

Abstract
In human carbonic anhydrase II (HCA II) the mutation of position 64 from histidine to alanine (H64A) disrupts the rate limiting proton transfer (PT) event, resulting in a reduction of the catalytic activity of the enzyme as compared to the wild-type. Potential of mean force (PMF) calculations utilizing the multistate empirical valence bond (MS-EVB) methodology for H64A HCA II give a PT free energy barrier significantly higher than that found in the wild-type enzyme. This high barrier, determined in the absence of exogenous buffer and assuming no additional ionizable residues in the PT pathway, indicates the likelihood of alternate enzyme pathways that utilize either ionizable enzyme residues (self-rescue) and/or exogenous buffers (chemical rescue). It has been shown experimentally that the catalytic activity of H64A HCA II can be chemically rescued to near wild type levels by the addition of the exogenous buffer 4-methylimidazole (4MI). Crystallographic studies have identified two 4MI binding sites, yet site specific mutations intended to disrupt 4MI binding have demonstrated these sites to be non-productive. In the present work MS-EVB simulations show that binding of 4MI near Thr199 in the H64A HCA II mutant, a binding site determined by NMR spectroscopy, results in a viable chemical rescue pathway. Additional viable rescue pathways are also identified where 4MI acts as a proton transport intermediary from the active site to ionizable residues on the rim of the active site, revealing a probable mode of action for the chemical rescue pathway
Keywords: carbonic anhydrase, human carbonic anhydrase II, proton transport, chemical rescue, 4MI, multistate empirical valence bond
1. Introduction
Proton transport is fundamentally important for many biologically relevant processes.1–4 The carbonic anhydrases (CAs) rely on a rate limiting proton transfer (PT) step and therefore have been extensively utilized as prototypical systems to gain insight into the underlying biophysics that mediate the PT event.5–20 CAs, which can be structurally classified into five families (α, β, γ, δ, and ε), are important metalloenzymes that are ubiquitous in nature. The CAs catalyze the relatively rapid reversible conversion of carbon dioxide to bicarbonate and an excess proton.21–24 Catalyzing this particular reaction has made the β-CAs central to photosynthesis in plants, and the α-CAs central to various physiological processes in mammals such as respiration and pH maintenance.21–26
A great deal of attention has been directed towards human carbonic anhydrase II (HCA II),4,14,17 which is one of the fastest enzymes known with a turnover ~ 0.8 μs−1. The reaction catalyzed by HCA II proceeds in two distinct stages, where the first stage equation (eq) 1 is the conversion of CO2 into bicarbonate by the nucleophilic attack of the zinc-bound hydroxide on CO2 and the subsequent displacement of the bicarbonate by an active site water:
| (1) |
The second stage in eqs 2, 3 below is the transfer of a proton to solution thereby regenerating the zinc-bound hydroxide.27–35 This is the rate limiting stage in maximum velocity and has two parts, the PT between the zinc-bound water and His64 (the active site PT shuttling residue), eq 2, and the PT between His64 and an exogenous buffer, eq 3:
| (2) |
| (3) |
Experimental21,36 and computational19 work indicate that eq 2 is rate limiting under physiological conditions where the exogenous buffers are abundant, while eq 3 is rate limiting at low exogenous buffer concentrations.19,21,28,29,36 Three important features of the reaction in eq 2 are the active site water cluster that connects the zinc-bound water to His64, the two different orientations of His64 side chain, and the pKa difference between the proton donor and acceptor.22,37,38 The two orientations of His64 are the inward ( ) with the imidazole ring of His64 inside the active site pointing toward the zinc, and the outward ( ), with the imidazole ring outside the active site. While the reaction in eq 2 depends primarily on properties inside the active site, the reaction in eq 3 depends on the exogenous buffer concentration that acts as a proton sink or reservoir. It is the presence of the proton sink that creates a favorable free energy environment for the deprotonation of His64H+ while in the outward orientation and this, in turn, eliminates/reduces the probability of the back reaction from occurring (the dehydration direction).
The distance for the intramolecular PT event, which spans 8 to 10 Å, is too large for direct PT even with His64 in the inward orientation and is therefore dependent on the stability of the active site water cluster.18–20 Computational studies indicate that short non-branched water clusters connecting the zinc-bound water and an inward oriented His64 play a major role in the elevated catalytic rate seen in the Y7F mutant, while disorder in the active site water cluster is linked to a reduction in the catalytic rate of the N67L mutant.18,20 In addition to the active site water cluster’s stability and size, the orientation of His64 has recently been shown to have an influence on the rate limiting PT event with the inward orientation more favorable than the outward.19 For additional information on the computational studies of HCA II the readers are referred to a recent review.17
The importance of His64 to the catalytic rate of HCA II was clearly demonstrated in the experimental studies34 carried out on the H64A mutant, where His64 was replaced by alanine, a non-ionizable amino acid. This experiment revealed a 20 to 50 fold decrease in the reaction rate, thus identifying His64 as the prominent proton donor/acceptor residue. The presence of a reduced rate indicates that the mutant is using an alternate pathway(s) for PT in the absence of His64, which is less favorable for PT (higher free energy barrier). Subsequent studies using the exogenous buffer 4-methylimidazole (4MI) showed the catalytic rate of H64A HCA II could be chemically rescued to near WT levels, demonstrating the importance of an imidazole containing molecule acting as a proton donor/acceptor in the active site cavity with a similar pKa to that of the zinc bound water.35 Two binding sites for 4MI have been identified in the active site of H64A HCA II using x-ray crystallography.39,40 One of the binding sites showed 4MI in a π-stacking interaction with the indole ring of Trp5, occupying a similar position as His64 in the outward orientation in the WT enzyme (rZn-N4MI ≈ 10 Å). The other binding site of 4MI was located near Asn67, Glu69, Asp72 and Ile91 (rZn-N4MI ≈ 13 Å). Kinetic studies have revealed that these binding sites are non-productive, indicating the use of additional viable transient proton transport pathway(s), which working alone or in combination are responsible for the chemical rescue event.41–43 In addition to the x-ray structures an NMR study conducted using cobalt H64A HCA II identified an additional binding site where 4MI resides deep in the active site near the catalytic cobalt (rCo-N4MI ≈ 5 Å).43 These experiments highlight the need for an atomistic level understanding of the mechanism by which the 4MI chemical rescue agent takes part in the rate limiting PT event.
In order for the 4MI molecule to chemically rescue the H64A HCA II mutant, either 4MI acts as a surrogate for His64 or it mediates an as of yet unknown alternate PT pathway. For 4MI to be acting as a surrogate, it would need to bind/occupy a region of space that is favorable to PT. The optimal region corresponds to a separation distance that would place the imidazole ring at a distance no greater than ~ 8 Å from the catalytic zinc.17,19,43 This separation distance favors smaller, more stable water clusters connecting the proton donor/acceptor and would therefore have a positive impact on the PT event.9,17,18 Computational studies of plausible hydrogen bonded water cluster pathways for H64A HCA II, with and without 4MI, have been conducted on the static crystallographic structure and its variants. To reveal the impact of 4MI on the hydrogen bonding network these systems were modeled with reorganized water structures at and near the active site, as predicted by the potential of mean force (PMF) method.44,45 It was found that when 4MI is absent from the active site no proton pathways are found that connect the zinc-bound water to any ionizable enzyme residues at the protein surface. However, when 4MI is present in the x-ray binding sites, several complete hydrogen bonded pathways are detected. This analysis indicates that the x-ray binding sites may be productive chemical rescue pathways either by directly participating in the rate liming PT event or by having a favorable impact on the formation of hydrogen bonded water networks that may shuttle the excess proton to other ionizable residues.
Clearly, there are many unanswered questions pertaining to how 4MI chemically rescues the H64A mutant of HCA II. To address these issues the present study focuses on the characterization of the rate limiting PT event in H64A HCA II in addition to the chemical rescue properties of the exogenous buffer 4MI. The calculation of the PMF for the PT event in H64A HCA II without 4MI and in the absence of any additional ionizable enzyme residues was conducted, as well as PMF calculations in the presence of 4MI to elucidate the many contributing factors to the chemical rescue phenomena. The multi-state empirical valence bond (MS-EVB) methodology3,4 was utilized for these PMF calculations due to its capability of modeling the delocalized nature of the excess proton and capturing the underlying physics critical to the proper representation of an excess proton in bulk water,46–50 weak acid ionization reactions,51–53 and complex biomolecules.3,4 In addition, experimental kinetic studies were conducted to further evaluate the role of the binding site near Glu69, as determined by x-ray crystallography, in the overall chemical rescue of H64A HCA II by 4MI. The simulations identify the viable PT pathway(s) that give rise to the chemical rescue of the H64A HCA II mutant by the exogenous buffer 4MI. These finding may be used to gain a greater understanding and interpretation of the impact mutations have on reaction mechanisms and the role of rescue agents. In particular, the information gained on the chemical rescue phenomena may be used to address small ion rescue of anionic residue mutations, since small ions are virtually impossible to exclude from experiments.54
2. Methodology
Multi-State Empirical Valence Bond (MS-EVB) Methodology
A full description of the MS-EVB methodology can be found in refs3,4,46,47 and will only be briefly summarized here. In the MS-EVB formalism the lowest energy solution of the MS-EVB Hamiltonian matrix defines the potential energy surface upon which the system nuclei propagate via Newtonian mechanics. This matrix is expressed in terms of a dynamically adaptive basis set of empirical valence bond11,55 (EVB) state,|i〉, which form the MS-EVB Hamiltonian matrix elements from the operator
| (4) |
where the vector r represents the positions of the complete set of nuclear degrees of freedom. The diagonal elements of the EVB Hamiltonian, hii, represent a specific bonding topology of the system with the corresponding potential energy as determined by the underlying classical empirical force field for each EVB state |i〉 (in this case parm99 of AMBER56). Figure 1 illustrates the MS-EVB complex and the resulting EVB states |i〉 created for the simple example of 4MI, three water molecules, and one excess proton. The off-diagonal elements, hij, represent the coupling between MS-EVB basis states |i〉 and |j〉. The functional form of the off-diagonal elements are represented by53
Figure 1.
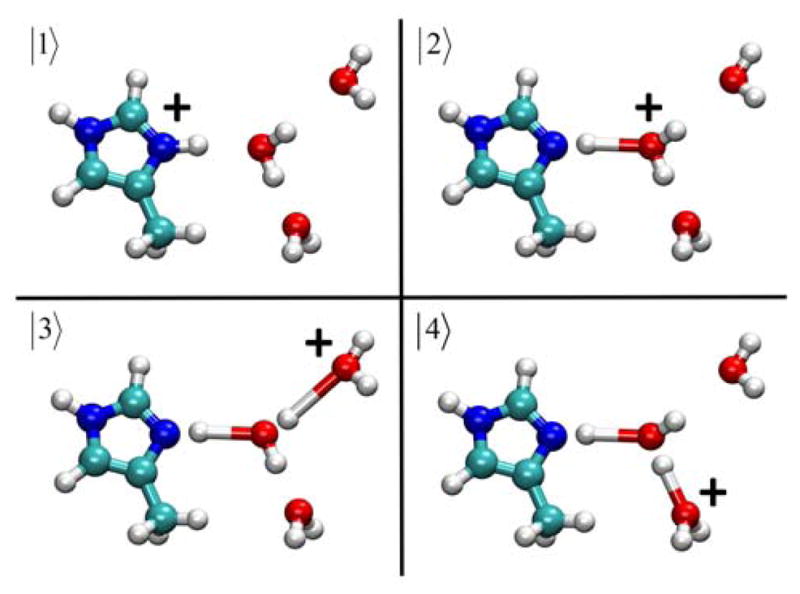
Schematic picture of the 4MIH+-(H2O)3 (|1〉) and 4MI-H3O+-(H2O)2 (|2〉, |3〉, and |4〉) EVB states used in the creation the MS-EVB Hamiltonian matrix.
| (5) |
where q is the PT coordinate, RDA is the distance between donor and acceptor atoms, and is an adjustable parameter. The functional form of f(RDA) for the ionizable residue-hydronium pair is given by53
| (6) |
and g(q) in Eq. (5) is defined as
| (7) |
where C, α, β, γ, ε, aDA, bDA, and cDA are adjustable parameters. The PT reaction coordinate, q, is a geometric reaction coordinate described by
| (8) |
where
| (9) |
and , λ and are additional adjustable parameters.
An additional modification made to the underlying amino acid MS-EVB force field is the substitution of the standard harmonic approximation by a Morse potential, which allows for a more accurate description of the dissociable bond between the excess proton and the donating atom
| (10) |
were a0, a1, and a2 are parameters and r corresponds the equilibrium separation distance. The parameterization process for the MS-EVB methodology has been previously described in detail in refs.19,46,53
Due to the delocalized nature of the charge defect associated with the hydrated excess proton, the center of excess charge (CEC) is used to follow the charge defect. The CEC is defined as48
| (11) |
where ci are the coefficients obtained after the diagonalization of the MS-EVB matrix at each time step and the center of charge (COC) of each MS-EVB basis state is given by
| (12) |
where the summation is over all the atoms contained in the particular EVB state “i” with their respective partial charges qk. The PT reaction coordinate ξ that is used in the current study defines the distance between the donor molecule of interest and the CEC as determined by
| (13) |
were D is the donor molecule such as the catalytic zinc or the Nδ of 4MI.
Parameterization of the 4MI/4MIH+ Moiety
Four ionizable moieties are contained in the present study: hydronium, zinc-bound water, glutamic acid, and 4MI. The parameterization of the hydronium, zinc-bound water and glutamic acid has been reported previously in the literature19,48,53 and therefore only 4MI will be discussed here. The parameterization process used for the 4MI ionizable residue is different than the previously reported procedures due to the similarities between 4MI and histidine. Both residues contain an imidazole ring and only differ in the chemical group attached to the imidazole ring. In addition both residues have similar pKa values, with histidine possessing a lower pKa value than 4MI, 6.0457 and 7.4558, respectively. Therefore, the histidine MS-EVB parameters53 were used for the 4MI model with the exception of the term, which represents the relative diabatic shift between donor and acceptor species (i.e., the models have different pKa values), and the charge distribution which was taken from the underlying force field. The new term was determined by calculating deprotonation PMFs of the 4MI molecule in a box of water molecules and subsequently determining the resulting pKa. The method for calculating the pKa is discussed in ref 53 and incorporates the contributions due to the standard state correction.59 The term was then systematically changed and the PMF and pKa recalculated until the pKa of the 4MI ionizable residue matched the experimentally determined value. The resulting term for 4MI is −105.1 kcal/mol as compared to histidine’s value of −101.4 kcal/mol. The new term is more negative than that of histidine, indicating a larger diabatic well shift between the ionizable residue and the hydronium cation, which results in a more basic residue (i.e., higher pKa). The 4MI model parameters are given in Supplemental Information Table 1S and deprotonation PMFs shown in Supplemental Information Figure 1S. The 4MI pKa value in bulk water was 7.6, accurately reproducing the experimental value of 7.45.58 Details of the 4MI MS-EVB simulations are also given in the Supplemental Information.
MS-EVB Simulations of the Proton Transport
The x-ray crystal structure (PDB accession # 1MOO) with its original 308 waters was used as the initial coordinates for the H64A HCA II system while the two x-ray binding sites (also from PDB accession # 1MOO) and 1 NMR binding site was used for the initial 4MI coordinates.39,40,43 The three 4MI binding site systems were each solvated in a box of modified TIP3P48 water with dimensions of 76 Å × 66 Å × 66 Å. The system waters were then relaxed for 50 steps of steepest descent followed by 950 steps of conjugated gradient minimization with the geometry of the protein and 4MI constrained. The relaxation procedure was then repeated with no constraints. The relaxed system was next subjected to an annealing simulation of 100 ps where the temperature of the system was brought from 0 K to 300 K. A post-annealing equilibration simulation was then carried out for 500 ps in the constant NPT ensemble. Following the equilibration simulation a production run of approximately 3 ns in the constant NPT ensemble was conducted for each of the four systems (H64A HCA II, and H64A HCA II with 4MI in each of the three known binding sites). The equilibration protocol was found to be sufficient in length by evaluating the convergence properties of several system observables (Supplemental Information Figures 3S and 4S). The constant NPT simulations were simulated at 300 K and used periodic boundary conditions with long-range Coulombic interactions calculated by the Ewald summation.60 The MD time integration step was 1 fs and used the leapfrog Verlet integrator while the short-ranged nonbonded interactions and forces were subject to a 9 Å cutoff. The simulations were run at 1 atm and utilized Langevin dynamics for the thermostat. All MD simulations were performed with the AMBER 10 package and used the parm99 force field when not otherwise specified56,61 The MS-EVB systems for the H64A HCA II, and H64A HCA II plus 4MI in the NMR binding site were taken from the equilibrated binding site study described above (i.e. after the 500 ps constant NPT equilibration simulation and before the 3 ns production simulation). The two systems were then transferred to a modified DL-POLY 2.15 software62 package incorporating the MS-EVB2 algorithm and equilibrated for 200 ps in the constant NVT ensemble to accommodate the delocalized nature of the excess hydrated proton. The two equilibrated systems were used for PMF calculations of various aspects of the PT event in H64A HCA II and the chemical rescue process.
The simulation of the H64A HCA II system without 4MI and in the absence of any additional ionizable enzyme residues was conducted to study the underlying system without a proton shuttle group (i.e. no chemical or self-rescue). The PMF calculation for the deprotonation event of the zinc-bound water consisted of 70 umbrella sampling windows spanning the range of , where is defined as the distance between the catalytic zinc and the excess proton CEC. An umbrella force constant, kn = 40 kcal mol−1 Å−2, was used to restrain the CEC to ensure adequate sampling along the reaction coordinate. Each window was equilibrated in the constant NVT ensemble for 250 ps followed by a data collection period of 1 ns.
A simulation of the H64A system without 4MI and incorporating an ionizable Glu69 was also carried out to study the possibility of enzyme self-rescue. The PMF calculation for the deprotonation event of the zinc-bound water through the intramolecular water cluster and to Glu69 consisted of 41 umbrella sampling windows spanning the range of , where is again defined as the distance between the catalytic zinc and the excess proton CEC. An umbrella force constant, kn = 40 kcal mol−1 Å−2, was again used to restrain the CEC to ensure adequate sampling along the reaction coordinate. Each window was equilibrated in the constant NVT ensemble for 250 ps followed by a data collection period of 1 ns.
The PT event between the catalytic zinc-bound water and 4MI residing at the NMR binding site was studied to help determine if this binding site may result in a viable chemical rescue pathway. The PMF calculation for the PT event between the zinc-bound water and 4MI consisted of 30 umbrella sampling windows spanning the range of , where is defined as before. The umbrella sampling was also carried out as described earlier.
The PT process between 4MI and Glu69 was investigated to evaluate the role of ionizable enzyme residues in the overall chemical rescue process. The coordinates for this calculation came from the simulation involving the PT event between the catalytic zinc-bound water and 4MI. Coordinates corresponding to conditions when the 4MI molecule was involved in a size 2 or 3 water bridge with Glu69 were selected as the starting configurations for a subsequent PMF calculation that involved the incorporation of the Glu69 residue into the MS-EVB model. The PMF calculation for the PT process between the 4MI and Glu69 consisted of 15 umbrella sampling windows spanning the range of , the definitions and sampling protocol was as described previously.
All biased MS-EVB simulations were run at 300 K using a Nosé-Hoover thermostat. Periodic boundary conditions were imposed with long-range Columbic interactions calculated by the Ewald summation, with a 9.5 Å cutoff for the short-ranged nonbonded interactions and a Verlet integrator step of 1fs. The weighted histogram analysis method63,64 (WHAM) was used to match the umbrella sampling windows together to form a continuous PMF. The error in the PMFs was calculated using the Monte Carlo bootstrap error analysis method and was found to be ≤ 0.3 kcal/mol.
Experimental Site-Directed Mutagenesis and Purification
Site-specific mutants of HCA II at residues His64, Glu69, and Asp72 (H64A-E69A and H64A-D72A) were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and a plasmid vector containing the HCA II coding region. Mutations were verified by sequencing of the entire HCA II coding region. Plasmids were transformed into E. coli BL21(DE3)pLysS expression cells. Following large scale growth and induction with 0.5 mM isopropyl-β-D-thiogalactopyranoside, the cells were pelleted and lysed with lysozyme. The lysate was passed through an affinity gel column consisting of an agarose matrix conjugated to p-(aminomethyl)-benzene-sulfonamide.65 Variants were eluted with 0.1M Tris-HCl, 0.4M sodium azide, pH 7.0. Extensive buffer exchange against 15mM Tris-HCl, pH 8.0 followed, and the samples were concentrated to approximately 10mg/mL as measured by UV spectroscopy (ε = 54,800 M−1 cm−1). SDS-PAGE and Coomassie staining showed >95% purity.
18O Exchange
This method is based on the exchange of 18O from species of CO2 into water measured by membrane inlet mass spectrometry.66 The CO2 passing across a silicon rubber membrane entered a mass spectrometer (Extrel EXM-200) providing a measure of the isotopic content of the CO2. The dehydration of the labeled bicarbonate has a probability of transiently labeling the active site zinc with 18O (eq 14). The subsequent protonation the zinc-bound hydroxide by exogenous buffer BH+ produces H218O, which is then released into bulk solvent (eq 15).
| (14) |
| (15) |
This method was used to obtain RH2O, the rate of release from the active site of water that bears 18O, as in eq 15. It is this component of 18O exchange that is dependent on the donation of protons to the 18O-labeled zinc-bound hydroxide (eq 15).
| (16) |
The value of this rate can be interpreted in terms of the rate constant for proton transfer from the proton donor to the zinc-bound hydroxide according to eq 16. In this equation, kBobs is the maximal turnover for proton transfer and kBobs/KeffB is an apparent second-order rate constant for proton transfer from buffer to enzyme that enhances catalysis. To determine the kinetic constant kBobs/KeffB, nonlinear least-squares methods were used (Enzfitter, Biosoft) to fit data obtained by the 18O exchange method. We do not report kBobs because of inhibition of catalysis at the high buffer concentrations required to determine the kBobs value. is the value in the absence of added buffer.
The measurements for catalyzed and uncatalyzed 18O exchange were made at 25°C in the presence of a total substrate concentration (all species of CO2) of 25 mM. Additionally, the total ionic strength of the solution was kept at 0.2 M by the addition of sodium sulfate.
3. Results and Discussion
Structural Properties of the Active Site Waters in H64A
In H64A HCA II, the amino acid that stabilizes the intramolecular water cluster and acts as a proton donor/acceptor is replaced by Ala, a small hydrophobic residue that does not act as a proton donor/acceptor. This mutation has a large effect on the rate limiting PT event as seen by the experimentally determined 20 to 50 fold reduction in the observed rate.34 In an effort to understand the effect of the H64A HCA II mutation on catalysis, the structural properties of the active site intramolecular waters are evaluated.
The spatial occupancy plots for the water oxygens in the H64A HCA II simulation indicate the average oxygen position and reveal the impact of amino acid mutations on the stability and position of the active site waters. The occupancy plots for the H64A HCA II mutant, Figure 2, indicate active site water positions that are similar to those reported in the x-ray structure (PDB accession # 1MOO).39 While the x-ray structure for the H64A HCA II mutant and the WT show similar active site water positions (W1, W2, W3a, and W3b), the occupancy plots, which are averaged over thermal fluctuations, reveal that W2, W3a, and W3b for H64A HCA II have an increased mobility (i.e., decreased stabilization and localization) when compared to the WT water occupancy data.18 In the WT system W2 and W3a have clear spherical densities present at the 75% iso-surface and W3b has a clear spherical 50% iso-surface while in the H64A mutant W2 is only present at the 50% iso-surface, W3a has much smaller 75% iso-surface when compared to WT, and W3b only appears at iso-surfaces less than 50%. The decreased stabilization of the W2 water is of interest because W2 corresponds to the branching point in the intramolecular water cluster and is involved in the rate limiting proton transport step as determined in the WT system.19 The destabilization of W2 is accompanied by a destabilization of W3b a water believed to be an integral participant in the rate limiting proton transfer process (destabilization relative to the WT system). It is noted that in Figure 2 (top panel) W3b is absence from the iso-surfaces. This does not mean that W3b is not present, only that it is more mobile and samples a larger volume of space (i.e., a reduced iso-surface). At lower iso-surface values density for water in this region is present (data not shown). In the WT system the rate limiting PT event when His64 is in the inward orientation corresponds to the excess proton transferring between W2 and W3a, or between W2 and W3b, while when His64 is in the outward orientation the rate limiting step corresponds to the excess proton transferring through waters around W3b. Therefore, it is thought that the stabilization of W2, W3a, and W3b is necessary for the efficiency of the rate limiting PT event.14,17
Figure 2.

Occupancy data for the active site water oxygens inside H64A HCA II for the ZnH2O2+ (top) and ZnOH+ (bottom) systems. The gray regions represent the 50% occupancy iso-surface and the red regions represent the 75% occupancy iso-surface for the active site waters.
In addition to the occupancy data, the radial distribution function (RDF) from the catalytic zinc to the surrounding water oxygens, g(RZn-OW), was evaluated (Figure 3). The RDF’s were considered converged due to the difference in the RDFs calculated from the first half and second half of the simulations possessing a Δg(r) ≤ 0.025. Inspection of the g(RZn-OW) for the zinc-bound water (ZnH2O2+) system reveals that there are major changes to the active site solvation structure as compared to the WT system. It is apparent that in the H64A HCA II ZnH2O2+ system the first peak (corresponding to W1) has a similar magnitude and resides at a similar distance from the zinc as the H64A HCA II zinc-bound hydroxide (ZnOH+) system. The greater distance between the zinc and W1 in the H64A HCA II ZnH2O2+ system as compared to the WT ZnH2O2+ system with H64 in the inward orientation is due to the absence of the His64 residue, which creates a first solvation shell similar to that seen in the WT as ZnOH+ well as the ZnH2O2+ case when His64 is in the outward orientation. While the magnitude and location of the first peak does not provide a reason for the 20 to 50 fold decrease in the rate, the second peak in the ZnH2O2+ system illuminates the structural defects in the active site water structure that may contribute, in addition to the absence of a proton shuttle group, to the inefficient PT in H64A HCA II.
Figure 3.
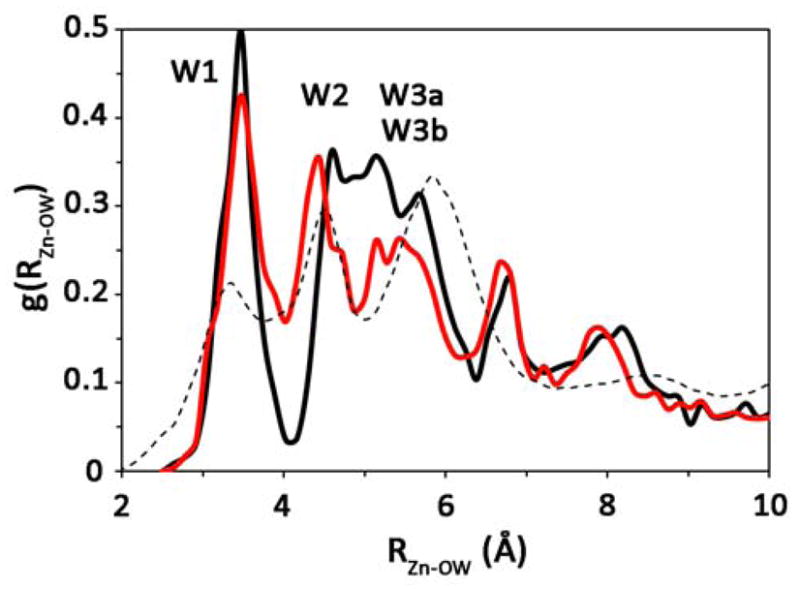
Radial distribution function of the active site waters for the H64A HCA II ZnH2O2+ (black) and ZnOH+ (red) systems. For reference purposes the WT ZnH2O2+ system with His64 in the outward orientation is included (black dotted).18
The second peak in the g(RZn-OW) for the ZnH2O2+ system consists of a merged second and third peak (corresponding to W2, W3a, and W3b). The peak location indicates that in H64A HCA II the W2 water has shifted to a larger separation distance from the zinc, while the W3a and W3b waters have shifted to a shorter zinc separation distance resulting in a relatively broad peak with minor structural features. The three small peaks on the overall broad peak are not significantly larger than the error in the RDF and therefore cannot be distinguished from the background noise. The broad peak structural feature is significantly different than that seen in the WT and indicates that the intramolecular water cluster is more disordered in the H64A mutant than in the WT system where there exists a clear enzyme stabilized water cluster tuned for PT. While major structural differences are seen for the g(RZn-OW) for the H64A HCA II ZnH2O2+ system, the g(RZn-OW) for the H64A HCA II ZnOH+ system is indistinguishable from that for the WT ZnOH+ system. This similarity is not surprising given the small difference between the corresponding occupancy plots and the reported x-ray structure. The similarity between the simulated structural data and the x-ray structure supports the hypothesis that the x-ray structure is indicative of the ZnOH+ system.
The occupancy and radial distribution data for the H64A HCA II mutant indicate that His64 is important to the stability of the active site waters due to its ability to participate in the hydrogen bonding network, thereby adding structural integrity to the transient active site water clusters as seen by water cluster analysis of WT and mutant HCA II systems.18 The stability of the active site water cluster plays an important role in the rate limiting PT event as seen in the 7-fold faster Y7F HCA II and 4-fold slower N67L HCA II mutants, which increase or decrease the relative stability of the active site water clusters as compared to WT, respectively.18,20 Experimentally, PT in H64A HCA II is 20 to 50 fold slower than in WT, which is due in part to the fact that Ala64 cannot act as a proton donor/acceptor, that W2, W3a, and W3b are destabilized, and that H64A HCA II modifies the active site water cluster such that there are no discernable pre-determined hydrogen bonded paths that would allow for the excess proton to be transferred/transported through the active site.45
Rate Limiting Proton Transport Event in H64A HCA II
Intramolecular water cluster
The rate limiting PT event in the WT is strongly influenced by the intramolecular water cluster, the identity of the proton donor/acceptor (i.e. pKa), and the location of the proton donor/acceptor. In the WT system the proton donor/acceptor is His64, which has a similar pKa to the zinc-bound water, near 7, and resides 8 to 10 Å from the zinc. The inward orientation of His64 has a more favorable free energy barrier for PT than the outward orientation due to the shorter separation distance between the catalytic zinc and His64 and the structure of the intramolecular active site waters.19 Removing the primary proton donor/acceptor (His64) results in a significant reduction in the observed rate (increase in the free energy barrier). In the previous section it has been shown that this mutation disrupts the stability of critical active site waters. This observation subsequently raises several other questions. What is the underlying PMF and associated atomistic description of the rate limiting PT event? What (if any) proton acceptor/donors are necessary to facilitate PT in the H64A HCA II mutant? How does the enzyme manage to maintain the rate limiting PT event in the absence of His64 (although at a reduced rate)? To address these issues, simulations were conducted to determine the PMF for the H64A HCA II mutant in the absence of exogenous buffers and without any additional ionizable residues (i.e. all residues present in the protein contribute to the electrostatic interactions but did not take part in the proton transfer part of the MS-EVB Hamiltonian in an ionizable fashion: A− + H+ ⇌ AH). In addition to this underlying PMF, the occupancy of the hydrated proton CEC was evaluated in order to probe the paths that the CEC utilizes to enter/exit the active site. These paths are then used to identify possible ionizable residues that may participate in the PT event. With the information from the CEC occupancy plots plausible self-rescue pathways are identified.
The PMF for the rate limiting PT event from the zinc-bound water to the surrounding bulk environment in the absence of exogenous buffers or self-rescue pathways for H64A HCA II is found in Figure 4 (Validation of the zinc bound water/hydroxide MS-EVB model can be found in Ref. 19). It is clear from the figure that the calculated barrier to PT, ΔF‡ = 21.9 ± 0.3 kcal/mol, is significantly elevated when compared to the experimental value for H64A HCA II (11.2 to 11.8 kcal/mol as determined by a 20 to 50 fold reduction in the rate from WT and transition state theory). Interestingly, this significant overestimation of the PT barrier was also reported by the work of Riccardi et al. using the SCC-DFTB method.67
Figure 4.
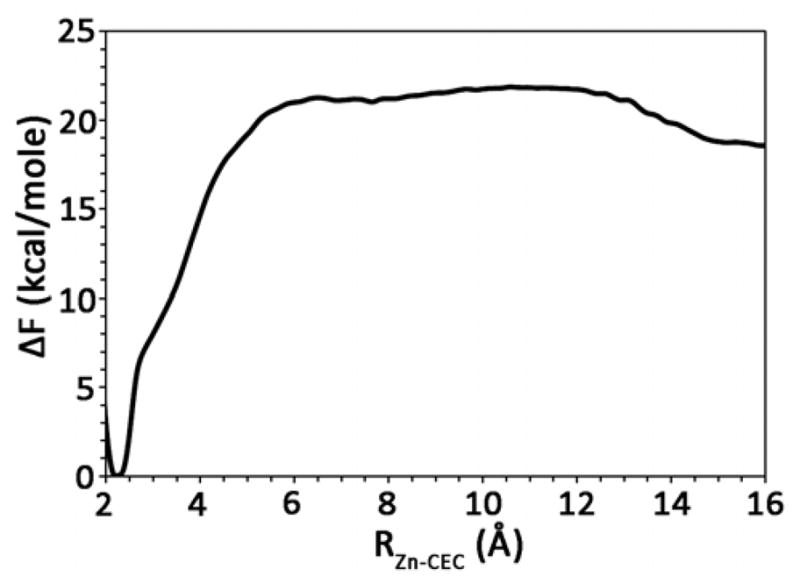
Free energy profile (PMF) for the PT event in H64A HCA II with no other ionizable residues included in the model and no chemical rescue agents. The thickness of the line denotes the standard deviation, which was evaluated to be ≤ 0.3 kcal/mol.
Comparing the H64A PT PMF with the WT PT PMFs reveals that the H64A mutant has an increased slope and free energy value as the excess proton passes through the zundel cation between the zinc-bound water and W1. This unfavorable free energy trend continues as the excess proton progresses through the region of the active site where the destabilized W2 and W3b reside. The destabilization of W2 and W3b has reduced the ability of the excess proton to delocalize into the active site water cluster, which in the WT has a stabilizing effect on the excess proton. Therefore, the reduced ability of the excess proton to delocalize has resulted in an unfavorable environment and an elevated PMF when compared to the WT system. This result strongly suggests that this PT process is not utilized in H64A HCA II. That is to say, in the H64A HCA II mutant the excess proton is not transported in/out of the active site solely by the active site waters.
Examination of the hydrated proton CEC pathways reveals two main possibilities that may account for the observed rate in the H64A HCA II. The first possibility is that exogenous buffers in the solution interact with the enzyme and acting as surrogate proton donor/acceptors by chemically rescuing the enzyme. This possibility, supported by the intersection of the CEC density with known binding sites of 4MI (Figure 5), will be discussed below in the chemical rescue section. The second possibility is that other ionizable residues in the enzyme act as proton donor/acceptors in a process known as self-rescue.
Figure 5.
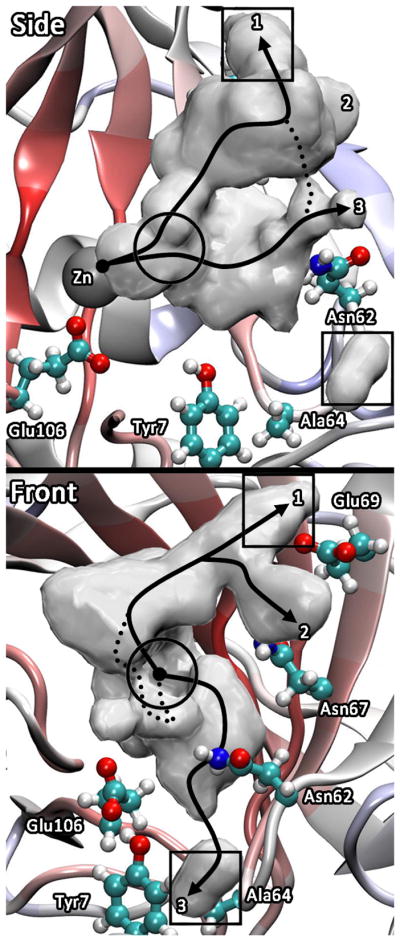
Spatial occupancy plots of the CEC for the hydrated excess proton in H64A HCA II. The squares indicate 4MI binding sites determined from x-ray data39,40 and the circle indicates the binding site determined from NMR data.43
Self-rescue
Self-rescue is a viable concept given that the simulated PT process in the absence of chemical and/or self-rescue pathways indicates a significantly elevated free energy barrier to PT that is not in agreement with experimental data. Moreover, that experimental data (Figure 6) indicates a measurable rate for the H64A HCA II based mutants in the absence of exogenous buffers (it is noted that bicarbonate is present at 25 mM and is a buffer with an apparent pKa of 10.3). Therefore, it may be concluded that some other ionizable moiety is participating in the rate limiting PT event, which we assume here is either bicarbonate and/or an ionizable residue in the active site (such as His, Glu, or Asp) and/or some other process is occurring such as the dissociation of the 18O labeled hydroxide from the catalytic zinc, which would appear in the experiment as an activated enzyme. For the remainder of the discussion on self-rescue we will ignore the impact of bicarbonate as a viable chemical rescue agent and focus attention on other ionizable moieties. Bicarbonate has a pKa of 10.3, which seems too basic to be a potent contributor to chemical rescue.
Figure 6.
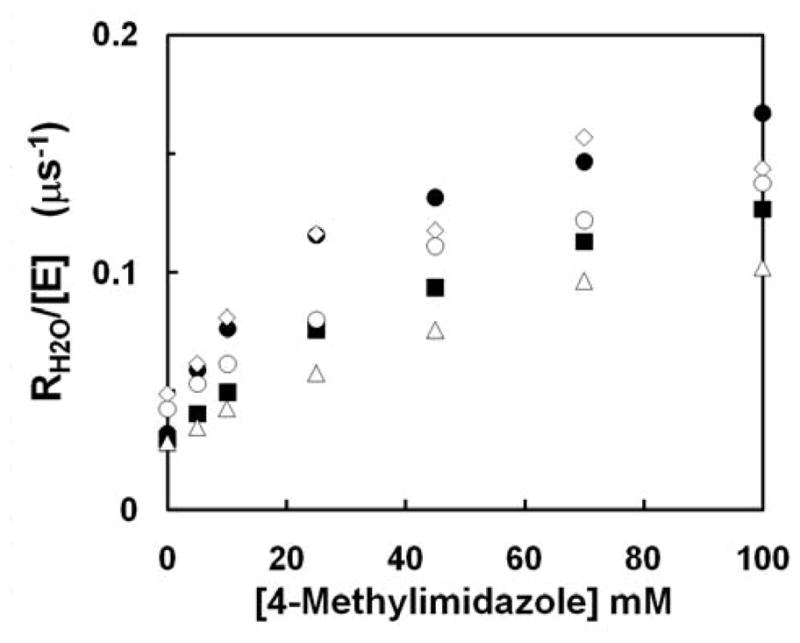
Dependence of RH2O/[E] (μs−1) on the concentration of the exogenous proton donor 4-methylimidazole in catalysis by the following variants of HCA II: (●) H64A35; (◇) W5A-H64A40; (△) H64A-E69A; (■) H64A-D72A; and (○) H64A-E69A-D72A43. Data were obtained by 18O exchange at pH 7.8 with ionic strength maintained at 0.2 M by addition of sodium sulfate. Temperature was 25 °C. The following values of kBobs/KeffB in units of μM−1s−1 were determined by a fit of eq 16 to the data: H64A, 6.7 ± 1.0; W5A-H64A, 5.2 ± 1.2; H64A-E69A, 1.8 ± 0.2; H64A-D72A, 3.3 ± 0.6; and H64A-E69A-D72A, 2.4 ± 0.3.
To study the possibility of self-rescue, ionizable residues that have an appropriate pKa (close to the pKa of the zinc-bound water, ~7), reside at an acceptable distance from the catalytic zinc, and are in the path of the hydrated proton CEC as it enters/leaves the active site were identified. The path(s) by which the excess proton (CEC) enters/leaves the active site for the H64A HCA II is seen in Figure 5. Much like the case of the WT,19 the CEC utilizes three predominant pathways to enter/leave the active site. The most probable pathway takes the CEC within the vicinity of Val134 and Phe130 and continues on to Glu69 and Asp72 (x-ray binding site of 4MI). The second most probable pathway utilizes the same path up to Val134 and Phe130 at which point the pathway branches and the CEC continues on towards the other side of Glu69, near Asn67. These pathways are similar to the CEC pathways found in WT when His64 is not considered as a donor/acceptor.19 They also closely correspond to the alternate pathways (labeled as Alt-2 and 4) found by analysis of the x-ray structure of WT (and also in some of its mutants) coupled to the fluctuations of non-ionizable amino acid side chains such as Asn-62 and possible long-time disordering of water molecules in and around the active site cavity.68,69 These two pathways reveal a utilization of the interface between the water/enzyme to move the excess proton in/out of the active site. This interfacial preference of the excess proton has also been seen for liquid/vapor and liquid/hydrophobic interfaces.70–75 The least populated pathway takes the excess proton near residues Asn62, Asn67, and Tyr7 near Ala64 and Trp5 (x-ray binding site of 4MI).
Using the excess proton CEC pathways it is possible to identify ionizable residues along the pathways that may participate in the PT event. An important component to identifying viable self-rescue amino acids is the separation distance between the ionizable residue and the catalytic zinc. From Figure 4 it is clear that the maximal free energy barrier is reached at ~ 6 Å from the catalytic zinc. Therefore, residues that may participate in self-rescue should be capable of interacting (stabilizing) the excess proton in this region. From previous computational work53 it is known that glutamic acid has a relatively large interaction distance of ~ 6 Å (i.e., the distance at which the ionizable residue favorably interacts with the proton CEC, Supplemental Information Figure 1S). This means that Glu69, which is ~ 13 Å from the catalytic zinc, corresponds to the upper bound for the likely zinc ionizable residue separation distances. A calculated PMF describing the PT event between the zinc-bound water and Glu69 yielded only a slight reduction ( ~ 2 kcal/mol) in the barrier to PT as compared to the free energy barrier in Figure 4. The major difference between the two PMFs is that in the self-rescue PMF there exists significant stabilization of the excess proton ≥ 10 Å corresponding to the proton CEC strongly interacting and subsequently residing on Glu69 (Supplemental Information Figure 8S). This PMF suggests that Glu69 does not participate in self-rescue and that ionizable residues farther away are at too great a distance to adequately couple with the active site waters that can lower the barrier to PT. Therefore, other residues that lie in the path of the CEC at greater separation distances were not considered as viable candidates for self-rescue. Residues such as Asn62, Asn67, Tyr7, and Thr199 which reside ≤ 8 Å from the zinc, were not considered due to the side chains being too basic (pKa ≫12 for Asn and Thr, and pKa = 10 for Tyr). In addition, Glu106 was not considered because it does not reside in, or h-bond to, the intramolecular water cluster. Glu106 throughout the simulations is h-bonded to WD (which is h-bonded to Tyr7) and to Thr199 (Figure 2). While it may be possible for a PT event from ZnH2O2+→Thr199→Glu106→WD→Tyr7 the high pKa of Thr199 and Tyr7 and the need for significant rearrangement of Glu106 and WD makes this pathway an unlikely candidate. However, new structural data indicates Tyr7 may be unprotonated.76 This new discovery opens the possibility of Tyr7 acting in a self-rescue pathway utilizing the intramolecular water cluster (ZnH2O2+→W1→W2→W3a→Tyr7). The development of a tyrosine MS-EVB model and the subsequent self-rescue simulations are a target for future studies.
While Glu69 has been eliminated as a possible self-rescue residue here, the overall question of self-rescue is still unanswered and left to future simulations that incorporate Tyr7 as an ionizable residue and/or experimental studies of the Y7F-H64A double mutant. It can be reasoned that if Tyr7 is participating in the rate limiting PT event then the Y7F-H64A mutant would eliminate this pathway and result in a decrease in the observed rate in the absence of exogenous buffers (y-intercept decreases). With the elimination of all but one self-rescue candidate, the possibility that the chemical rescue phenomena is responsible (in part or fully) for the residual rate observed in the H64A mutant is investigated.
Chemical Rescue Pathways
Self-rescue is not the only means by which the H64A HCA II mutant may continue the rate limiting PT step. Chemical rescue experiments have clearly identified the possibility of exogenous buffers to act as proton donor/acceptors.35,40 This section will study the role of the specific exogenous buffer, 4MI, in the chemical rescue phenomena. It is stressed that while these simulations only utilize 4MI, other buffers in solution with appropriate pKa values may participate in similar processes to facilitate the rate limiting PT event and provide chemical rescue.
X-ray binding sites
The simulations of 4MI with the H64A HCA II mutant indicate that the binding of 4MI in either x-ray determined binding sites (i.e., near Glu69 and/or near Trp5) are non-productive for chemical rescue, in line with kinetic experiments (Figure 6).41–43 This conclusion was reached by inspection of the 3 ns simulations of 4MI associating with the H64A HCA II mutant in the three known binding sites and evaluating the average position of 4MI (affinity for binding site and distance from the catalytic zinc), impact of 4MI on the active site solvent, water cluster formation, and looking at the PT PMF (Supplemental Information Figures 2S and 7S). From the PMF for self-rescue by Glu69 it is likely that ionizable residues at relatively large distances (≥ 13 Å) from the catalytic zinc are too far away to effectively stabilize the rate limiting PT event. In addition, simulations of the deprotonation of glutamic acid and 4MI in water indicate that 4MI has a smaller effective interaction distance with an excess proton than glutamate (Supplemental Information Figure 1S) and therefore would be less capable of significantly interacting/stabilizing the hydrated excess proton as it is transported through the intramolecular water cluster.53 Furthermore, the impact of the 4MI bound near Glu69 on the structure of the intramolecular water cluster indicates that while 4MI influences the water structure deep in the active site it does not create a water structure similar to the WT (Supplemental Information Figure 2S), nor stabilize the formation of hydrogen bonded water clusters to the same extent as the WT system (Supplemental Information Figure 6S). In fact, while 4MI is associating near Glu69 it rectifies some deficiencies in the water cluster structure seen in the H64A HCA II mutant (i.e., no-resolved structure for W2, W3a, and W3b becomes more resolved although W2 still appears to be perturbed when compared to WT water structure), it does not significantly influence the formation of complete water clusters connecting the zinc bound water and 4MI (Supplemental Figure 6S). That is to say, inspection of water cluster distributions indicates infrequent formation of short lived water clusters connecting the zinc bound water and 4MI by a hydrogen bonded network, one that could be capable of transporting an excess proton through the Grotthuss mechanism. Similar computational results are also seen when 4MI is bound near Trp5 even though 4MI in this binding site is closer to the catalytic zinc (Supplemental Information Figure 2S).
In addition to the distance and water structure arguments in the previous paragraph, it has been shown through both the experimentally determined 4MI binding constant and the present simulations of 4MI in complex with HCA II that 4MI weakly associates/binds to HCA II. In fact, with a binding energy of ~ 2.2 kcal/mol ( 43), 4MI is found to weakly associate with the active site cavity while freely diffusing and intermittently associating for longer periods of time near Glu69, Tyr7, and the NMR determined binding site. In light of these results it may be concluded that 4MI bound near Glu69 and/or Trp5 is largely ineffective in chemical rescue. In addition to the computational study of these binding sites several mutation experiments (H64A, W5A-H64A, H64A-E69A, H64A-D72A, and H64A-E69A-D72A) that influence the binding of 4MI near Glu69 and Trp5, and perturb possible self-rescue residues have also been conducted and are discussed below.
Catalysis
The chemical rescue kinetics for mutations that disrupt 4MI binding to the x-ray binding sites has been previously evaluated.41–43 The experiments in this section provide further information on the chemical rescue kinetics due to mutations near Glu69, which disrupt the binding of 4MI and eliminate ionizable residues near the mouth of the active site.
The rate constant in catalysis RH2O/[E] was determined from mass spectrometric measurement of the catalyzed exchange of 18O between CO2 and water and is found in Figure 6. RH2O/[E] describes the proton transfer dependent release of 18O-labeled water from the active site. From Figure 6 it is apparent that in the absence of exogenous buffer (y-intercept), and in the presence of bicarbonate, there remains an underlying rate of reaction that is similar between H64A,35 W5A-H64A,40 H64A-E69A, H64A-D72A, and H64A-E69A-D72A43 HCA II. This rate may be attributed to the combination of self-rescue (possible Tyr7), chemical rescue by bicarbonate and/or H3O+, dissociation of the zinc bound hydroxide, and/or an as of yet unknown process. Upon addition of 4MI all systems show an enhancement in the RH2O/[E], Figure 6, with inhibition at 4-MI concentrations in excess of 100 mM. This inhibition is most likely due to the binding of buffer to the zinc and/or increased ‘traffic’ in the limited space of the active site cavity. Due to the small 4-MI binding constant, 25 ± 4 mM43, and the weak association of 4MI with HCA II seen in the simulation component of this work, a hypothesis can be formulated that increased 4MI concentrations leads to crowding of buffer in the active site region resulting in steric conflicts and possible reduced 4MI diffusion.
The activation of RH2O/[E] shown in Figure 6 was fit to eq 16 and the apparent second-order rate constant, kBobs/KeffB in units of μM−1s−1, was determined. The kBobs/KeffB for H64A, W5A-H64A, H64A-E69A, H64A-D72A, and H64A-E69A-D72A HCA II were 6.7 ± 1, 5.2 ± 1.2, 1.8 ± 0.2, 3.3 ± 0.6, and 2.4 ± 0.3 μM−1s−1, respectively. From these results it is clear that H64A and W5A-H64A have very similar chemical rescue kinetics, strongly indicating that binding of 4MI near residue 5 (known x-ray binding site) is non-productive. The mutants H64A-E69A, H64A-D72A, and H64A-E69A-D72A all possess reduced apparent second-order rate constants when compared to H64A. Due to inhibition by 4-MI at high concentrations the maximal rates of rescue, kBobs, was not directly evaluated. However, if the RH2O/[E] at 100 mM 4-MI is used as the maximal rate of rescue a range of between 0.167 to 0.10 μs−1 is observed between H64A (fastest) and H64A-E69A (slowest). This range equates to a free energy difference of around 0.3 kcal/mol (using transition state theory) and indicates very similar overall maximal rates. Utilizing these rates it is also possible to estimate the KeffB for 4MI that is in the range of 25 mM for H64A and 55 mM for H64A-E69A. Again, these small binding constants indicate that in all of these systems 4MI weakly associates with the active site and support the hypothesis that the apparent inhibition of 4MI at very large concentrations of buffer is due to impaired diffusion of buffers as buffer molecules crowd into the cavity.
The similar maximal rates of rescue for the H64A-E69A, H64A-D72A, and H64A-E69A-D72E mutant as compared to H64A HCA II (Δ0.067 μs−1 or ≤ 0.3 kcal/mol) indicates that the 4MI binding near Glu69, like the binding site near Trp5, is non-productive. The apparent small but noticeable decrease in the maximal rate of rescue may have implications as to Glu69 and/or Asp72; participating in the chemical rescue pathway (i.e ZnH2O2+→W1→4MI→Glu69/Asp72, this pathway will be discussed below). With the elimination of both x-ray binding sites as major productive contributors to the chemical rescue process an alternative pathway needs to be generated. In the subsequent section the association of 4MI near the NMR site will be studied in addition to the idea that there is no one particular binding site for exogenous buffers but instead an exogenous buffer associates with the active site relatively close to the catalytic zinc (< 8 Å), setting up stabilized water clusters, and facilitating PT.
NMR binding site
The calculated PMF corresponding to the PT event between the zinc-bound water and 4MI, residing ~ 6 Å from the zinc, is found in Figure 7. It is evident that the resulting chemical rescue PMF is very similar to the WT PMF when His64 resides in the inward orientation.19 Examination of the PMF indicates a free energy barrier of 9.3 ± 0.3 kcal/mol for the deprotonation of the zinc-bound water to the first water (W1), followed by the transfer of the excess proton from W1 to 4MI with a ΔF = 3.7 ± 0.3 kcal/mol. This is a drastic reduction in the free energy barrier as compared to the H64A HCA II PMF in the absence of 4MI (Fig. 4) and reveals a viable pathway for the chemical rescue of H64A HCA II by 4MI. Once 4MI has accepted the excess proton, the free energy profile indicates that the most favorable pathway is then for protonated 4MIH+ to diffuse out of the active site (PMF region between 6 and 10 Å). In other words, the umbrella sampling of the excess proton CEC in this region involves the proton remaining bound to 4MI (i.e., 4MIH+), but with the 4MIH+ then diffusing out of the active site. Possession of the CEC is tracked by following the square of the coefficients determined by diagonalizing the MS-EVB matrix. 4MIH+ is concluded to possess the CEC due to the majority of the CEC amplitude residing on the 4MI molecule.
Figure 7.
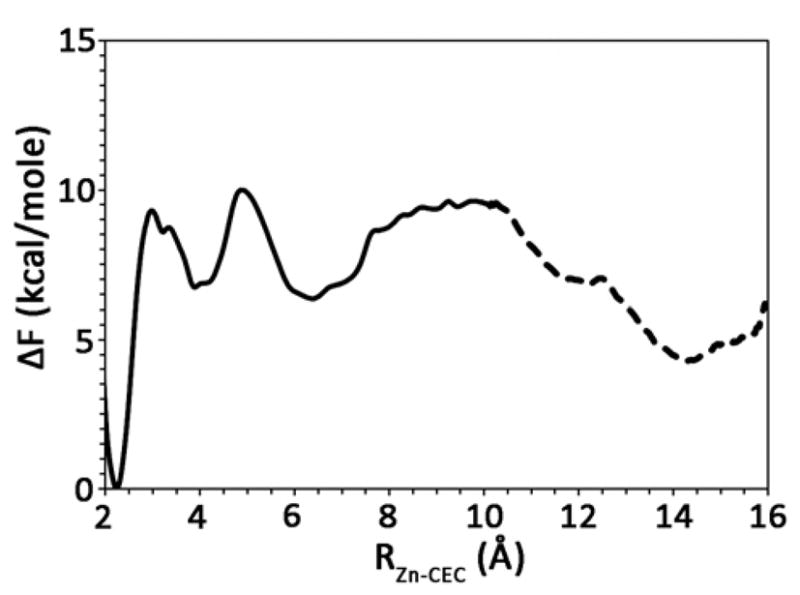
Free energy profile (PMF) for the chemical rescue of the H64A HCA II mutant by 4MI. The solid line indicates the proton transfer from the zinc-bound water to 4MI and the subsequent diffusion of the 4MI inside the active site. The dotted line indicates the proton transfer event between the 4MI and Glu69. The thickness of the line denotes the standard deviation, which was evaluated to be ≤ 0.3 kcal/mol.
During the diffusion of 4MIH+ out of the active site, > 9 Å from the zinc, the 4MIH+ passes within 2 to 4 Å of Glu69, and a small water cluster forms between the 4MIH+ and Glu69. It is found that the waters that participate in the small water cluster connecting the 4MIH+ and Glu69 allow for the proton CEC to delocalize across the waters, 4MI, and Glu69 thereby stabilizing the excess proton. The delocalization was monitored by watching the CEC amplitude for the participating molecules. It was found that the CEC amplitude on 4MIH+ decreased while the intervening waters and Glu69’s amplitude increased, ultimately resulting in the majority of the CEC amplitude resided on Glu69H+. The PMF for the PT event between 4MI and Glu69 is found in Figure 7 and is represented by the dotted black line between 10 and 16 Å. Once the coupling with Glu69 is sufficiently large, the free energy cost for the deprotonation of 4MIH+ is lowered, and the excess proton transports through the small water cluster to Glu69. As indicated in Figure 7, the PT event of the excess proton from 4MI to Glu69 is favorable. These results certainly indicate another viable pathway for chemical rescue where 4MI acts as a transporter of the excess proton from the zinc-bound water to ionizable residues on the rim of the active site, which then transfer the proton to solution. The release of the proton by 4MI and its return to the active site to accept a new proton mimics at some extent the rotation of His64 between the inward and outward orientation. The existence of an amino acid such as Glu69 and/or Asp72 that can relay the excess proton by accepting it from 4MI may be an important component to explaining the rescue of the catalytic activity of H64A HCA II to near WT levels. This is seen in the chemical rescue kinetics of H64A-E69A and H64A-D72A HCA II, which is less pronounced than the H64A HCA II (Figure 6).
4. Conclusions
In this paper, a detailed characterization of the proton transport event in the H64A HCA II mutant has been presented as well as insights into the chemical rescue of its catalytic activity. Simulations of H64A HCA II in the absence of exogenous buffers and/or self-rescue pathways indicate a free energy barrier in line with the auto dissociation of water. This high free energy barrier clearly indicates the necessity for a proton donor/acceptor in the form of an exogenous buffer and/or self-rescue residue to properly describe the observed rate in H64A HCA II. Analysis of the active site waters suggests that the mutation of His to Ala at position 64 disrupts the stability of waters W2, W3a, and W3b, which are known to be important to efficient PT. In an effort to identify probable self-rescue residues the behavior of the excess proton CEC was used to identify the possible paths utilized by the proton to enter/leave the active site. Subsequent PMF simulations revealed that Glu69 is too far away to adequately stabilize the PT event in the active site, so Glu69 was found to not be a viable self-rescue residue. Recent experimental data has indicated that Tyr7, which also resides along a proton CEC pathway, may contribute to the PT. An investigation into the possibility of Tyr7 acting as a self-rescue residue is currently underway.
The occupancy plots of the excess proton CEC density also reveal important information into the chemical rescue of H64A HCA II by 4MI. The occupancy plots show three main PT pathways where the first two pathways guide the excess proton toward Glu69, a known 4MI binding site, while the third pathway leads toward Tyr7 and Trp5, another known binding site of 4MI. All pathways leading out of the active site also intersect the NMR determined binding site of 4MI. Kinetic analysis of various H64A HCA II based mutants (W5A-H64A, H64A-E69A, H64A-D72A, and H64A-E69A-D72A) and simulations of 4MI binding to H64A HCA II indicate that the x-ray determined binding sites are non-productive and therefore do not represent viable chemical rescue pathways.
Simulations on the NMR determined binding site of 4MI resulted in a PT free energy barrier in line with the WT system, indicating a viable pathway for chemical rescue. Analysis of the PT event with 4MI in the NMR binding site also revealed the possibility of 4MI acting as an intermediary that carries the excess proton from deep in the active site to ionizable residues on the rim of the active site, such as Glu69. The computational and experimental studies of the chemical rescue phenomena thus indicate that the 4MI molecule may act as a direct proton donor/acceptor (i.e., accept the proton and diffuse in/out of the active site) and/or as an intermediary by passing the excess proton from the active site to various ionizable residues on the rim of the active site. In addition, the combined computational and experimental results indicate that 4MI weakly associates with HCA II and the classification of specific binding sites may be misleading. A more accurate depiction is that 4MI or other buffers associate with the enzyme but diffuse around the active site region and when the buffer resides ≤ 8 Å from the catalytic zinc the probability of forming a stable water cluster and facilitating PT increases (as was seen for 4MI in the NMR site). This description of chemical rescue also helps to explain the high apparent Km value for buffer activation and the inhibition patterns at high exogenous buffer concentrations where multiple exogenous buffers near the active site may become hindered (from steric clashing, reduced diffusion, etc). While the present work has focused specifically on chemical rescue mediated by 4MI, the mechanism and pathways used by 4MI are not limited to this particular exogenous buffer. In fact, any exogenous buffer with an appropriate pKa that is capable of being associated in the active site (< 8 Å from the zinc) may stabilize the PT event and result in an observed increase in the rate, i.e., chemical rescue.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01-GM53148 to GAV and R01-GM25154 to DNS and RM). ST acknowledges a fellowship grant from Indo-US Science & Technology Forum (IUSSTF), New Delhi, India.
Abbreviations
- 4MI
4-methylimidazole
- PT
proton transfer
- MD
molecular dynamics
- HCA
human carbonic anhydrase
- WT
wild-type
- H64A HCA II
HCA II site-specific mutant at position 64 from histidine to alanine
- PDB
protein data bank
- PMF
potential of mean force
- WHAM
weighted histogram analysis method
- MS-EVB
multi-state empirical valence bond
Footnotes
Supplemental Information Available. Table 1S, Figures 1S, 2S, 3S, 4S, 5S, 6S, 7S, 8S, and complete refs 60. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Decoursey TE. Physiol Rev. 2002;83:475. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- 2.Wraight CA. Biochimica et Biophysica Acta. 2006;1757:886. doi: 10.1016/j.bbabio.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 3.Voth GA. Acc Chem Res. 2006;39:143. doi: 10.1021/ar0402098. [DOI] [PubMed] [Google Scholar]
- 4.Swanson JMJ, Maupin CM, Chen H, Petersen MK, Xu J, Wu Y, Voth GA. J Phys Chem B. 2007;111:4300. doi: 10.1021/jp070104x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warshel A, Hwang JK, Åqvist J. Faraday Discuss. 1992;22 doi: 10.1039/fd9929300225. [DOI] [PubMed] [Google Scholar]
- 6.Lu D, Voth GA. Proteins. 1998;33:119. [PubMed] [Google Scholar]
- 7.Lu D, Voth GA. J Am Chem Soc. 1998;120:4006. [Google Scholar]
- 8.Toba S, Colombo G, Merz KM., Jr J Am Chem Soc. 1999;121:2290. [Google Scholar]
- 9.Cui Q, Karplus K. J Phys Chem B. 2003;107:1071. [Google Scholar]
- 10.Smedarchina Z, Siebrand W, Fernandez-Ramos A, Cui Q. J Am Chem Soc. 2003;125:243. doi: 10.1021/ja0210594. [DOI] [PubMed] [Google Scholar]
- 11.Braun-Sand S, Strajbl M, Warshel A. Biophys J. 2004;87:2221. doi: 10.1529/biophysj.104.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schutz CN, Warshel A. J Phys Chem B. 2004;108:2066. [Google Scholar]
- 13.Riccardi D, Schaefer P, Yang Y, Yu H, Ghosh N, Prat-Resina X, König P, Li G, Xu D, Guo H, Elstner M, Cui Q. J Phys Chem B. 2006;110:6458. doi: 10.1021/jp056361o. [DOI] [PubMed] [Google Scholar]
- 14.Silverman DN, McKenna R. Acc Chem Res. 2007;40:669. doi: 10.1021/ar7000588. [DOI] [PubMed] [Google Scholar]
- 15.Fisher SZ, Maupin CM, Govindasamy L, Budayova-Spano M, Govindasamy L, Tu C, Agbandje-McKenna M, Silverman DN, Voth GA, McKenna R. Biochemistry. 2007;46:2930. doi: 10.1021/bi062066y. [DOI] [PubMed] [Google Scholar]
- 16.Maupin CM, Voth GA. Biochemistry. 2007;46:2938. doi: 10.1021/bi062170f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maupin CM, Voth GA. BBA Proteins and Proteomics. 2009;1804:332. [Google Scholar]
- 18.Maupin CM, Saunders MG, Thorpe IF, McKenna R, Silverman DN, Voth GA. J Am Chem Soc. 2008;130:11399. doi: 10.1021/ja802264j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maupin CM, McKenna R, Silverman DN, Voth GA. J Am Chem Soc. 2009;131:7598. doi: 10.1021/ja8091938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maupin CM, Zheng J, Tu C, McKenna R, Silverman DN, Voth GA. Biochemistry. 2009;48:7996. doi: 10.1021/bi901037u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindskog S, Behravan G, Engstrand C, Forsman C, Jonsson B, Liang Z, Ren X, Xue Y. In: Carbonic Anhydrase--From Biochemistry and Genetics to Physiology and Clinical Medicine. Botre F, Gros G, Storey BT, editors. VCH; Weinheim, Germany: 1991. p. 1. [Google Scholar]
- 22.Silverman DN, Lindskog S. Acc Chem Res. 1988;21:30. [Google Scholar]
- 23.Davies DR. Ann Rev Biophys Biophys Chem. 1990;19:189. doi: 10.1146/annurev.bb.19.060190.001201. [DOI] [PubMed] [Google Scholar]
- 24.Fersht A. Enzyme Structure and Mechanism. W. H. Freeman and Sons; New York: 1985. [Google Scholar]
- 25.Tashian RE. BioEssays. 1989;10:186. doi: 10.1002/bies.950100603. [DOI] [PubMed] [Google Scholar]
- 26.Maren TH. Physiol Rev. 1967;47:595. doi: 10.1152/physrev.1967.47.4.595. [DOI] [PubMed] [Google Scholar]
- 27.Christianson DW. Adv Prot Chem. 1991;42:281. doi: 10.1016/s0065-3233(08)60538-0. [DOI] [PubMed] [Google Scholar]
- 28.Lindskog S, Coleman JE. Proc Natl Acad Sci USA. 1973;70:2505. doi: 10.1073/pnas.70.9.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindskog S. In: Zinc enzymes. Spiro TG, editor. John Wiley & Sons; New York: 1983. p. 78. [Google Scholar]
- 30.Liljas A, Kannan KK, Bergsten PC, Waara I. Nature. 1972;235:131. doi: 10.1038/newbio235131a0. [DOI] [PubMed] [Google Scholar]
- 31.Nair SK, Christianson DW. J Am Chem Soc. 1991;113:9455. [Google Scholar]
- 32.Fisher Z, Hernandez Prada JA, Tu C, Duda D, Yoshioka C, An H, Govindasamy L, Silverman DN, McKenna R. Biochemistry. 2005;44:1097. doi: 10.1021/bi0480279. [DOI] [PubMed] [Google Scholar]
- 33.Steiner H, Jonsson B-H, Lindskog SJ. Eur J Biochem. 1975;59:253. doi: 10.1111/j.1432-1033.1975.tb02449.x. [DOI] [PubMed] [Google Scholar]
- 34.Tu C, Silverman DN, Forsman C, Jonsson B-H, Lindskog S. Biochemistry. 1989;28:7913. doi: 10.1021/bi00445a054. [DOI] [PubMed] [Google Scholar]
- 35.Duda D, Tu C, Qian M, Laipis P, Agbandje-McKenna M, Silverman DN, McKenna R. Biochemistry. 2001;40:1741. doi: 10.1021/bi002295z. [DOI] [PubMed] [Google Scholar]
- 36.Silverman DN, Vincent SH. CRC Crit Rev Biochem. 1983;14:207. doi: 10.3109/10409238309102794. [DOI] [PubMed] [Google Scholar]
- 37.Erikson AE, Jones AT, Liljas A. Proteins. 1988;4:274. doi: 10.1002/prot.340040406. [DOI] [PubMed] [Google Scholar]
- 38.Erikson AE, Kylsten PM, Jones TA, Liljas A. Prot Struct Funct Gen. 1988;4:283. doi: 10.1002/prot.340040407. [DOI] [PubMed] [Google Scholar]
- 39.Duda D, Govindasamy L, Agbandje-McKenna M, Tu C, Silverman DN, McKenna R. Acta Crystallogr Sect D. 2003;59:93. doi: 10.1107/s0907444902019455. [DOI] [PubMed] [Google Scholar]
- 40.An H, Tu C, Duda D, Montanez-Clemente I, Math K, Laipis PJ, McKenna R, Silverman DN. Biochemistry. 2002;41:3235. doi: 10.1021/bi0120695. [DOI] [PubMed] [Google Scholar]
- 41.Haiqian A, Tu C, Duda D, Montanez-Clemente I, Math K, Laipis PJ, McKenna R, Silverman DN. Biochemistry. 2002;41:3235. doi: 10.1021/bi0120695. [DOI] [PubMed] [Google Scholar]
- 42.Bhatt D, Fisher SZ, Tu C, McKenna R, Silverman DN. Biophys J. 2007;92:562. doi: 10.1529/biophysj.106.093203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elder I, Tu C, Ming L-J, McKenna R, Silverman DN. Arch Biochem Biophys. 2005;437:106. doi: 10.1016/j.abb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 44.Garcia AE, Hummer G, Soumpasis DM. Prot Struct Funct Gen. 1997;27:471. [PubMed] [Google Scholar]
- 45.Roy A, Taraphder S. Biopolymers. 2006;82:623. doi: 10.1002/bip.20516. [DOI] [PubMed] [Google Scholar]
- 46.Schmitt UW, Voth GA. J Phys Chem B. 1998;102:5547. [Google Scholar]
- 47.Schmitt UW, Voth GA. J Chem Phys. 1999;111:9361. [Google Scholar]
- 48.Day TJ, Soudackov AV, Cuma M, Schmitt UW, Voth GA. J Chem Phys. 2002;117:5839. [Google Scholar]
- 49.Kim J, Schmitt UW, Gruetzmacher JA, Voth GA. J Chem Phys. 2002;116:737. [Google Scholar]
- 50.Wu YHCFWFP, Voth GA. J Phys Chem B. 2008;112:467. doi: 10.1021/jp076658h. [DOI] [PubMed] [Google Scholar]
- 51.Cuma M, Schmitt UW, Voth GA. Chem Phys. 2000;258:187. [Google Scholar]
- 52.Cuma M, Schmitt UW, Voth GA. J Phys Chem A. 2001;105:2814. [Google Scholar]
- 53.Maupin CM, Wong KF, Soudackov AV, Kim S, Voth GA. J Phys Chem A. 2006;110:631. doi: 10.1021/jp053596r. [DOI] [PubMed] [Google Scholar]
- 54.Gao J. Current Opinion in Structural Biology. 2003;13:184. doi: 10.1016/s0959-440x(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 55.Warshel A, Weiss J. J Am Chem Soc. 1980;102:6218. [Google Scholar]
- 56.Wang J, Cieplak P, Kollman PA. J Comput Chem. 2000;21:1049. [Google Scholar]
- 57.CRC Handbook of Chemistry and Physics. 90. CRC Press/Taylor and Francis; Boca Raton, FL: 2010. [Google Scholar]
- 58.Bruice TC, Schmir GL. J Am Chem Soc. 1957;80:148. [Google Scholar]
- 59.Chen J, Brooks CL, III, Scheraga HA. J Phys Chem B. 2008;112:242. doi: 10.1021/jp074355h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hummer G, Pratt LR, Garcia AE. J Phys Chem. 1996;100:1206. [Google Scholar]
- 61.Case DA, et al. 10. San Francisco: 2008. [Google Scholar]
- 62.Smith W, Forester TR. CCLRC. Daresbury Laboratory; Daresbury, Warrington, England: 1999. [Google Scholar]
- 63.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. J Comp Chem. 1992;13:1011. [Google Scholar]
- 64.Roux B. Comput Phys Commun. 1995;91:275. [Google Scholar]
- 65.Khalifah RG, Strader DJ, Bryant SH, Gibson SM. Biochemistry. 1977;16:2241. doi: 10.1021/bi00629a031. [DOI] [PubMed] [Google Scholar]
- 66.Silverman DN. Methods Enzymol. 1982;87:732. doi: 10.1016/s0076-6879(82)87037-7. [DOI] [PubMed] [Google Scholar]
- 67.Riccardi D, König P, Guo H, Cui Q. Biochemistry. 2008;47:2369. doi: 10.1021/bi701950j. [DOI] [PubMed] [Google Scholar]
- 68.Roy A, Taraphder S. J Phys Chem B. 2007;111:10563. doi: 10.1021/jp073499t. [DOI] [PubMed] [Google Scholar]
- 69.Roy A, Taraphder S. J Phys Chem B. 2008;112:13597. doi: 10.1021/jp0757309. [DOI] [PubMed] [Google Scholar]
- 70.Petersen MK, Iyengar SS, Day TJF, Voth GA. J Phys Chem B. 2004;108:14804. [Google Scholar]
- 71.Petersen PB, Saykally RJ. Ann Rev Phys Chem. 2006;57:333. doi: 10.1146/annurev.physchem.57.032905.104609. [DOI] [PubMed] [Google Scholar]
- 72.Petersen PB, Saykally RJ. J Phys Chem B. 2005;109:7976. doi: 10.1021/jp044479j. [DOI] [PubMed] [Google Scholar]
- 73.Tian C, Ji N, Waychunas GA, Shen YR. J Am Chem Soc. 2008;130:13033. doi: 10.1021/ja8021297. [DOI] [PubMed] [Google Scholar]
- 74.Petersen MK, Voth GA. J Phys Chem B. 2006;110:7085. doi: 10.1021/jp060698o. [DOI] [PubMed] [Google Scholar]
- 75.Iuchi S, Chen H, Paesani F, Voth GA. J Phys Chem B. 2009;113:4017. doi: 10.1021/jp805304j. [DOI] [PubMed] [Google Scholar]
- 76.Fisher ZS, Kovalevsky AY, Domsic JF, Mustyakimov M, McKenna R, Silverman DN, Langan PA. Biochemistry. 2010;49:415. doi: 10.1021/bi901995n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.