

Abstract
PURPOSE
Determine the efficacy and toxicity of higher dose versus standard dose intravenous methotrexate and pulses of high dose cytosine arabinoside with asparaginase versus standard dose cytosine arabinoside and teniposide during intensified continuation therapy for higher risk pediatric B-precursor acute lymphoblastic leukemia (ALL).
PATIENTS AND METHODS
From 1994–1999, the Pediatric Oncology Group conducted a randomized Phase III clinical trial in higher risk pediatric B-precursor ALL. 784 patients were randomized in a 2 × 2 factorial design to receive methotrexate 1 gm/m2 versus 2.5 gm/m2 and to cytosine arabinoside/teniposide versus high dose cytosine arabinoside/asparaginase during intensified continuation therapy.
RESULTS
Patients receiving standard dose methotrexate had 5-yr DFS of 71.8 ± 2.4%; patients receiving higher dose methotrexate had 5-yr DFS of 71.7 ± 2.4% (p=0.55). Outcomes on cytosine arabinoside/teniposide (DFS of 70.4 ± 2.4) were similar to higher dose cytosine arabinoside/asparaginase (DFS of 73.1 ± 2.3%) (p=0.41). OS rates were not different between methotrexate doses or cytosine arabinoside/teniposide versus cytosine arabinoside/asparaginase.
CONCLUSION
Increasing methotrexate dosing to 2.5 gm/m2 did not improve outcomes in higher risk pediatric B-precursor ALL. Giving high dose cytarabine and asparaginase pulses instead of standard dose cytarabine and teniposide produced non-significant differences in outcomes, allowing for teniposide to be removed from ALL therapy.
Introduction
Survival of children with acute lymphoblastic leukemia (ALL) has increased dramatically over the past fifty years.1–14 Multiagent systemic chemotherapy, prophylactic central nervous system therapy and intensive supportive care have contributed to this progress.15 In addition, the ability to better identify children at higher risk of relapse has led to risk-stratified treatment protocols. Using factors such as patient age, white blood cell count (WBC) at diagnosis, cytogenetics, and DNA index, a group of patients with B-precursor ALL with a higher risk of relapse can be selected to receive intensified therapy.16
Early Pediatric Oncology Group (POG) protocols demonstrated that anti-metabolite therapy was inadequate for many higher risk patients.4, 17 In POG protocol 8602 (1986–1991) 5-year event free survival for higher risk patients was 60%, versus 80% for standard risk patients.4, 18 This study examined whether adding Cytarabine (cytosine arabinoside, ara-C) or L-asparaginase to methotrexate (MTX)-based intensification therapy improved overall outcomes.18, 19 Overall outcomes improved, but it was unclear whether ara-C had any independent effect.
POG 9006 (1991–1994) tested the Goldie-Coldman hypothesis of using rotating combinations of anti-leukemic drugs (including ara-C) versus intensified intravenous mercaptopurine (6-MP) plus MTX (1 gm/m2) alone during early consolidation.20 Early interim analysis showed that rotating intensified consolidation appeared to be more effective and the study was closed.20 After data maturation, however, there was no significant difference in leukemia-free survival between the two treatments.4 Although not seen in POG 9006, intensification with intermediate dose MTX (1 gm/m2) has improved event-free survival in children with ALL.21–24
In 1994, POG opened a group-wide randomized Phase III clinical trial (POG 9406) to study the role of intensified chemotherapy in children with higher risk B-precursor ALL. The primary objectives of the study were to (1) determine the efficacy of higher dose (2.5 gm/m2 over 24 hours) versus standard dose (1 gm/m2 over 24 hours) intravenous MTX during intensified continuation therapy; and (2) determine whether pulses of high dose ara-C (3 gm/m2 × 4 doses) with asparaginase were superior to pulses of teniposide and ara-C (150 mg/m2/day × 72 hours) during intensified continuation therapy. We report the results of this trial.
Patients and methods
Patients
POG 9406 enrolled patients between November 15, 1994 and November 15, 1999. Local institutional review board approval and written informed consent from the patient and/or a parent were required prior to enrollment.
Eligibility
Eligibility included (1) newly diagnosed B-precursor ALL; (2) enrollment on the POG 9400 classification study; and (3) meeting the criteria for high risk B-precursor ALL. Those criteria were (1) age 10.00–21.99 years without trisomies of chromosomes 4 and 10 [if cytogenetic studies were informative (karyotypically abnormal)] or with DNA index ≤1.16 if cytogenetic studies were uninformative; (2) any age with presence of t(1;19), t(4;11), t(9;22), CNS leukemia or testicular disease; or (3) age 1.001- 9.99 years with initial WBC ≥50,000/μl without trisomies of chromosomes 4 and 10 or with DNA index ≤1.16 (if cytogenetic studies were uninformative). Patients < 12 months of age were not eligible.
Definition of disease and response
CNS leukemia was defined as (1) cerebral spinal fluid (CSF) WBC ≥ 5 WBC/μl with leukemic blasts without peripheral blood contamination; or (2) any involvement of the eye, brain or spinal cord parenchyma, or cranial nerves. Complete remission (CR) was defined as < 5% leukemic blasts in a cellular bone marrow and no evidence of leukemia involvement elsewhere. Relapse was defined as recurrence of disease whether in marrow (>25 % blasts) or an extra-medullary site (biopsy confirmed).
Randomization
POG 9406 randomized patients in a 2 × 2 factorial design to MTX, 1 gm/m2 (Regimens A/B) versus 2.5 gm/m2 (Regimens C/D) and to teniposide/ara-C (Regimens A/C) versus high dose ara-C/asparaginase (Regimens B/D). Patients with t(4;11) or t(9;22) were excluded from randomization and were assigned to Regimen A. Patients with Down syndrome were randomized to receive only Regimens A or B (lower MTX dosing). Patients with induction failure were not eligible to receive post-Induction therapy.
Treatment
Treatment regimens are detailed in Table 1. Induction was standard and risk-based in all patients. Patients aged 1–9.99 years with initial WBC <50,000/μl received 3-drug induction. All other patients received 4-drug induction therapy. Intrathecal therapy was given. If the day 29 bone marrow had 5–25% blasts, two weeks of extended induction was given with prednisone, vincristine, and L-asparaginase. Patients with >25% marrow blasts at day 29 or ≥ 5% blasts at day 43 were considered to be induction failures. Intensified continuation therapy was started after remission and count recovery.
Table 1.
Therapy on POG 9406 Protocol
|
Induction (4 weeks)
| |||
| Prednisone 40 mg/m2/d po divided tid days 1–28 | |||
| Vincristine 1.5 mg/m2 IV days 1,8,15,22 | |||
| Daunomycina 30 mg/m2 IV days 8,15,22 | |||
| L-asparaginase 6000 IU/m2 IM days 2,5,8,12,15,19 | |||
| Intrathecal therapyb IT days 1,8c,15,22c | |||
|
| |||
| Intensive Phase (30 weeks) | |||
|
| |||
| Regimen A | Regimen B | ||
| Drug pair #1 | Week 1 | Drug Pair #1 | Same as Regimen A |
| Methotrexate 1 g/m2 IV hours 0–24 | Drug Pair #2 | Ara-C 3 g/m2/dose every 12 hours × 4 doses | |
| 6-mercaptopurine 1 g/m2 IV hours 24–30 | PEG-asparaginase 1000 IU/m2 IM hour 42 | ||
| Leucovorin 10 mg/m2 po every 6 hours × 5 doses hours 42–72 | Drug Pair #3 | Same as Regimen A | |
| Week 2 | |||
|
|
|||
| Methotrexate 20 mg/m2 IM day 1 | Regimen C | ||
| 6-mercaptopurine 75 mg/m2 po days 1–7 | Drug Pair #1 | Methotrexate 2.5 g/m2 IV hours 0–24 | |
| Repeat weeks 6–7, 11–12, 16–17, 21–22, 26–27 | Remainder as in Regimen A | ||
| Drug pair #2 | Week 3 | Drug Pair #2 | Same as Regimen A |
| Teniposide 165 mg/m2 IV days 1,2 | Drug Pair #3 | Same as Regimen A | |
| Ara-C 150 mg/m2 IV/SC days 1–3 | |||
|
|
|||
| Repeat weeks 13,23 | Regimen D | ||
| Drug pair #3 | Week 8 | Drug Pair #1 | Same as Regimen C |
| Daunomycin 30 mg/m2/d days 1,4 | Drug Pair #2 | Same as Regimen B | |
| Ara-C 150 mg/m2 IV/SC days 1–3 | Drug Pair #3 | Same as Regimen A | |
| Vincristine 1.5 mg/m2 days 1,8 | |||
| Prednisone 40 mg/m2 po days 1–7 | |||
| PEG-L-asparaginase 2500 IU/m2 IM day 1 | |||
| Repeat weeks 18,28 | |||
|
| |||
| Maintenance (until 130 weeks from beginning therapy) | |||
|
| |||
| Methotrexate 20 mg/m2 IM weekly | |||
| 6-mercaptopurine 75 mg/m2 po daily | |||
|
| |||
| Post-Induction Intrathecal Therapyb,d | |||
|
| |||
| Given weeks 1,2,4,6,11,16,21,25,26,34 then every 8 weeks through week 106 | |||
= NCI high-risk patients only;
= age-adjusted dosing; triple intrathecal therapy (methotrexate, ara-C, hydrocortisone) prior to study amendment, methotrexate post- amendment;
= CNS2 and CNS3 patients only;
= post-amendment leucovorin 5 mg/m2 × 2 doses was given after intrathecal therapy weeks 4, 6, and after each maintenance intrathecal dose; tid: 3 times per day; IV: intravenous; IM: intramuscular; IT: intrathecal; SC: subcutaneous; Patients with CNS leukemia at diagnosis received craniospinal radiation (2400 cGy in twelve fractions to the brain and 1400 cGy in 10 fractions to the spine) at the beginning of year 2 of therapy. No IT therapy was given after completing irradiation. Patients with testicular leukemia received 2400 cGy of radiation in 12 fractions to the testes in the intensive phase of therapy
Patients who achieved a complete remission were randomized in a 2 × 2 factorial design to 30 weeks of intensification with Regimens A, B, C, or D. Regimens A and B had standard MTX dosing (1 gm/m2), while Regimens C and D had a higher dose of MTX (2.5 gm/m2). Leucovorin dosing was the same for all regimens. Regimens A and C used teniposide/standard dose ara-C, while Regimens B and D contained high dose ara-C/asparaginase. Teniposide was temporarily unavailable (March 26, 1999 – June 11, 1999). During this time etoposide 330 mg/m2 was substituted for teniposide. Each intensification course began when: (1) absolute neutrophil count (ANC) ≥500/μl; (2) platelet count ≥75,000/μl; (3) total bilirubin <1.7 mg/dl; (4) alanine aminotransferase (ALT) < 10–20 × upper limit of normal for age; and (5) creatinine normal for age.
Initially, triple intrathecal therapy (TIT) was used for CNS prophylaxis. There was an excess of neurotoxicity, especially seizures. Neurotoxicity included subcategories of mood, headache, cortical (includes seizures), sensory, motor, cerebellar, hearing, and vision. After a July 29, 1999 amendment, intrathecal (IT) therapy was changed to MTX alone. Further analysis indicated the neurotoxicity resulted from the interaction of several factors, including the repetitive use of IV MTX and the timing of IT injections in relation to IV MTX with leucovorin rescue. Therefore, on November 15, 1999, the study was amended to add leucovorin after all intensive continuation lumbar punctures that did not coincide with IV MTX and after all maintenance lumbar punctures.
Interim analyses by the Data Monitoring Committee revealed outcomes on the higher dose MTX arms were inferior to the standard dose MTX arms, and it was unlikely that the higher dose arm could ever prove to be superior to the standard dose arm. Therefore, on November 15, 1999, all patients in intensification were switched to the lower dose of MTX.
Patients with CNS leukemia at diagnosis received craniospinal radiation (2400 cGy in twelve fractions to the brain and 1400 cGy in 10 fractions to the spine) at the beginning of year 2 of therapy. No IT therapy was given after completing irradiation. Patients with testicular leukemia received 2400 cGy of radiation in 12 fractions to the testes in the intensive phase of therapy
Statistical analysis
POG 9406 was originally designed to enroll 673 patients to detect an improvement in 4-year continuous complete remission rates between treatment arms from 60% to 68.75% with 80% power and alpha at 5% using a 1-sided log-rank test. Accrual was extended since there were fewer events than projected in the statistical section which would have resulted in lower power than originally projected. Down syndrome patients were not included in the power calculations.
Analyses in this report are based on data frozen as of January 21, 2009 when collection of follow-up data was completed for the study. Disease-free survival (DFS) is defined as the time from complete remission to the time of first event (relapse, second malignancy, or death). Relapse-free survival (RFS) is defined as the time from complete remission to date of first relapse. Overall survival (OS) is defined as the time from complete remission to the date of death or date last seen for those who did not experience an event. Estimates for DFS, RFS, and OS were calculated using the Kaplan-Meier method, and standard errors of the estimates were obtained by the method of Peto and Peto.25 The log-rank test was used to compare survival curves among groups. Cumulative incidence rates were estimated using Gray’s method.26 The Chi-square test was used to compare proportions. Statistical significance was defined as a p-value less than 0.05. All analyses were performed using SAS® software. All graphics were generated using R (http://www.R-project.org, version 2.13.1).
Results
Induction
910 patients were enrolled. Three patients were ineligible and 2 Down syndrome patients were made inevaluable after enrollment. Of the 905 eligible patients, 35 were removed from protocol therapy prior to intensified continuation due to induction failure (n=15), death (n=7), toxicity (n=12), and refusal of randomization (n=1). Twenty-four patients did not achieve CR (7 early deaths, 1 partial response, 14 progressive disease, 2 patients not evaluable for response and off Induction therapy for toxicity). The remission rate was 97.3% (881 out of 905).
Randomization
A total of 805 patients were randomized; 784 patients without Down syndrome were randomized in a 2 × 2 factorial design to post-induction therapy on this trial: Regimen A (n=198); Regimen B (n=197); Regimen C (n=193); Regimen D (n=196). Eighteen patients with t(4;11) and 47 patients with t(9;22) were excluded from randomization and received Regimen A. Twenty-one patients had Down syndrome (9 randomized to Regimen A, 12 to Regimen B). Baseline characteristics, by regimen, are summarized in Table 2.
Table 2.
Baseline Characteristics by Regimen (n=870)
| Regimen A n (%) |
Regimen B n (%) |
Regimen C n (%) |
Regimen D n (%) |
|
|---|---|---|---|---|
| Number of patients | 272 | 209 | 193 | 196 |
| Median age | 10.78 | 10.98 | 10.48 | 11.26 |
| Median WBC | 32.25 | 23.20 | 27.50 | 28.20 |
|
| ||||
| Race/Ethnicity
| ||||
| White | 165 (60.7) | 135 (64.6) | 127 (65.8) | 120 (61.2) |
| Black | 28 (10.3) | 21 (10.1) | 23 (11.9) | 27 (13.8) |
| Hispanic | 57 (21) | 36 (17.2) | 34 (17.6) | 35 (17.9) |
| Other | 22 (8.1) | 17 (8.1) | 9 (4.7) | 14 (7.1) |
|
| ||||
| Gender
| ||||
| Male | 155 (57) | 124 (59.3) | 92 (47.7) | 117 (59.7) |
| Female | 117 (43) | 85 (40.7) | 101 (52.3) | 79 (40.3) |
|
| ||||
| Disease
| ||||
| CNS disease | 17 (6.3) | 10 (4.8) | 14 (7.3) | 13 (6.6) |
| Testicular disease | 4 (1.5) | 2 (1) | 1 (0.5) | 0 (0) |
|
| ||||
| Cytogenetics
| ||||
| Down syndrome | 9 (3.3) | 12 (5.7) | 0 (0) | 0 (0) |
| t(1;19) | 27 (9.9) | 29 (13.9) | 26 (13.5) | 29 (14.8) |
| t(4;11) | 18 (6.6) | 0 (0) | 0 (0) | 0 (0) |
| t(9;22) | 47 (17.3) | 0 (0) | 0 (0) | 0 (0) |
Outcome
The 5-year DFS and OS in all patients were 69 ± 1.6% and 80.4 ± 1.4%, respectively (Figure 1). Five-year cumulative incidence rates were 14.9 ± 1.2 % for isolated bone marrow relapse, 3.9 ± 0.66% for isolated CNS relapse, 1.1 ± 0.35% for isolated testicular relapse, and 7.2 ± 0.9% for relapse at other sites (including combined relapse). There were 3.7% (32 out of 870) remission deaths (Table 3); the 5-year cumulative incidence rate was 3.2 ± 0.6%.
Figure 1. Overall Survival (OS) and Disease-Free Survival (DFS).

The 5-year OS and DFS for POG 9406 patients is 80.4 ± 1.4% and 69 ± 1.6%, respectively.
POG=Pediatric Oncology Group
Table 3.
Remission deaths (as first event): regimen by reason for death for all eligible patients (n=870)
| Regimen | Reason for death
|
Total | |||||
|---|---|---|---|---|---|---|---|
| Unknown | Tumor | Infection | Hemorrhage | Tumor & Infection | Other | ||
| Regimen A | 1 | 4 | 5 | 2 | 0 | 4 | 16 |
|
| |||||||
| t(4;11) | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| t(9;22) | 1 | 2 | 4 | 2 | 0 | 3 | 12 |
|
| |||||||
| Regimen B | 0 | 0 | 8 | 0 | 0 | 3 | 11 |
|
| |||||||
| CNS | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| Down Syndrome | 0 | 0 | 2 | 0 | 0 | 1 | 3 |
| t(1;19) | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
|
| |||||||
| Regimen C | 0 | 0 | 2 | 0 | 1 | 1 | 4 |
|
| |||||||
| Regimen D | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
|
| |||||||
| Total | 2 | 4 | 15 | 2 | 1 | 8 | 32 |
Patients who received standard dose MTX (Regimens A/B; n=395) had 5-yr DFS of 71.8 ± 2.4% while patients treated with higher dose MTX (Regimens C/D; n=389) had 5-yr DFS of 71.7 ± 2.4% (Hazard Ratio [HR] =1.1; 95% CI: 0.84, 1.4; p=0.55; Figure 2a). Outcomes for patients on ara-C/teniposide (Regimens A/C: DFS of 70.4 ± 2.4%; n=391) were similar to patients on higher dose ara-C/asparaginase (Regimens B/D: DFS of 73.1 ± 2.3%; n=393) (HR=1.1; 95% CI: 0.86, 1.4; p=0.41; Figure 2b). DFS for Regimens A, B, C, and D were 68 ± 3.5%, 75.5 ± 3.2%, 72.7 ± 3.3%, and 70.7 ± 3.3%, respectively (p=0.55). However, this trial was not designed as a four arm study and has insufficient power to determine which regimen is superior. The interaction between the two randomizations was not significant (p=0.32). Survival rates were not significantly different between patients receiving standard versus higher dose MTX or high dose ara-C/asparaginase versus standard dose ara-C/teniposide.
Figure 2.


Figure 2a. DFS for MTX comparison: AB vs. CD (n=784)
The 5-year DFS for patients who received Regimens A/B (standard dose of MTX at 1 gm/m2) was 71.8 ± 2.4% versus 71.7 ± 2.4% for patients who received Regimens C/D (the higher dose of MTX at 2.5 gm/m2).
Figure 2b. DFS for AraC comparison: AC vs. BD (n=784)
The 5-year DFS for patients who received Regimens A/C (ara-C/teniposide) was 70.4 ± 2.4% versus 73.1 ± 2.3% for patients who received Regimens B/D (HD ara-C/asparaginase).
A total of 221 first event relapses occurred among the 784 patients randomized to one of the four regimens: 67 on Regimen A, 46 on Regimen B, 52 on Regimen C, and 56 on Regimen D (Table 4). The 5-year relapse-free survival was 69.3 ± 3.5% for Regimen A, 78.5 ± 3.1% for Regimen B, 74.3 ± 3.3% for Regimen C, and 72.1 ± 3.3% for Regimen D (p=0.15). Time to relapse was slightly shorter for higher dose MTX than standard dose MTX (mean 869.8 ± 562.4 days and 1029.3 ± 630.1 days, respectively, p=0.049). There was no significant difference in time to relapse for the ara-C regimens (p=0.373).
Table 4.
Site of Relapse as a First Event by Regimen (n=784)
| Site of Relapse | Regimen A n |
Regimen B n |
Regimen C n |
Regimen D n |
Total n |
|---|---|---|---|---|---|
| Isolated CNS | 11 | 7 | 7 | 9 | 34 |
| CNS/Testicular | 0 | 1 | 0 | 1 | 2 |
| Eye, Lung, Mediastinal, Other | 1 | 2 | 1 | 1 | 5 |
| Marrow | 38 | 22 | 31 | 31 | 122 |
| Marrow + CNS | 2 | 4 | 7 | 5 | 18 |
| Marrow + CNS + Testicular | 0 | 0 | 0 | 1 | 1 |
| Marrow + Other | 3 | 0 | 1 | 1 | 5 |
| Marrow + Other + Testicular | 0 | 0 | 1 | 1 | 2 |
| Marrow + Testicular | 5 | 6 | 3 | 3 | 17 |
| Not specified | 2 | 2 | 1 | 0 | 5 |
| Other + Testicular | 1 | 0 | 0 | 0 | 1 |
| Testicular | 4 | 2 | 0 | 3 | 9 |
| Total | 67 | 46 | 52 | 56 | 221 |
Outcomes for special subgroups are shown in Figure 3. Down syndrome patients had a DFS of 65.2 ± 10.7%. The t(1;19) translocation was considered a poor prognostic factor at the time this study was initiated. Patients with this translocation had a DFS of 81.1 ± 3.9% (n=111) versus 70.2 ± 1.8% (n=673) for those without the translocation. Patients with a t(4;11) translocation had a DFS of 66.7 ± 11.1%. Of these 18 patients, 4 went off therapy for a BMT. The DFS for patients with t(9;22) was 25.5 ± 6.4%; 26 were taken off study to receive a bone marrow transplant, while the remaining 21 patients had a DFS of 19.1 ± 8.6%. Patients with CNS disease (n=54) had a DFS of 57.4 ± 6.8%, while those without CNS disease (n=816) had a DFS of 69.7 ± 1.7% (p=0.018) (Figure 4). Patients with CNS disease were more likely to relapse in the bone marrow (15 of 18 relapses) than the CNS (3 of 18 relapses).
Figure 3. DFS curves for biologic subgroups (n=197).

The 5-year DFS for DS, t(1;19), t(4;11), and t(9;22) patients was 65.2 ± 10.7%, 81.1 ± 3.9%, 66.7 ± 11.1%, and 25.5 ± 6.4%, respectively.
Figure 4. DFS curves for CNS disease.
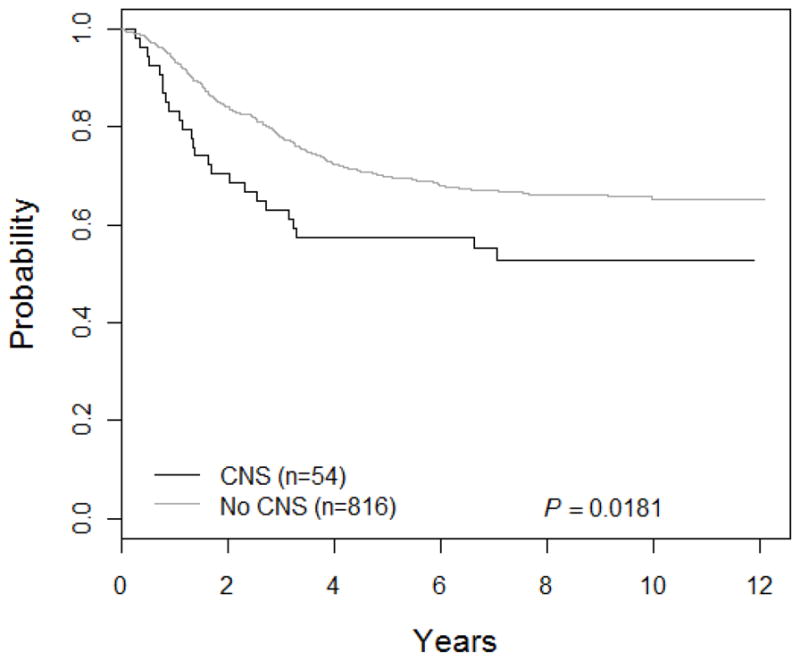
The 5-year DFS for patients with CNS was 57.4 ± 6.8% versus 69.7 ± 1.7% for those without CNS disease.
Toxicities
Therapy was discontinued due to toxicity in 4 (1.5%) patients on Regimen A, 7 (3.4%) on Regimen B, 5 (2.6%) on Regimen C, and 7 (3.6%) on Regimen D (p=0.44). Table 5 describes the common grade 3–4 toxicities by treatment regimen. Grade 3–4 bacterial sepsis, zoster, and thrombocytopenia occurred at a significantly higher rate in the high-dose ara-C/asparaginase regimens. Stomatitis occurred at a higher rate in the 2.5 gm/m2 MTX arms. Eighty-one percent of Down syndrome patients developed grade 3–4 stomatitis, despite all receiving 1 gm/m2 MTX.
Table 5.
Grade 3–4 Post-Induction Toxicities (n=784)
| Toxicity | N | Total | % | MTX Comparison: A/B vs. C/D |
AraC comparison: A/C vs. B/D |
|
|---|---|---|---|---|---|---|
| Neutropenia | Regimen A | 198 | 192 | 97.0 | 0.8427 | 0.8076 |
| Regimen B | 197 | 193 | 98.0 | |||
| Regimen C | 193 | 189 | 97.9 | |||
| Regimen D | 196 | 191 | 97.4 | |||
|
| ||||||
| Total | 784 | 765 | 97.6 | |||
|
| ||||||
| Thrombocytopenia | Regimen A | 198 | 172 | 86.9 | 0.3025 | 0.0004 |
| Regimen B | 197 | 190 | 96.4 | |||
| Regimen C | 193 | 177 | 91.7 | |||
| Regimen D | 196 | 187 | 95.4 | |||
|
| ||||||
| Total | 784 | 726 | 92.6 | |||
|
| ||||||
| Anemia | Regimen A | 198 | 175 | 88.4 | 0.2631 | 0.3468 |
| Regimen B | 197 | 184 | 93.4 | |||
| Regimen C | 193 | 181 | 93.8 | |||
| Regimen D | 196 | 181 | 92.3 | |||
|
| ||||||
| Total | 784 | 721 | 92.0 | |||
|
| ||||||
| Bacterial Sepsis | Regimen A | 198 | 58 | 29.3 | 0.3198 | 0.0207 |
| Regimen B | 197 | 77 | 39.1 | |||
| Regimen C | 193 | 54 | 28.0 | |||
| Regimen D | 196 | 66 | 33.7 | |||
|
| ||||||
| Total | 784 | 255 | 32.5 | |||
|
| ||||||
| Varicella | Regimen A | 198 | 10 | 5.1 | 0.9604 | 0.1082 |
| Regimen B | 197 | 10 | 5.1 | |||
| Regimen C | 193 | 5 | 2.6 | |||
| Regimen D | 196 | 15 | 7.7 | |||
|
| ||||||
| Total | 784 | 40 | 5.1 | |||
|
| ||||||
| Zoster | Regimen A | 198 | 9 | 4.5 | 0.5528 | <0.0001 |
| Regimen B | 197 | 25 | 12.7 | |||
| Regimen C | 193 | 7 | 3.6 | |||
| Regimen D | 196 | 22 | 11.2 | |||
|
| ||||||
| Total | 784 | 63 | 8.0 | |||
|
| ||||||
| Fungal Stomatitis | Regimen A | 198 | 1 | 0.5 | 0.4639 | 0.4817 |
| Regimen B | 197 | 2 | 1.0 | |||
| Regimen C | 193 | 2 | 1.0 | |||
| Regimen D | 196 | 3 | 1.5 | |||
|
| ||||||
| Total | 784 | 8 | 1.0 | |||
|
| ||||||
| Allergy | Regimen A | 198 | 28 | 14.1 | 0.8492 | 0.7423 |
| Regimen B | 197 | 31 | 15.7 | |||
| Regimen C | 193 | 33 | 17.1 | |||
| Regimen D | 196 | 27 | 13.8 | |||
|
| ||||||
| Total | 784 | 119 | 15.2 | |||
|
| ||||||
| Drug Fever | Regimen A | 198 | 2 | 1.0 | 0.9850 | 0.4089 |
| Regimen B | 197 | 1 | 0.5 | |||
| Regimen C | 193 | 2 | 1.0 | |||
| Regimen D | 196 | 1 | 0.5 | |||
|
| ||||||
| Total | 784 | 6 | 0.8 | |||
|
| ||||||
| Stomatitis | Regimen A | 198 | 44 | 22.2 | <0.0001 | 0.3157 |
| Regimen B | 197 | 35 | 17.8 | |||
| Regimen C | 193 | 70 | 36.3 | |||
| Regimen D | 196 | 67 | 34.2 | |||
|
| ||||||
| Total | 784 | 216 | 27.6 | |||
|
| ||||||
| Cortical Neurotoxicity | Regimen A | 198 | 11 | 5.6 | 0.2117 | 0.1999 |
| Regimen B | 197 | 13 | 6.6 | |||
| Regimen C | 193 | 5 | 2.6 | |||
| Regimen D | 196 | 11 | 5.6 | |||
|
| ||||||
| Total | 784 | 40 | 5.1 | |||
MTX=methotrexate
The study was amended due to excessive neurotoxicity. Seventy-four patients experienced 122 CNS events prior to the amendments, including 33 seizures in 29 patients. Fifteen patients had 30 CNS events after the amendments, with 5 seizures in 4 patients. Study records do not indicate how many patients actually received altered therapy after the amendments. There were 9 first event second malignancies (SMNs) (5-year cumulative incidence rate 0.69 ± 0.28%).
Discussion
This trial used a 2 × 2 factorial design to explore whether intensification of methotrexate or rotating pairs of chemotherapy would result in better leukemia free survival in patients with higher risk ALL. Neither the increased dose of MTX, nor the pulses of high dose ara-C with asparaginase improved outcomes.
The study as initially designed demonstrated increased neurotoxicity as compared to prior studies, leading to two amendments removing cytarabine from IT therapy and intensifying leucovorin rescue.14, 16, 17, 18, 19, 20, 21 The timing of these changes affected patients in a variable way depending on the portions of therapy received before and after the amendment implementations. There was an unknown effect on acute neurotoxicity, as the study records are unclear which patients received altered therapy after the amendments. Therefore, a comparison of CNS events before and after the amendments is impossible. The increased neurotoxicity may be due to a disproportionate delivery of MTX to the CNS, combined with prolonged excretion of MTX, resulting in higher systemic drug exposure over time.
Consistent with other trials, patients with CNS disease or t(9;22) translocations had worse outcomes. In contrast, patients with the t(1;19) and t(4;11) translocations were not more likely to relapse. Intensification of MTX or araC did not reduce the risk of relapse in patients with CNS disease.1, 4, 27
A comparatively higher dose of methotrexate in this trial (1 gm/m2 vs. 2.5 gm/m2) was not associated with a significant improvement in outcome. Given the recent demonstration that the addition of courses of even higher dose methotrexate (5 gm/m2) was associated with a decrease in both CNS and marrow relapses in patients with high risk ALL treated on the Children’s Oncology Group (COG) trial AALL0232, it is possible that the increase to 2.5 gm/m2 was not high enough to alter the steady state level in significantly more patients.28 In addition, a small number of patients initially randomized to receive the higher MTX dose were switched to the lower dose after the amendments. There is a possibility, although unlikely due to the low numbers, that these crossover patients decreased the apparent effect in the intent-to-treat analysis. Alternatively, the necessary threshold steady state level may be higher than was achieved in this trial. COG reported that serum MTX levels > 14 μm improved outcomes.29 Results from St. Jude trials suggest that 16 μm is an important cut point and show that patients treated with individualized MTX dosing to maintain a steady state level > 20 μm and ≤ 30 μm are significantly more likely to achieve a continuous complete remission than patients given conventional dosing with 1.5 gm/m2.30–31 Increasing the MTX dose from 0.5 gm/m2 in BFM-83 to 5 gm/m2 in BFM-86 did not improve EFS in patients with B-precursor ALL, but these trials were sequential, the methotrexate doses were not the only variables between the trials, and patients were not randomized between the different doses.32 Despite the fact that there has never been a study directly comparing 2.5 gm/m2 vs. 5 gm/m2, COG study AALL0232 demonstrated the importance of higher dose MTX. In that study, high risk B-cell precursor ALL patients were randomized to receive either high dose MTX at 5 gm/m2 over 24 hours with leucovorin rescue or escalating doses of MTX without rescue plus PEG asparaginase (Capizzi regimen).28 The high dose MTX regimen produced significantly superior EFS. Even though a direct comparison has never been done, the 5 gm/m2 dose is now the standard of care for children with B-cell precursor high risk ALL treated in COG ALL trials.
Pulses of high dose cytarabine and asparaginase were equivalent to pulses of cytarabine and teniposide. Cytarabine in combination with asparaginase is effective in relapsed ALL.33 Teniposide is also effective in ALL therapy, but causes many toxicities including secondary leukemia.34 Since asparaginase with ara-C was effective, teniposide could be replaced. However, the asparaginase/ara-C regimen was associated with higher risk of infections. Therefore, caution should be exercised when adding this combination to future therapy.
EFS for this study was better than the previous POG higher risk ALL trial, 9006, which had 4-yr EFS of 61.6±3.3% and 69.4±3.1% for its regimens.20 The improvement is in part due to the incorporation of the best regimen of 9006 as the standard regimen of this trial and better supportive care. Although there were variations in higher risk determinants between groups, contemporaneous studies involving higher risk patients showed similar EFS (71.8±1.1%, 70±4%, 76.7±4.6%, and 68.1±2.1%).1, 2, 35, 36 Treatment regimens with superior outcomes were more likely to include a delayed intensification (protocol II) phase of reinduction and reconsolidation. Thus, the augmented BFM (aBFM) therapy backbone used by many groups was selected for use in COG high risk ALL trials.
The follow-up study, P9906, used a BFM therapy. Outcomes were not significantly better than 9406.37 Outcomes in higher risk ALL continue to improve over those from 9406. The reasons are multiple. Superior EFS in younger higher risk patients has been achieved by using dexamethasone in Induction therapy, whereas 9406 used prednisone.38 The dose of prednisone used in Induction therapy has increased since 9406, from 40 mg/m2/day to 60 mg/m2/day. Most importantly, high risk groups are better able to be identified using techniques not available for 9406, including minimal residual disease testing and better genetic profiling.39–42
In summary, outcomes were better than 9006, but not better than contemporaneous trials. Increasing methotrexate dosing to 2.5 gm/m2 over 24 hours did not improve outcomes in higher risk pediatric patients with ALL. Giving high dose cytarabine and asparaginase pulses instead of standard dose cytarabine and teniposide produced equivalent outcomes, allowing for teniposide to be removed from ALL therapy.
Acknowledgments
The authors would like to thank all of the participating institutions, their data managers, and the patients and families who took part in the study.
Footnotes
Conflicts of interest and source of funding: Supported by the following NIH grants:
COG Chairs grant U10CA98543
POG Statistical Center grant U10CA30969
COG Statistics and Data Center grant U10CA98413
No conflicts were declared.
References
- 1.Gaynon PS, Angiolillo AL, Carroll WL, et al. Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: A Children’s Oncology Group Report. Leukemia. 2010;24:285–297. doi: 10.1038/leu.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moricke A, Zimmermann M, Reiter A, et al. Long-term results of five consecutive trials in childhood acute lymphoblastic leukemia performed by the ALL-BFM Study Group from 1981 to 2000. Leukemia. 2010;24:265–284. doi: 10.1038/leu.2009.257. [DOI] [PubMed] [Google Scholar]
- 3.Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:371–382. doi: 10.1038/leu.2009.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salzer WL, Devidas M, Carroll WL, et al. Long-term results of the Pediatric Oncology Group studies for childhood acute lymphoblastic leukemia 1984–2001: A report from the Children’s Oncology Group. Leukemia. 2010;24:355–370. doi: 10.1038/leu.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverman LB, Stevenson KE, O’Brien JE, et al. Long-term results of Dana-Farber Cancer Institute ALL Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1985–2000) Leukemia. 2010;24:320–334. doi: 10.1038/leu.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conter V, Arico M, Basso G, et al. Long-term results of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Studies 82, 87, 88, 91 and 95 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:255–264. doi: 10.1038/leu.2009.250. [DOI] [PubMed] [Google Scholar]
- 7.Kamps WA, van der Pal-de Bruin KM, Veerman AJ, et al. Long-term results of Dutch Childhood Oncology Group studies for children with acute lymphoblastic leukemia from 1984 to 2004. Leukemia. 2010;24:309–319. doi: 10.1038/leu.2009.258. [DOI] [PubMed] [Google Scholar]
- 8.Liang DC, Yang CP, Lin DT, et al. Long-term results of Taiwan Pediatric Oncology Group studies 1997 and 2002 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:397–405. doi: 10.1038/leu.2009.248. [DOI] [PubMed] [Google Scholar]
- 9.Schmiegelow K, Forestier E, Hellebostad M, et al. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia. 2010;24:345–354. doi: 10.1038/leu.2009.251. [DOI] [PubMed] [Google Scholar]
- 10.Stark B, Nirel R, Avrahami G, et al. Long-term results of the Israeli National Studies in childhood acute lymphoblastic leukemia: INS 84, 89 and 98. Leukemia. 2010;24:419–424. doi: 10.1038/leu.2009.254. [DOI] [PubMed] [Google Scholar]
- 11.Stary J, Jabali Y, Trka J, et al. Long-term results of treatment of childhood acute lymphoblastic leukemia in the Czech Republic. Leukemia. 2010;24:425–428. doi: 10.1038/leu.2009.255. [DOI] [PubMed] [Google Scholar]
- 12.Tsuchida M, Ohara A, Manabe A, et al. Long-term results of Tokyo Children’s Cancer Study Group trials for childhood acute lymphoblastic leukemia, 1984–1999. Leukemia. 2010;24:383–396. doi: 10.1038/leu.2009.260. [DOI] [PubMed] [Google Scholar]
- 13.Tsurusawa M, Shimomura Y, Asami K, et al. Long-term results of the Japanese Childhood Cancer and Leukemia Study Group studies 811, 841, 874 and 911 on childhood acute lymphoblastic leukemia. Leukemia. 2010;24:335–344. doi: 10.1038/leu.2009.259. [DOI] [PubMed] [Google Scholar]
- 14.Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the Children’s Oncology Group. J Clin Oncol. 2012;30:1663–1669. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 16.Schultz KR, Pullen DJ, Sather HN, et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: A combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG) Blood. 2007;109:926–935. doi: 10.1182/blood-2006-01-024729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Progress against childhood cancer. The Pediatric Oncology Group experience. Pediatrics. 1992;89:597–600. [PubMed] [Google Scholar]
- 18.Harris MB, Shuster JJ, Pullen J, et al. Treatment of children with early pre-B and pre-B acute lymphocytic leukemia with antimetabolite-based intensification regimens: A Pediatric Oncology Group Study. Leukemia. 2000;14:1570–1576. doi: 10.1038/sj.leu.2401886. [DOI] [PubMed] [Google Scholar]
- 19.Land VJ, Shuster JJ, Crist WM, et al. Comparison of two schedules of intermediate-dose methotrexate and cytarabine consolidation therapy for childhood B-precursor cell acute lymphoblastic leukemia: A Pediatric Oncology Group study. J Clin Oncol. 1994;12:1939–1945. doi: 10.1200/JCO.1994.12.9.1939. [DOI] [PubMed] [Google Scholar]
- 20.Lauer SJ, Shuster JJ, Mahoney DH, Jr, et al. A comparison of early intensive methotrexate/mercaptopurine with early intensive alternating combination chemotherapy for high-risk B-precursor acute lymphoblastic leukemia: A Pediatric Oncology Group phase III randomized trial. Leukemia. 2001;15:1038–1045. doi: 10.1038/sj.leu.2402132. [DOI] [PubMed] [Google Scholar]
- 21.Mahoney DH, Jr, Shuster J, Nitschke R, et al. Intermediate-dose intravenous methotrexate with intravenous mercaptopurine is superior to repetitive low-dose oral methotrexate with intravenous mercaptopurine for children with lower-risk B-lineage acute lymphoblastic leukemia: A Pediatric Oncology Group phase III trial. J Clin Oncol. 1998;16:246–254. doi: 10.1200/JCO.1998.16.1.246. [DOI] [PubMed] [Google Scholar]
- 22.Abromowitch M, Ochs J, Pui CH, et al. Efficacy of high-dose methotrexate in childhood acute lymphocytic leukemia: analysis by contemporary risk classifications. Blood. 1988;71:866–869. [PubMed] [Google Scholar]
- 23.Freeman AI, Weinberg V, Brecher ML, et al. Comparison of intermediate-dose methotrexate with cranial irradiation for the post-induction treatment of acute lymphocytic leukemia in children. N Engl J Med. 1983;308:477–484. doi: 10.1056/NEJM198303033080902. [DOI] [PubMed] [Google Scholar]
- 24.Green DM, Brecher ML, Blumenson LE, et al. The use of intermediate dose methotrexate in increased risk childhood acute lymphoblastic leukemia. A comparison of three versus six courses. Cancer. 1982;50:2722–2727. doi: 10.1002/1097-0142(19821215)50:12<2722::aid-cncr2820501204>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 25.Peto R, Peto J. Asymptotically efficient rank invariant test procedure. J Royal Stat. 1972;135:185–198. [Google Scholar]
- 26.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. The Annals of Statistics. 1988;16:1141–1154. [Google Scholar]
- 27.Arico M, Schrappe M, Hunger SP, et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol. 2010;28:4755–4761. doi: 10.1200/JCO.2010.30.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larsen EC, Salzer WL, Devidas M, et al. High dose methotrexate (HD-MTX) as compared to capizzi methotrexate plus asparaginase (C-MTX/ASNase) improves event-free survival (EFS) in children and young adults with high-risk acute lymphoblastic leukemia (HR-ALL): A report from the Children’s Oncology Group study AALL0232. J Clin Oncol. 2011;29:6s. (suppl; abstr 3; ASCO 2011 Plenary Session presentation) [Google Scholar]
- 29.Salzer WL, Winick NJ, Wacker P, et al. Plasma methotrexate, red blood cell methotrexate, and red blood cell folate values and outcome in children with precursor B-acute lymphoblastic leukemia: A report from the Children’s Oncology Group. J Pediatr Hematol Oncol. 2012;34:e1–7. doi: 10.1097/MPH.0b013e31820ee239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans WE, Crom WR, Abromowitch M, et al. Clinical pharmacodynamics of high-dose methotrexate in acute lymphocytic leukemia. Identification of a relation between concentration and effect. N Engl J Med. 1986;314:471–477. doi: 10.1056/NEJM198602203140803. [DOI] [PubMed] [Google Scholar]
- 31.Evans WE, Relling MV, Rodman JH, et al. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med. 1998;338:499–505. doi: 10.1056/NEJM199802193380803. [DOI] [PubMed] [Google Scholar]
- 32.Reiter A, Schrappe M, Ludwig WD, et al. Chemotherapy in 998 unselected childhood acute lymphoblastic leukemia patients. Results and conclusions of the multicenter trial ALL-BFM 86. Blood. 1994;84:3122–3133. [PubMed] [Google Scholar]
- 33.Wells RJ, Feusner J, Devney R, et al. Sequential high-dose cytosine arabinoside-asparaginase treatment in advanced childhood leukemia. J Clin Oncol. 1985;3:998–1004. doi: 10.1200/JCO.1985.3.7.998. [DOI] [PubMed] [Google Scholar]
- 34.Pui CH, Ribeiro RC, Hancock ML, et al. Acute myeloid leukemia in children treated with epipodophyllotoxins for acute lymphoblastic leukemia. N Engl J Med. 1991;325:1682–1687. doi: 10.1056/NEJM199112123252402. [DOI] [PubMed] [Google Scholar]
- 35.Moghrabi A, Levy DE, Asselin B, et al. Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95–01 for children with acute lymphoblastic leukemia. Blood. 2007;109:896–904. doi: 10.1182/blood-2006-06-027714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pui CH, Sandlund JT, Pei D, et al. Improved outcome for children with acute lymphoblastic leukemia: Results of Total Therapy Study XIIIB at St Jude Children’s Research Hospital. Blood. 2004;104:2690–2696. doi: 10.1182/blood-2004-04-1616. [DOI] [PubMed] [Google Scholar]
- 37.Bowman WP, Larsen EL, Devidas M, et al. Augmented therapy improves outcome for pediatric high risk acute lymphocytic leukemia: Results of Children’s Oncology Group trial P9906. Pediatr Blood Cancer. 2011;57:569–577. doi: 10.1002/pbc.22944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winick N, Salzer WL, Devidas M, et al. Dexamethasone (DEX) versus prednisone (PRED) during induction for children with high risk acute lymphoblastic leukemia (HR-ALL): A report from the Children’s Oncology Group study AALL0232. J Clin Oncol. 2011;29:586s. (suppl; abstr 9504) [Google Scholar]
- 39.Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: Correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–4884. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang H, Chen IM, Wilson CS, et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood. 2010;115:1394–1405. doi: 10.1182/blood-2009-05-218560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen IM, Harvey RC, Mullighan CG, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: A Children’s Oncology Group study. Blood. 2012;119:3512–3522. doi: 10.1182/blood-2011-11-394221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’s Oncology Group study. Blood. 2008;111:5477–5485. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]