therefore, an interpretation of “uncoupled” mutants may be that the re-protonation process of Glu286 from the D-pathway is slowed and its pKa is shifted. The free energy barrier is likely caused by the change of electrostatic environment due to the introduced negatively charged residue. To confirm this hypothesis, the same MS-EVB simulation was conducted with the Asp139 in neutral/protonated form [48]. Its PMF changed back to downhill and was very similar to the WT result. This suggests that the electrostatic environment inside the channel may control the deprotonation dynamics of Glu286, even if an amino acid residue that is very far away (further than 10Å) from it. The simulation with neutral Asp139 may also suggest the reason why, in experiments, the swapped double mutants Asn139Asp/Asp132Asn can recover the proton pumping [104-106].
In addition to Asp132, another possible candidate for a proton donor in the D-pathway, Tyr35, was reported recently [107, 108]. It was found that this residue may become the proton donor in the Asp132Asn mutant, and is able to provide a proton to Glu286 when the D-pathway is blocked by the mutant. Therefore, the apparent pKa in the Asp132Asn (>11.0) mutant may be a result of the deprotonation of Tyr35, but not Glu286.
3.3. Proton pumping from Glu286 to PRDa3
Because the D-pathway conducts both pumped protons and chemical protons, it is believed that the Glu286 residue plays the role of a branching point. Four electrons will be transported to the active site in a complete catalytic cycle and, for each transported electron, one proton is pumped and one is passed to the active site (Fig. 1). The delicate mediation of proton translocation implies the importance of the mechanism of Glu286 deprotonation, as proposed in the “kinetic gating” model [37, 109]. An important pathway of proton translocation in the hydrophobic cavity is from Glu286 to the PLS, where the proton is linked to the exit pathway. Although there are different suggestions for the identity of the PLS, PRDa3 may be a possible candidate supported by experiments [33, 34].
How is the proton pumping driven by the electron translocation through the metal centers in CcO? To answer this question, computer simulations of the explicit proton transport process in the hydrophobic cavity are necessary; however, two main challenges exist in the preparation of the simulated systems. The first challenge is that the redox states and the partial charges on the atoms of the CcO catalytic center need to be carefully selected in order to construct a correct electrostatic environment along the proton pathways. These atoms include the metal sites (CuA, CuB and heme a/a3) and surrounding residues coordinated to them, i.e., His333 and His334 in subunit I. The conventional approach is to calculate the electronic structures of the atoms at the catalytic center based on their coordinates in crystal structures utilizing ab-initio methods, then fit the charges by ESP or RESP methods [47, 70, 110, 111]. The second challenge is that, so far, no crystal structure of CcO has revealed water molecules in the hydrophobic cavity. Despite this, the available volume and proton translocation in this region suggest the presence of water molecules. Therefore, before any theoretical studies of the hydrophobic cavity can be conducted, its hydration structure must be carefully prepared and computed with appropriate methods [42, 70, 73, 112].
MS-EVB simulations of proton pumping in the CcO hydrophobic cavity have been conducted in three different redox states, OO, RO and OR, in which each letter here denotes the oxidized (O) or reduced (R) state of heme a/a3, respectively [53, 85]. The free energy profiles of proton translocation were computed when the catalytic center was in one of these three states. The results show that the excess proton is most likely to leave the Glu286 in the RO state, translocating to the second or third solvation shell of Glu286. It is then modulated by a downhill free energy landscape in the RO state and transferred to the PLS. In the OR or OO states, the proton pumping encounters a large free energy barrier, so it is likely to stay at Glu286. This MS-EVB simulation has supported the idea that the driving force for the proton pumping is due to the electron transport along the metal sites [113, 114].
The free energy barriers found in the MS-EVB simulations indicate a high pumping rate for this process, which is in agreement with the “kinetic gating” theory [37, 40]. Because the excess proton PMF is very flat for proton pumping, a mechanism to prevent proton back leak may be important. The MS-EVB simulations also revealed that after the deprotonation in its “up” state, Glu286 could transition to its “down” state only in the RO electron state [85]. Therefore, Glu286 can quickly reorient into the D-pathway and accept another proton in order to prevent the proton leak [55, 115]. This mechanism was questioned recently by the study conducted by Cui and co-workers [99]. In their standard MD simulations, the Glu286 always favors the “down” state, and thus could be reprotonated from the D-pathway without the re-orientation. However, their simulations did not include explicit proton transport and an excess proton in the water mediated pathway. MS-EVB simulations also showed that the proton pumping PMF is not affected by the species bound to BNC (i.e., O2, OH- or H2O) [85], which suggests that the four pumped protons in one catalytic cycle are controlled by the same pumping mechanism.
In the MS-EVB simulations, the excess proton ultimately reaches PRDa3, which is about 8Å above Glu286. PRDa3 is salt-bridged with nearby Arg481 both in crystal structures and in MD simulations when the excess proton is absent [53]. When the proton is present, PRDa3 becomes detached from Arg481; a structure was discovered that showed the excess proton is solvated by two water molecules and the charged carboxyl group (Fig. 2B). This is similar to the situation when the excess proton is near Glu286. MD simulations also reveal that the PRDa3/Arg481 pair is in loose contact and can be easily dissociated when the excess proton is present. The PMF calculations show that the rotation of the PRDa3 dihedral away from Arg481 requires only about 5kcal/mol energy, therefore supporting the PRDa3 as a potential PLS.
4. Conclusions
The CcO protein drives transmembrane proton pumping by an electron flux in the opposite direction. The protein achieves the charge separation across the membrane, which is used later in ATP synthesis or other biological processes. Understanding the mechanism of the redox-coupled proton pump can give insight into biological energy translocation and perhaps contribute to the design of new materials, drugs, nanodevices, etc. The MS-EVB method, which is an efficient and accurate reactive MD approach for the simulation of proton transport processes, has been applied in computational studies of CcO and has revealed the structural and dynamical properties of excess protons in the pumping pathway. The MS-EVB simulations of CcO, which were mainly focused on the proton uptake in the D-pathway from Asp132 to Glu286 and the gated translocation from Glu286 to PRDa3, a potential PLS, have been discussed and summarized in this article. Explicit proton transport via the Grotthuss mechanism through the water HBC was simulated, and the free energy profiles (PMFs) along the translocation pathways were computed. It was shown that the excess protonic charge defect can be greatly delocalized across multiple water molecules in CcO, and its dynamics are significantly affected by Grotthuss shuttling along the hydrogen bonded water pathways. The polar and charged residues, and also the redox states of the metal complex, modulate the proton transport process through the electrostatic environment, hydration structures, and water dynamics.
In the future, more subunits in CcO and the membrane environment will be considered in the MS-EVB simulations. In most computer simulations in the past, only subunit I was used (in very few cases subunit II was included) due to the reduced computational cost. However, it is believed that subunit III also impacts the proton uptake in the D-pathway [116-118]. Another extension of the methodology in future will be to achieve greater accuracy in modeling the protonatable groups [92, 119]. These groups, such as titratable residues, D-propionate, and hydroxide, can behave as proton acceptors/donors involved in the proton transport and charge translocation. Furthermore, these future studies will be more focused on the proton transport pathways in CcO that have not been simulated by MS-EVB before: 1) the proton exits from PLS to the P-side of the membrane and 2) the “chemical” pathway, which corresponds to the Pr →F transition [37, 98, 120-122] in the catalytic cycle and transports a proton from Glu286 to the active site [60]. These MS-EVB studies will further contribute to our overall understanding of the mechanism of CcO and ultimately to that of other redox-driven proton pumps.
Highlights.
Reactive molecular dynamics studies of proton transport in cytochrome c oxidase are reported.
The multi-state empirical valence bond (MS-EVB) methodology is used.
Proton transport via the shuttling mechanism through water molecules in CcO was thus studied.
Two CcO proton transport pathways were studied (D-channel and hydrophobic cavity past E286).
Molecular insight is provided how the electronic state modulates the proton transport.
Acknowledgements
This research was supported by the National Institutes of Health (NIH grant R01-GM053148). The contributions of present and former members from the Voth Group, who participated in the development of MS-EVB methodology and studies on cytochrome c oxidase referred to in this article, must also be acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Belevich I, Verkhovsky MI, Wikstrom M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 2.Brzezinski P, Gennis RB. Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosler JP, Ferguson-Miller S, Mills DA. Energy transduction: Proton transfer through the respiratory complexes. Annu Rev Biochem. 2006;75:165–187. doi: 10.1146/annurev.biochem.75.062003.101730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaila VRI, Verkhovsky MI, Wikstrom M. Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 5.Vanverseveld HW, Krab K, Stouthamer AH. Proton Pump Coupled to Cytochrome-C Oxidase in Paracoccus-Denitrificans. Biochim. Biophys. Acta. 1981;635:525–534. doi: 10.1016/0005-2728(81)90111-0. [DOI] [PubMed] [Google Scholar]
- 6.Wikstrom M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim. Biophys. Acta. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Wikstrom MKF. Proton Pump Coupled to Cytochrome-C Oxidase in Mitochondria. Nature. 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 8.Zaslavsky D, Gennis RB. Proton pumping by cytochrome oxidase: progress, problems and postulates. Biochim. Biophys. Acta. 2000;1458:164–179. doi: 10.1016/s0005-2728(00)00066-9. [DOI] [PubMed] [Google Scholar]
- 9.Brzezinski P, Faxen K, Gilderson G, Adelroth P. A mechanistic principle for proton pumping by cytochrome c oxidase. Nature. 2005;437:286–289. doi: 10.1038/nature03921. [DOI] [PubMed] [Google Scholar]
- 10.Cukierman S. Et tu, Grotthuss! and other unfinished stories. Biochim. Biophys. Acta. 2006;1757:876–885. doi: 10.1016/j.bbabio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 11.de Grotthuss CJT. Sur la décomposition de l'eau et des corps qu'elle tient en dissolution à l'aide de l'électricité galvanique. Anal. Chim. 1806;58:54–73. [Google Scholar]
- 12.Schmitt UW, Voth GA. The computer simulation of proton transport in water. J Chem Phys. 1999;111:9361–9381. [Google Scholar]
- 13.Marx D, Tuckerman ME, Hutter J, Parrinello M. The nature of the hydrated excess proton in water. Nature. 1999;397:601–604. [Google Scholar]
- 14.Agmon N, Markovitch O, Chen H, Izvekov S, Paesani F, Voth GA. Special pair dance and partner selection: Elementary steps in proton transport in liquid water. J Phys Chem B. 2008;112:9456–9466. doi: 10.1021/jp804018y. [DOI] [PubMed] [Google Scholar]
- 15.Swanson JMJ, Simons J. Role of Charge Transfer in the Structure and Dynamics of the Hydrated Proton. J Phys Chem B. 2009;113:5149–5161. doi: 10.1021/jp810652v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onsager L. Motion of Ions - Principles and Concepts. Science. 1969;166:1359–1364. doi: 10.1126/science.166.3911.1359. [DOI] [PubMed] [Google Scholar]
- 17.DeCoursey TE. Voltage-Gated Proton Channels and Other Proton Transfer Pathways. Physiol. Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- 18.Nagle JF, Tristramnagle S. Hydrogen-Bonded Chain Mechanisms for Proton Conduction and Proton Pumping. J Membrane Biol. 1983;74:1–14. doi: 10.1007/BF01870590. [DOI] [PubMed] [Google Scholar]
- 19.Dellago C, Naor MM, Hummer G. Proton transport through water-filled carbon nanotubes. Phys. Rev. Lett. 2003;90 doi: 10.1103/PhysRevLett.90.105902. [DOI] [PubMed] [Google Scholar]
- 20.Voth GA, Brewer ML, Schmitt UW. The formation and dynamics of proton wires in channel environments. Biophys J. 2001;80:1691–1702. doi: 10.1016/S0006-3495(01)76140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan TY, Cao Z, Peng YX, Li S, Li AL, Voth GA. Mechanism of Fast Proton Transport along One-Dimensional Water Chains Confined in Carbon Nanotubes. J. Am. Chem. Soc. 2010;132:11395–11397. doi: 10.1021/ja1046704. [DOI] [PubMed] [Google Scholar]
- 22.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, ShinzawaItoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 angstrom. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 23.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8-Angstrom Resolution of Cytochrome-C-Oxidase from Paracoccus-Denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 24.Svensson-Ek M, Abramson J, Larsson G, Tornroth S, Brzezinski P, Iwata S. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 25.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. P Natl Acad Sci USA. 1997;94:9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fetter JR, Qian J, Shapleigh J, Thomas JW, Garciahorsman A, Schmidt E, Hosler J, Babcock GT, Gennis RB, Ferguson-Miller S. Possible Proton Relay Pathways in Cytochrome-C-Oxidase. P Natl Acad Sci USA. 1995;92:1604–1608. doi: 10.1073/pnas.92.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimokata K, Katayama Y, Murayama H, Suematsu M, Tsukihara T, Muramoto K, Aoyama H, Yoshikawa S, Shimada H. The proton pumping pathway of bovine heart cytochrome c oxidase. P Natl Acad Sci USA. 2007;104:4200–4205. doi: 10.1073/pnas.0611627104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamiya K, Boero M, Tateno M, Shiraishi K, Oshiyama A. Possible mechanism of proton transfer through peptide groups in the H-pathway of the bovine cytochrome c oxidase. J. Am. Chem. Soc. 2007;129:9663–9673. doi: 10.1021/ja070464y. [DOI] [PubMed] [Google Scholar]
- 29.Yoshikawa S, Muramoto K, Shinzawa-Itoh K, Aoyama H, Tsukihara T, Shimokata K, Katayama Y, Shimada H. Proton pumping mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta. 2006;1757:1110–1116. doi: 10.1016/j.bbabio.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Yoshikawa S, Shimokata K, Katayama Y, Murayama H, Suematsu M, Tsukihara T, Muramoto K, Aoyama H, Shimada H. The proton pumping pathway of bovine heart cytochrome c oxidase. P Natl Acad Sci USA. 2007;104:4200–4205. doi: 10.1073/pnas.0611627104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshikawa S, Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyoma H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. P Natl Acad Sci USA. 2003;100:15304–15309. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HJ, Ojemyr L, Vakkasoglu A, Brzezinski P, Gennis RB. Properties of Arg481 Mutants of the aa(3)-Type Cytochrome c Oxidase from Rhodobacter sphaeroides Suggest That neither R481 nor the Nearby D-Propionate of Heme a(3) Is Likely To Be the Proton Loading Site of the Proton Pump. Biochemistry. 2009;48:7123–7131. doi: 10.1021/bi901015d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma V, Wikstrom M, Kaila VRI. Redox-coupled proton transfer in the active site of cytochrome cbb(3) Biochim. Biophys. Acta. 2010;1797:1512–1520. doi: 10.1016/j.bbabio.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Kaila VRI, Sharma V, Wikstrom M. The identity of the transient proton loading site of the proton-pumping mechanism of cytochrome c oxidase. Biochim. Biophys. Acta. 2011;1807:80–84. doi: 10.1016/j.bbabio.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 35.Belevich I, Bloch DA, Belevich N, Wikstrom M, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. P Natl Acad Sci USA. 2007;104:2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muramoto K, Hirata K, Shinzawa-Itoh K, Yoko-O S, Yamashita E, Aoyama H, Tsukihara T, Yoshikawa S. A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. P Natl Acad Sci USA. 2007;104:7881–7886. doi: 10.1073/pnas.0610031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popovic DM, Stuchebrukhov AA. Proton pumping mechanism and catalytic cycle of cytochrome c oxidase: Coulomb pump model with kinetic gating. Febs Lett. 2004;566:126–130. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 38.Henry RM, Yu CH, Rodinger T, Pomes R. Functional Hydration and Conformational Gating of Proton Uptake in Cytochrome c Oxidase. J Mol Biol. 2009;387:1165–1185. doi: 10.1016/j.jmb.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 39.Lee HJ, Svahn E, Swanson JM, Lepp H, Voth GA, Brzezinski P, Gennis RB. Intricate role of water in proton transport through cytochrome c oxidase. J. Am. Chem. Soc. 2010;132:16225–16239. doi: 10.1021/ja107244g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brzezinski P, Malmstrom BG. Electron-Transport-Driven Proton Pumps Display Nonhyperbolic Kinetics - Simulation of the Steady-State Kinetics of Cytochrome-COxidase. P Natl Acad Sci USA. 1986;83:4282–4286. doi: 10.1073/pnas.83.12.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Backgren C, Hummer G, Wikstrom M, Puustinen A. Proton translocation by cytochrome c oxidase can take place without the conserved glutamic acid in subunit I. Biochemistry. 2000;39:7863–7867. doi: 10.1021/bi000806b. [DOI] [PubMed] [Google Scholar]
- 42.Zheng XH, Medvedev DM, Swanson J, Stuchebrukhov AA. Computer simulation of water in cytochrome c oxidase. Biochim. Biophys. Acta. 2003;1557:99–107. doi: 10.1016/s0005-2728(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 43.Cukier RI. Quantum molecular dynamics simulation of proton transfer in cytochrome c oxidase. Biochim. Biophys. Acta. 2004;1656:189–202. doi: 10.1016/j.bbabio.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 44.Olkhova E, Hutter MC, Lill MA, Helms V, Michel H. Dynamic water networks in cytochrome c oxidase from Paracoccus denitrificans investigated by molecular dynamics simulations. Biophys J. 2004;86:1873–1889. doi: 10.1016/S0006-3495(04)74254-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olsson MHM, Warshel A. Simulating redox coupled proton transfer in cytochrome c oxidase. Biophys J. 2005;88:548A–548A. doi: 10.1016/j.febslet.2005.02.051. [DOI] [PubMed] [Google Scholar]
- 46.Xu J, Voth GA. Computer simulation of explicit proton translocation in cytochrome c oxidase: the D-pathway. P Natl Acad Sci USA. 2005;102:6795–6800. doi: 10.1073/pnas.0408117102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tashiro M, Stuchebrukhov AA. Thermodynamic properties of internal water molecules in the hydrophobic cavity around the catalytic center of cytochrome c oxidase. J Phys Chem B. 2005;109:1015–1022. doi: 10.1021/jp0462456. [DOI] [PubMed] [Google Scholar]
- 48.Xu JC, Voth GA. Free energy profiles for H+ conduction in the D-pathway of Cytochrome c Oxidase: A study of the wild type and N98D mutant enzymes. Biochim. Biophys. Acta. 2006;1757:852–859. doi: 10.1016/j.bbabio.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 49.Olsson MHM, Warshel A. Monte Carlo simulations of proton pumps: On the working principles of the biological valve that controls proton pumping in cytochrome c oxidase. P Natl Acad Sci USA. 2006;103:6500–6505. doi: 10.1073/pnas.0510860103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J, Sharpe MA, Qin L, Ferguson-Miller S, Voth GA. Storage of an excess proton in the hydrogen-bonded network of the d-pathway of cytochrome C oxidase: identification of a protonated water cluster. J. Am. Chem. Soc. 2007;129:2910–2913. doi: 10.1021/ja067360s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warshel A, Olsson MHM, Slegbahn PEM, Blomberg MRA. Exploring pathways and barriers for coupled ET/PT in cytochrome c oxidase: A general framework for examining energetics and mechanistic alternatives. Biochim. Biophys. Acta. 2007;1767:244–260. doi: 10.1016/j.bbabio.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tuukkanen A, Kaila VRI, Laakkonen L, Hummer G, Wikstrom M. Dynamics of the glutamic acid 242 side chain in cytochrome c oxidase. Biochim. Biophys. Acta. 2007;1767:1102–1106. doi: 10.1016/j.bbabio.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 53.Xu J, Voth GA. Redox-coupled proton pumping in cytochrome c oxidase: further insights from computer simulation. Biochim. Biophys. Acta. 2008;1777:196–201. doi: 10.1016/j.bbabio.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pisliakov AV, Sharma PK, Chu ZT, Haranczyk M, Warshel A. Electrostatic basis for the unidirectionality of the primary proton transfer in cytochrome c oxidase. P Natl Acad Sci USA. 2008;105:7726–7731. doi: 10.1073/pnas.0800580105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaila VRI, Verkhovsky MI, Hummer G, Wikstrom M. Glutamic acid 242 is a valve in the proton pump of cytochrome c oxidase. P Natl Acad Sci USA. 2008;105:6255–6259. doi: 10.1073/pnas.0800770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sugitani R, Stuchebrukhov AA. Molecular dynamics simulation of water in cytochrome c oxidase reveals two water exit pathways and the mechanism of transport. Biochim. Biophys. Acta. 2009;1787:1140–1150. doi: 10.1016/j.bbabio.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cui Q, Riccardi D, Yang S. Proton transfer function of carbonic anhydrase: Insights from QM/MM simulations. Biochim. Biophys. Acta. 2010;1804:342–351. doi: 10.1016/j.bbapap.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pisliakov A, Sugita Y, Warshel A. Computer simulations of proton transfer in cytochrome c oxidase and nitric oxide reductase. Biochim. Biophys. Acta. 2010;1797:99–100. [Google Scholar]
- 59.Daskalakis V, Farantos SC, Guallar V, Varotsis C. Regulation of Electron and Proton Transfer by the Protein Matrix of Cytochrome c Oxidase. J Phys Chem B. 2011;115:3648–3655. doi: 10.1021/jp1115993. [DOI] [PubMed] [Google Scholar]
- 60.Johansson AL, Chakrabarty S, Sioberg CB, Hogbom M, Warshel A, Brzezinski P. Proton-transport mechanisms in cytochrome c oxidase revealed by studies of kinetic isotope effects. Biochim. Biophys. Acta. 2011 doi: 10.1016/j.bbabio.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chakrabarty S, Namslauer I, Brzezinski P, Warshel A. Exploration of the cytochrome c oxidase pathway puzzle and examination of the origin of elusive mutational effects. Biochim. Biophys. Acta. 2011;1807:413–426. doi: 10.1016/j.bbabio.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dziekonski P, Sokalski WA. The nature of catalytic activity of molecular environment on proton transfer reactions: Testing reliability of QM/MM results. J Mol Graph Model. 1998;16:279–279. [Google Scholar]
- 63.Gready JE, Cummins PL. Application of free energy perturbation QM/(MM + MD) methods to the study of proton affinities in enzyme-active sites. Abstr Pap Am Chem S. 1999;218:U527–U527. [Google Scholar]
- 64.Hillier IH, Burton NA, Nicoll RM, Sheppard DW. Proton and hydride ion transfers in biological systems modeled by QM/MM methods. Abstr Pap Am Chem S. 1999;218:U527–U527. [Google Scholar]
- 65.Jensen JH, Kairys V. QM/MM boundaries across covalent bonds: Proton affinities of amino acids and small peptides. Abstr Pap Am Chem S. 1999;218:U510–U510. [Google Scholar]
- 66.Hayashi S, Ohmine I, Schulten K. Ab initio QM/MM and molecular dynamics study on proton pump mechanism of bacteriorhodopsin. Biophys J. 2001;80:603A–604A. [Google Scholar]
- 67.Galvan IF, Volbeda A, Fontecilla-Camps JC, Field MJ. A QM/MM study of proton transport pathways in a [NiFe] hydrogenase, Proteins: Struct. Funct. Bioinform. 2008;73:195–203. doi: 10.1002/prot.22045. [DOI] [PubMed] [Google Scholar]
- 68.Altarsha M, Wang DQ, Benighaus T, Kumar D, Thiel W. QM/MM Study of the Second Proton Transfer in the Catalytic Cycle of the D251N Mutant of Cytochrome P450cam. J Phys Chem B. 2009;113:9577–9588. doi: 10.1021/jp809838k. [DOI] [PubMed] [Google Scholar]
- 69.Riccardi D, Yang S, Cui Q. Proton transfer function of carbonic anhydrase: Insights from QM/MM simulations. Biochim. Biophys. Acta. 2010;1804:342–351. doi: 10.1016/j.bbapap.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghosh N, Prat-Resina X, Gunner MR, Cui Q. Microscopic pK(a) Analysis of Glu286 in Cytochrome c Oxidase (Rhodobacter sphaeroides): Toward a Calibrated Molecular Model. Biochemistry. 2009;48:2468–2485. doi: 10.1021/bi8021284. [DOI] [PubMed] [Google Scholar]
- 71.Li GH, Zhang XD, Cui Q. Free energy perturbation calculations with combined QM/MM Potentials complications, simplifications, and applications to redox potential calculations. J Phys Chem B. 2003;107:8643–8653. [Google Scholar]
- 72.Popovic DM, Stuchebrukhov AA. Two conformational states of Glu242 and pK(a)s in bovine cytochrome c oxidase. Photoch Photobio Sci. 2006;5:611–620. doi: 10.1039/b600096g. [DOI] [PubMed] [Google Scholar]
- 73.Song YF, Michonova-Alexova E, Gunner MR. Calculated proton uptake on anaerobic reduction of cytochrome c oxidase: Is the reaction electroneutral? Biochemistry. 2006;45:7959–7975. doi: 10.1021/bi052183d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sharp KA, Honig B. Electrostatic Interactions in Macromolecules - Theory and Applications. Annu. Rev. Biophys. Biophys. Chem. 1990;19:301–332. doi: 10.1146/annurev.bb.19.060190.001505. [DOI] [PubMed] [Google Scholar]
- 75.Schaefer M, Karplus M. A comprehensive analytical treatment of continuum electrostatics. J Phys Chem-Us. 1996;100:1578–1599. [Google Scholar]
- 76.Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophys J. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Warshel A, Sussman F, King G. Free-Energy of Charges in Solvated Proteins - Microscopic Calculations Using a Reversible Charging Process. Biochemistry. 1986;25:8368–8372. doi: 10.1021/bi00374a006. [DOI] [PubMed] [Google Scholar]
- 78.Hummer G, Szabo A. Calculation of free-energy differences from computer simulations of initial and final states. J Chem Phys. 1996;105:2004–2010. [Google Scholar]
- 79.Day TJF, Soudackov AV, Cuma M, Schmitt UW, Voth GA. A second generation multistate empirical valence bond model for proton transport in aqueous systems. J Chem Phys. 2002;117:5839–5849. [Google Scholar]
- 80.Schmitt UW, Voth GA. Multistate empirical valence bond model for proton transport in water. J Phys Chem B. 1998;102:5547–5551. [Google Scholar]
- 81.Swanson JM, Maupin CM, Chen H, Petersen MK, Xu J, Wu Y, Voth GA. Proton solvation and transport in aqueous and biomolecular systems: insights from computer simulations. J Phys Chem B. 2007;111:4300–4314. doi: 10.1021/jp070104x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Voth GA. Computer simulation of proton solvation and transport in aqueous and biomolecular systems. Acc. Chem. Res. 2006;39:143–150. doi: 10.1021/ar0402098. [DOI] [PubMed] [Google Scholar]
- 83.Wang F, Voth GA. A linear-scaling self-consistent generalization of the multistate empirical valence bond method for multiple excess protons in aqueous systems. J Chem Phys. 2005;122:144105. doi: 10.1063/1.1881092. [DOI] [PubMed] [Google Scholar]
- 84.Wu Y, Chen H, Wang F, Paesani F, Voth GA. An improved multistate empirical valence bond model for aqueous proton solvation and transport. J Phys Chem B. 2008;112:467–482. doi: 10.1021/jp076658h. [DOI] [PubMed] [Google Scholar]
- 85.Yamashita T, Voth GA. Heme a Reduction Effect on Proton Transport in Cytochrome c Oxidase: A molecular Dynamics Study. 2011. Submitted.
- 86.Warshel A, Weiss RM. An Empirical Valence Bond Approach for Comparing Reactions in Solutions and in Enzymes. J. Am. Chem. Soc. 1980;102:6218–6226. [Google Scholar]
- 87.Kamerlin SCL, Warshel A. The empirical valence bond model: theory and applications. Computational Molecular Science. 2011;1:30–45. [Google Scholar]
- 88.Chen H, Wu Y, Voth GA. Origins of proton transport behavior from selectivity domain mutations of the aquaporin-1 channel. Biophys J. 2006;90:L73–75. doi: 10.1529/biophysj.106.084061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen H, Wu Y, Voth GA. Proton transport behavior through the influenza A M2 channel: insights from molecular simulation. Biophys J. 2007;93:3470–3479. doi: 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang D, Voth GA. Proton transport pathway in the ClC Cl-/H+ antiporter. Biophys J. 2009;97:121–131. doi: 10.1016/j.bpj.2009.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smondyrev AM, Voth GA. Molecular dynamics simulation of proton transport near the surface of a phospholipid membrane. Biophys J. 2002;82:1460–1468. doi: 10.1016/S0006-3495(02)75500-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maupin CM, McKenna R, Silverman DN, Voth GA. Elucidation of the proton transport mechanism in human carbonic anhydrase II. J. Am. Chem. Soc. 2009;131:7598–7608. doi: 10.1021/ja8091938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Roux B. The Calculation of the Potential of Mean Force Using Computer-Simulations. Comput Phys Commun. 1995;91:275–282. [Google Scholar]
- 94.Torrie GM, Valleau JP. Monte-Carlo Free-Energy Estimates Using Non-Boltzmann Sampling - Application to Subcritical Lennard-Jones Fluid. Chem Phys Lett. 1974;28:578–581. [Google Scholar]
- 95.Riistama S, Hummer G, Puustinen A, Dyer RB, Woodruff WH, Wikstrom M. Bound water in the proton translocation mechanism of the haem-copper oxidases. Febs Lett. 1997;414:275–280. doi: 10.1016/s0014-5793(97)01003-x. [DOI] [PubMed] [Google Scholar]
- 96.Wikstrom M, Verkhovsky MI, Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta. 2003;1604:61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 97.Adelroth P, Ek MS, Mitchell DM, Gennis RB, Brzezinski P. Glutamate 286 in cytochrome aa(3) from Rhodobacter sphaeroides is involved in proton uptake during the reaction of the fully-reduced enzyme with dioxygen. Biochemistry. 1997;36:13824–13829. doi: 10.1021/bi9629079. [DOI] [PubMed] [Google Scholar]
- 98.Namslauer A, Aagaard A, Katsonouri A, Brzezinski P. Intramolecular proton-transfer reactions in a membrane-bound proton pump: The effect of pH on the peroxy to ferryl transition in cytochrome c oxidase. Biochemistry. 2003;42:1488–1498. doi: 10.1021/bi026524o. [DOI] [PubMed] [Google Scholar]
- 99.Yang SO, Cui Q. Glu-286 Rotation and Water Wire Reorientation Are Unlikely the Gating Elements for Proton Pumping in Cytochrome c Oxidase. Biophys J. 2011;101:61–69. doi: 10.1016/j.bpj.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pfitzner U, Hoffmeier K, Harrenga A, Kannt A, Michel H, Bamberg E, Richter OMH, Ludwig B. Tracing the D-pathway in reconstituted site-directed mutants of cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 2000;39:6756–6762. doi: 10.1021/bi992235x. [DOI] [PubMed] [Google Scholar]
- 101.Pawate AS, Morgan J, Namslauer A, Mills D, Brzezinski P, Ferguson-Miller S, Gennis RB. A mutation in subunit I of cytochrome oxidase from Rhodobacter sphaeroides results in an increase in steady-state activity but completely eliminates proton pumping. Biochemistry. 2002;41:13417–13423. doi: 10.1021/bi026582+. [DOI] [PubMed] [Google Scholar]
- 102.Han D, Namslauer A, Pawate A, Morgan JE, Nagy S, Vakkasoglu AS, Brzezinski P, Gennis RB. Replacing Asn207 by aspartate at the neck of the D channel in the aa(3)-type cytochrome c oxidase from Rhodobacter sphaeroides results in decoupling the proton pump. Biochemistry. 2006;45:14064–14074. doi: 10.1021/bi061465q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lepp H, Salomonsson L, Zhu JP, Gennis RB, Brzezinski P. Impaired proton pumping in cytochrome c oxidase upon structural alteration of the D pathway. Biochim. Biophys. Acta. 2008;1777:897–903. doi: 10.1016/j.bbabio.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Branden G, Pawate AS, Gennis RB, Brzezinski P. Controlled uncoupling and recoupling of proton pumping in cytochrome c oxidase. P Natl Acad Sci USA. 2006;103:317–322. doi: 10.1073/pnas.0507734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hosler J, Varanasi L. Alternative Initial Proton Acceptors for the D Pathway of Rhodobacter sphaeroides Cytochrome c Oxidase. Biochemistry. 2011;50:2820–2828. doi: 10.1021/bi102002v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pomes R, Henry RM, Caplan D, Fadda E. Molecular basis of proton uptake in single and double mutants of cytochrome c oxidase. J Phys-Condens Mat. 2011;23 doi: 10.1088/0953-8984/23/23/234102. [DOI] [PubMed] [Google Scholar]
- 107.Verkhovsky MI, Belevich I, Gorbikova E, Belevich NP, Rauhamaki V, Wikstrom M. Initiation of the proton pump of cytochrome c oxidase. P Natl Acad Sci USA. 2010;107:18469–18474. doi: 10.1073/pnas.1010974107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wikstrom M, Verkhovsky MI. The D-channel of cytochrome oxidase: An alternative view. Biochim. Biophys. Acta. 2011;1807:1273–1278. doi: 10.1016/j.bbabio.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 109.Kim YC, Wikstrom M, Hummer G. Kinetic gating of the proton pump in cytochrome c oxidase. P Natl Acad Sci USA. 2009;106:13707–13712. doi: 10.1073/pnas.0903938106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Autenrieth F, Tajkhorshid E, Baudry J, Luthey-Schulten Z. Classical force field parameters for the heme prosthetic group of cytochrome c. J Comput Chem. 2004;25:1613–1622. doi: 10.1002/jcc.20079. [DOI] [PubMed] [Google Scholar]
- 111.Johansson MP, Kaila VRI, Laakkonen L. Charge parameterization of the metal centers in cytochrome c oxidase. J Comput Chem. 2008;29:753–767. doi: 10.1002/jcc.20835. [DOI] [PubMed] [Google Scholar]
- 112.Woo HJ, Dinner AR, Roux B. Grand canonical Monte Carlo simulations of water in protein environments. J Chem Phys. 2004;121:6392–6400. doi: 10.1063/1.1784436. [DOI] [PubMed] [Google Scholar]
- 113.Wikstrom M, Krab K, Saraste M. Proton-Translocating Cytochrome Complexes. Annu Rev Biochem. 1981;50:623–655. doi: 10.1146/annurev.bi.50.070181.003203. [DOI] [PubMed] [Google Scholar]
- 114.Malmstrom BG. Hypothesis - Cytochrome-C Oxidase as a Proton Pump - a Transition-State Mechanism. Biochim. Biophys. Acta. 1985;811:1–12. [PubMed] [Google Scholar]
- 115.Kaila VRI, Verkhovsky M, Hummer G, Wikstrom M. Prevention of leak in the proton pump of cytochrome c oxidase. Biochim. Biophys. Acta. 2008;1777:890–892. doi: 10.1016/j.bbabio.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 116.Gilderson G, Salomonsson L, Aagaard A, Gray J, Brzezinski P, Hosler J. Subunit III of cytochrome c oxidase of Rhodobacter sphaeroides is required to maintain rapid proton uptake through the D pathway at physiologic pH. Biochemistry. 2003;42:7400–7409. doi: 10.1021/bi0341298. [DOI] [PubMed] [Google Scholar]
- 117.Mills DA, Tan Z, Ferguson-Miller S, Hosler J. A role for subunit III in proton uptake into the D pathway and a possible proton exit pathway in Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 2003;42:7410–7417. doi: 10.1021/bi0341307. [DOI] [PubMed] [Google Scholar]
- 118.Hosler JP. The influence of subunit III of cytochrorne c oxidase on the D pathway, the proton exit pathway and mechanism-based inactivation in subunit I. Biochim. Biophys. Acta. 2004;1655:332–339. doi: 10.1016/j.bbabio.2003.06.009. [DOI] [PubMed] [Google Scholar]
- 119.Maupin CM, Wong KF, Soudackov AV, Kim S, Voth GA. A multistate empirical valence bond description of protonatable amino acids. J Phys Chem A. 2006;110:631–639. doi: 10.1021/jp053596r. [DOI] [PubMed] [Google Scholar]
- 120.Brzezinski P, Larsson G. Redox-driven proton pumping by heme-copper oxidases. Biochim. Biophys. Acta. 2003;1605:1–13. doi: 10.1016/s0005-2728(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 121.Namslauer A, Brzezinski P. Structural elements involved in electron-coupled proton transfer in cytochrome c oxidase. Febs Lett. 2004;567:103–110. doi: 10.1016/j.febslet.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 122.Popovic DM, Stuchebrukhov AA. Electrostatic study of the proton pumping mechanism in bovine heart cytochrome c oxidase. J. Am. Chem. Soc. 2004;126:1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]