

Abstract
Schwann cells (SCs) are crucial for peripheral nerve development and regeneration; however, the intrinsic regulatory mechanisms governing post-injury responses are poorly understood. Activation and deacetylation of nuclear factor-κB (NF- κB) in SCs have been implicated as prerequisites for peripheral nerve myelination. Using GFAP-IκBα-dn mice in which NF- κB transcriptional activation is inhibited in SCs we found no discernable differences in the quantity or structure of myelinated axons in adult facial nerves. Following crush injury, axonal regeneration was impaired at 31 days and significantly enhanced at 65 days in transgenic animals. Compact re-myelination and Remak bundle organization were significantly compromised at 31 days and restored by 65 days post injury. Together, these data indicate that inhibition of NF- κB activation in SCs transiently delays axonal regeneration and compact re-myelination. Manipulating the temporal activation of nuclear factor-κB in Schwann cells may offer new therapeutic avenues for PNS and CNS regeneration.
Keywords: myelin, peripheral, nerve, injury
INTRODUCTION
In the peripheral nervous system (PNS) Schwann cells (SCs) myelinate axons, enabling efficient electrical conduction and protection from the extracellular environment. During mouse peripheral nerve development, neural crest cells undergo three primary transitions to become mature SCs. Neural crest cells give rise to Schwann cell precursors (E12–13) which associate with axons and quickly transition into immature Schwann cells (E 15–16) before birth (Jessen and Mirsky, 2005). Immature Schwann cells, pervading perinatal nerves, are partly characterized by expression of glial fibrillary acid protein (GFAP). Random association with axons of variable size dictates subsequent maturation; those associated with large diameter axons (>~1 μm) will adopt a myelinating phenotype and relinquish GFAP synthesis. Immature SCs associated with small diameter axons mature into non-myelinating (unmyelinating) SCs and maintain GFAP expression throughout adulthood (Jessen and Mirsky, 2005).
Following peripheral nerve injury, adult SCs cease myelination, ingest myelin and subsequently de-differentiate into a denervated phenotype permitting proliferation and re-association with regenerating axons. Denervated SCs adopt a phenotype reminiscent of perinatal, immature SCs and are partially characterized by re-expression of GFAP. SC de-differentiation and subsequent re-myelination is governed, at least in part, by positive (e.g. Krox-20, Oct-6) and negative (e.g. c-Jun, Notch) transcriptional regulators (Jessen and Mirsky, 2008). Perturbations in these regulatory programs can impair nerve regeneration and are associated with several elusive demyelinating neuropathies, including Dejerine-Sottas Syndrome, Charcot-Marie-Tooth disease and Congenital Hypomyelinating Neuropathy (Svaren and Meijer, 2008).
Nuclear factor-κB (NF- κB) is most appreciated for its transcriptional regulation of the immune response and is a target of many approved pharmaceuticals (Miller et al., 2010). Recent studies suggest that activation of NF- κB is essential for driving immature SCs into a pro-myelinating phenotype to ensheath axons during development in dorsal root ganglion-SC co-cultures (Nickols et al., 2003; Yoon et al., 2008; Limpert et al., 2010). Additionally, deacetylation of NF-κB was shown to be crucial for SC myelination using histone deacetylase 1/2 (HDAC 1/2) double knockout mice (Chen et al., 2011). Although peripheral nerve assault generates a robust upregulation of NF-κB activity, almost nothing is known regarding the effects of NF-κB activation in denervated SCs on peripheral nerve regeneration and re-myelination (Ma and Bisby, 1998; Fernyhough et al., 2005; Pollock et al., 2005; Smith et al., 2009; Fu et al., 2010).
Since NF-κB knock out animals die before birth (Beg et al., 1995) or suffer severe immunological complications (Sha et al., 1995), in vivo studies have been fairly limited. We sought to address these issues within the facial nerve of our previously described GFAP-IκBα-dn (transgenic) mice, in which NF-κB activity is functionally inhibited in GFAP-expressing cells, including SCs and astrocytes (Brambilla et al., 2005; Bracchi-ricard et al., 2008; Brambilla et al., 2009). Briefly, the cDNA encoding a truncated form of the human IκBα gene, driven by the human GFAP promoter (gfa2), was overexpressed in order to prevent NF-κB nuclear localization and subsequent gene regulation. Here, we show that inhibition of NF-κB activation in SCs transiently delays regeneration and re-myelination following facial nerve injury.
MATERIALS AND METHODS
Animals
2–4 month old, male mice were used throughout this study and were generated by breeding heterozygous GFAP-IκBα-dn males with WT females on a C57BL/6 background in order to produce progeny lacking functional activation of canonical NF-κB in GFAP-expressing cells (Brambilla et al., 2005). Animals were housed in a virus/antigen-free facility on a 12 hr light/dark cycle with food and water provisions ad libitum. Genotyping of tail DNA was performed as previously described (Brambilla et al., 2005; Bracchi-ricard et al., 2008).
Pinch Crush Injury
Animals were anesthetized with ketamine/xylazine (University of Miami, Division of Veterinary Resources) and the right facial nerve were uniformly crushed ~4mm rostral to trifurcation until completely transparent, using ground microforceps. Animals were sacrificed 1, 2, 4, 12, 31, or 65 days following injury. Surgeries were performed in the Animal and Surgical Core Facility at the Miami Project to Cure Paralysis under protocols approved by the University of Miami Animal Care and Use Committee.
Facial Nerve Axotomy to Assess Wallerian Degeneration
WT and transgenic littermates were anesthetized with ketamine/xylazine. Subsequent to facial nerve exposure, the buccal branch of the right facial nerve was completely transected ~4mm distal to trifurcation from the trunk. The proximal stump of the transected nerve was sutured (Ethicon 10-0 monofilament) to adjacent, underlying muscle tissue at a 90–120 angle to prevent reinnervation of the distal nerve segment. Animals were allowed to recover with food and water ad libitum for 4 or 12 days. At each respective time point, animals were sacrificed with a lethal dose of anesthesia and distal nerve stumps were removed and post-fixed for analysis of Wallerian degeneration. A 3mm segment distal to the site of transection was removed for CD11b (1:100; Serotec) immunostains and the remaining ~2mm nerve segment was collected for TB/PPD histology.
Semi-thin (1 μm), transverse sections of distal (5mm distal to transection) injured nerve stumps were collected, stained with PPD and counterstained with TB. Using StereoInvestigator, myelin rings undergoing demyelination, and intact myelin rings were counted from several random sites (25x25 μm2 counting frame; 75x75 μm2 grid). Myelin rings exhibiting severe lamellaer in/out-foldings, tethering, myelin debris, vacuolization, incisures and/or collapsed axoplasms were considered demyelinated. Total population projections of each identifier were compared between WT and transgenic littermates at the respective time points following injury.
Behavioral Testing
Following facial nerve transection ~1 mm caudal to trifurcation, vibrissae movement was completely abolished ipsilateral to injury and sustained contralateral to injury. Prior to Fluorogold (FG) administration, vibrissae behavior was carefully assessed 28 days following injury and scored on a scale from 0, indicating no movement, to 3, denoting robust, normal whisker sweeping, as previously described (Raivich et al., 2004). All animals demonstrated normal (3) vibrissae movement on the uninjured side.
Retrograde Tracing
Gel foam pads, pre-soaked in 20 μl of a 4% FG (Fluorochrom, Denver, CO) solution, were inserted for 20 minutes beneath the ipsilateral and contralateral whiskerpads 28 days after unilateral facial nerve transection, as previously described (Werner et al., 2000). Three days later, the total number of FG+ MNs within the facial motor nucleus (FMN) were counted in 6–8 sections by a single investigator, blinded to genotype and expressed as a ratio (injured/uninjured). Images were obtained using a 20X objective on a Zeiss Axiovert 200M fluorescent microscope (Zeiss, Thornwood, NY, USA) with Neurolucida software (MicroBrightField, Inc.).
Following buccal nerve crush injury, axonal sparing and whisker pad re-innervation were assessed by injecting 2 μl of a 4% FG solution subcutaneously into both whisker pads immediately or 9, 28, and 62 days following injury, respectively. To prevent labeling of non-buccal-associated motor neurons in the lateral and intermediate FMN, the right mandibular branches were surgically removed immediately before injections. After 48 or 72 hours, animals were transcardially perfused with a 4% paraformaldehyde solution in 0.1 M PBS, cryoprotected in 20% sucrose in 0.1M PBS, and cut into 20 μm coronal sections spanning the FMN; fluorescently labeled motoneurons within the FMN were quantified by a single investigator under double blind conditions using unbiased Stereo Investigator software (Stereo Investigator; MicroBrightField, Williston, VT, USA). The total number of FG+ MNs in the FMN ipsilateral to injury were compared following facial nerve crush. Images were obtained using a Leica TCS SP5 Confocal Microscope at 40X.
Immunohistochemistry
As previously described (Bracchi-ricard et al., 2008), animals were transcardially perfused and a ~4 mm segment containing the injury site from the buccal branch of the facial nerve was removed and fixed for 20 min prior to cryoprotection. Longitudinal sections were cut at 16 μm and incubated overnight at 4°C with a mouse antibody against NF-H (1:3000; Covance), p65, phosphoSer276 (1:400; Millipore), GFAP (1:1000; BD Pharmingen), MPZ (1:100; Abcam) or CD11b (1:100; Serotec) followed by a species specific secondary fluorescent antibody: Alexa Fluor 488 (1:750; Molecular Probes), Alexa Fluor 546 (1:750; Molecular Probes) for 1 hr at room temperature. Confocal images were acquired on a Zeiss LSM 510 confocal microscope with a 20X objective or 40X oil objective and LSM imaging software.
Facial Motor Neuron Counts
One month following transection, coronal sections spanning the FMN were prepared as described above (see Retrograde Tracing). Sections were incubated overnight in EtOH/Chloroform (1:1), rehydrated and placed in a 0.5% Cresyl Violet acetate (SIGMA) solution for 20 seconds to label MNs. Using Stereo Investigator software, the total number of Cresyl Violet MNs were quantified by a single investigator, under double blind conditions, in both facial motor nuclei and expressed as a ratio (Injured/Uninjured). The average motor neuron cell body size (area) was determined on each MN counted by measuring the soma diameter using the nucleator probe with 4 nodes and expressed in μm2.
Macrophage Quantification
To quantify the number of macrophages present in degenerating nerves, longitudinal fresh frozen sections (20 μm), distal to transection, were collected and stained with a rat-anti-CD11b antibody and DAPI. Using StereoInvestigator, CD11b+ cells whose nuclei were contained and/or overlapped with the “acceptance region” of a 25x25 μm2 counting frame were counted in 4–9 serial sections/animal/timepoint. The estimated total number of CD11b+ cells/mm2 were recorded in WT and transgenic animals. Images were acquired with a Zeiss LSM 510 confocal microscope and LSM imaging software.
Toluidine Blue and Electron Microscopy Tissue Preparation
Following perfusion, a 2 mm nerve segment distal to the crush injury site was fixed in EM fixative (2% Glutaraldehyde/0.05M PO4/100mM Sucrose) and post-fixed in 2% OSO4/0.1M PO4 for 1 hr at room temperature at the EM core facility in the Miami Project to Cure Paralysis as previously described (Xu et al., 1995). Following dehydration, tissues were placed in fresh resin molds and incubated overnight at 64°C. Using a Leica Ultracut E microtome, semi-thin and ultra-thin sections were cut transversely from the rostral portion corresponding to a cross section of the buccal nerve 4 mm distal to the injury site, and stained with Toluidine Blue. Toluidine Blue images were aquired on a Zeiss Axiovert 200M microscope with a 63X oil objective and Neurolucida Imaging software. Electron micrographs were obtained with a Philips CM-10 transmission electron microscope.
Morphometric Analysis
The total number of myelin rings were acquired using the optical fractionator probe (Stereo Investigator software) in transverse Toluidine Blue stained sections distal to injury with a 63X, oil objective. All ultrastructural analysis associated with myelin was determined by hand measurements (mm) from 20–28 randomly selected EM micrographs spanning the nerve and calibrated using an EM ultrastructural calculator. The vertical and horizontal diameter of the inner mesaxon were recorded and averaged to obtain axonal diameter (AxD). Similarly, the vertical and horizontal diameter of the outer lammellae were averaged to determine fiber diameter and ring thickness ((outer sheath diameter-axonal diameter)/2). For each myelinated axon, the gRatio was calculated: (axonal diameter)/(fiber diameter). All other ultrastructure descriptors were acquired by hand from 20–28 EM micrographs, including the number of SC nuclei, Remak bundles, umyelinated axons and organized Remak bundles. Values obtained were normalized to the total number of photos assessed prior to statistical analysis.
Western Blot Analysis
Distal tissues from the buccal nerve were disrupted in 200 μl of lysis buffer (50 mM Tris, 150 mM NaCl, 1% SDS, 0.5 mM dithiothreitol) containing complete protease inhibitor (Roche, Indianapolis, IN, USA) and phosphatase inhibitors (phosphatase inhibitor cocktails I and II, SIGMA) as previously described (Bracchi-ricard et al., 2008; Monje et al., 2008). SDS sample buffer was added to supernatants followed by 10 min incubation at 100°C. 10 μl of protein samples were resolved on a 12.5% SDS-polyacrylamide gel by electrophoresis and transferred to a nitrocellulose membrane (BioRad). All membranes were blocked in 5% BSA in TBS-T for 1 hr at room temperature and subsequently incubated with the following antibodies (1:1000) in TBS-T at 4°C overnight: anti-MBP (monoclonal rat; Millipore), Lamin A/C (polyclonal rabbit; Cell Signalling), c-Jun (polyclonal rabbit; Santa Cruz Biotechnology, Inc.), MPZ (Covance), Krox-20 (rabbit polyclonal; a kind gift from D. Meijer, Erasmus University Medical Center). Blots were incubated with horseradish peroxidase-conjugated anti-rabbit (Amersham Biosciences), anti-rat (SIGMA), or anti-chicken (Jackson ImmunoResearch Laboratories, Inc.) secondary antibodies (1:2000) in 3% BSA diluted in TBS-T for 1 hr at room temperature. Immunoreactive proteins were detected using a SuperSignal West Pico Chemiluminescent kit (Thermo Scientific). Densitometric quantification was performed using Quantity One software (BioRad) and normalized against Lamin A/C and expressed as a percentage of WT naïve. The total number of animals used for this procedure was between 3 and 7 per time point.
Real-time RT-PCR
Total RNA was extracted from distal, buccal nerve tissues with an RNeasy Micro Kit (Qiagen). cDNA was generated with a Sensiscript RT Kit (Qiagen), according to manufacturer’s instructions, using total RNA as a template. A Rotor-Gene 3000 Real Time Cycler (Corbett Research) was utilized for Real-time RT-PCR on cDNA, along with a Quantitect SYBR Green PCR kit (Qiagen). Relative gene expression was calculated upon comparison with a standard curve generated for each respective gene and subsequently normalized to 18S. The following primers were used: 18S (forward: 5′-GAACTGAGGCCATGATTAAGAG-3′; reverse: 5′-CATTCTTGGCAAATGCTTTC-3′); GFAP (forward: 5′-AGAAAGGTTGAATCGCTGGA-3′; reverse: 5′-CGGCGATAGTCGTTAGCTTC - 3′; SIGMA); IκBα-dn (forward: 5′-TTCATAAAGCCCTCGCATCC-3′; reverse: 5′-ACAGCCAGCTCCCAGAAGTG-3′); β-actin (forward: 5′-ATGGTGGGAATGGGTCAGA-3′; reverse: 5′-CACGCAGCTCATTGTAGAAGG-3′).
Statistical Analysis
For single comparisons, a two-tailed, unpaired Student’s t test was applied; p ≤ 0.05 determined significance. A one-way ANOVA was performed for multiple comparisons; p ≤ 0.05 determined significance.
RESULTS
Naïve GFAP-IκBα-dn and WT nerves display no discernable differences in adult mice
We first investigated whether inhibition of glial NF-κB affected axonal development or myelination within the facial nerve of naïve (unoperated), adult mice. Toluidine Blue (TB) stained sections and electron micrographs revealed no differences in the quantity (WT: 1108 ± 7.52; GFAP-IκBα-dn: 1067±38.43), caliber (WT: 2.28±0.34 μm; GFAP-IκBα-dn: 2.57±0.21μm) or quality of myelinated axons between WT (wild type) and transgenic nerves (Figs 1B–C,7A,C). Both groups exhibited the same distribution and thickness of compact myelin rings (data not shown), gRatio (axonal diameter/fiber diameter), and expression of myelin structural proteins MBP and MPZ (Figs 1D,7B,D). Surprisingly, all nerves contained Remak bundles encasing non-myelinated (unmyelinated) axons. We saw no differences in the total number of Remak bundles, unmyelinated axons, or SC nuclei between groups (Fig. 7E–G). Taken together, we found no morphological abnormalities within transgenic nerves that may preclude regeneration studies.
Figure 1.

Naïve GFAP-IκBα-dn nerves display no developmental aberrations. (A) Naive, WT facial nerve schematic showing the buccal (*) and mandibular (upper(U) and lower (L)) branches. Arrow indicates the cranial nerve exit site from the stylomastoid foramen, caudal to trifurcation. (B) Transverse, Toluidine Blue stains and electron micrographs (lower panels) of the buccal branch 8mm rostral to trifurcation. Scale bar: 20, 10, and 5 μm respectively from top to bottom panels. (C) Quantification of myelin rings (n=4). (D) Expression of MBP and MPZ proteins determined by Western Blot analysis (n=3–7). Results were obtained using stereology followed by the unpaired Student’s t-test and are expressed as the mean ± SEM.
Figure 7.

Effects of NF-κB inhibition on Schwann cell re-myelination. (A) Electron micrographs on transverse, ultrathin sections of the buccal branch 4 mm rostral to the crush injury site. *: intact, myelinated axon. Arrowhead: Remak bundle. SC: Schwann cell nuclei. Scale Bar: 5 μm. Quantification of intact, myelinated axons (B), average axonal diameter (C), gRatio (D), organized Remak bundles (E), unmyelinated axons (F) and Schwann cell nuclei (G) before and after injury. Geometric measurements were taken on all myelin rings encasing an axon regardless of ring morphology. X-axis: number of days post injury (dpi). N: naïve. Data were obtained from electron micrographs and are expressed as the mean ± SEM of 3–4 animals/group. *p<.05, **p<.01, unpaired Student’s t-test (WT vs transgenic). ###p<.001, one-way ANOVA.
Functional inhibition of NF-κB in GFAP-IκBα-dn, denervated Schwann cells following crush injury
To ensure that expression of our GFAP-IκBα-dn transgene results in functional inhibition of NF-κB activation in Schwann cell glia, we developed an injury model (Fig. 2A) whereby the buccal branch of the right facial nerve was crushed, leaving the epineurial sheath intact to foster regeneration and minimal motor neuron (MN) cell death. Injured facial nerves were immunostained for activated NF-κB [phospho-p65 (Ser276)] and GFAP, one day following crush injury (Fig. 2B). Confocal microscopy revealed clear nuclear expression of phospho-p65 (colocalized with DAPI) within GFAP expressing SCs in WT nerves. In contrast, transgenic nerves lacked NF-κB activation in SCs as demonstrated by the absence of nuclear phospho-p65; a finding consistent with our previous studies following sciatic nerve injury (Fu et al., 2010).
Figure 2.

Transgenic inhibition of NF-κB activation in denervated SCs following facial nerve crush injury. (A) Schematic representation of facial nerve crush model. (B) Immunostains of GFAP, phospho-p65, and DAPI within distal nerves 1 day following crush injury. Scale Bar: 20 μm. (C) Injured and uninjured buccal nerve. Arrow indicates crush site in the buccal branch. Neurofilament (NF) staining (top panels) of injured (ipsilateral) and uninjured (contralateral) nerves 2 days post injury. Scale Bar: 20 μm. (C) Fluorogold labeled motor neurons (bottom panels) in the facial motor nucleus 2 days post injury. Scale Bar: 50 μm.
Since the MN cell bodies of the facial nerve reside in the brainstem, we also assessed injury induced NF-κB activation in astrocytes. Sections from the brainstem, immunostained for phospho-p65 and GFAP, illustrated nearly no NF-κB activation in astrocytes within the facial motor nucleus (FMN) (Fig. 3) of both genotypes 1, 4 and 31 days post injury (dpi). Hence, we restricted our regenerative studies to the peripheral component of the facial nerve where NF-κB activation is functionally inhibited in SC glia.
Figure 3.

Minimal activation of astroglial NF-κB within the facial motor nucleus following crush injury. Representative immunostains of GFAP (green), phospho-p65 (red), and DAPI (blue) within WT and transgenic (IκBα-dn) nerves 1, 4, and 31 days post injury. Note: astrocytic activation is strongest at 4 dpi and activated NF-κB is robust at 1 and 31 dpi, but not in astrocytes, in both groups. Scale Bar: 20 μm.
Facial nerve crush injury model exhibits minimal axonal sparing, abrupt axonal degeneration, and elevated GFAP expression
Next, we assessed the efficacy of the crush injury on ablation of axon fibers and axonal retrograde transport. The mouse whiskerpad is innervated by the buccal and upper marginal mandibular branch of the facial nerve, which partially overlap in somatotopic organization within the lateral FMN (Ashwell, 1982). Immediately following injury, the mandibular branches were removed and Fluorogold (FG) was subcutaneously injected into the whisker pad. At 2 dpi, the facial nerve was stained for neurofilament, heavy isoform (NF-H), to illuminate the crush site and degenerating axons with punctate staining within an intact nerve sheath (Fig. 2C). No FG staining was observed in the FMN 2 dpi, indicating minimal axonal sparing (Fig. 2C) ipsilateral to injury. As an internal control, the contralateral, uninjured side showed FG uptake and labeling of facial motor neurons, indicating that the IκBα-dn transgene does not interfere with FG transport in uninjured nerves. A robust increase in GFAP gene expression was observed at 4 dpi (Fig. 4), which is indicative of SC de-differentiation into a denervated phenotype (Stoll and Muller; 1999). This explains the increase in GFAP-driven, IκBα-dn transgene expression (undetected in WT) seen in transgenic nerves following injury (Fig. 4).
Figure 4.
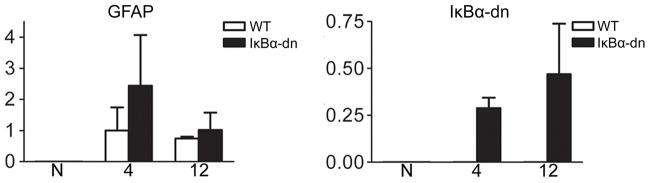
Quantification of gene expression using Real-time RT-PCR on cDNA (normalized to 18S) generated from buccal nerve tissue distal to the injury site. X-axis: number of days post injury. N: naive. Data are expressed as the mean ± SEM of 3 mice/group.
Inhibition of NF-κB activation transiently delays and then promotes regeneration
Using our pinch crush model (Fig. 2), we tested the hypothesis that activation of NF-κB is required for peripheral nerve regeneration (Fig. 5). Three days before sacrifice, the mandibular branches were surgically removed; subsequently, FG was injected subcutaneously into the ipsilateral whiskerpad to trace regenerating MNs. Brains were removed from sacrificed animals 12, 31, and 65 dpi and FG-positive (FG+) MNs were quantified (Fig. 5). There was no difference in regeneration at 12 dpi, but at 31 dpi transgenic animals had significantly fewer FG+ MNs (356.9 ± 56.50; n=7) compared to WT littermates (613.0 ± 27.53; n=5). Similar results were obtained following complete facial nerve transection, with no differences in facial motor neuronal death or cell body size 31 dpi (Fig. 6). Surprisingly, by 65 dpi transgenic animals had substantially more FG+ MNs (1305 ± 156.5; n=6) than WT (890.4 ± 78.37; n=9). Electron micrographs (Fig. 7A–B) revealed significantly less intact axons encased in compact myelin, 4 mm distal to the injury site, at 31 dpi within GFAP-IκBα-dn (40.96 ± 27.31/cm2; n=3) nerves compared to WT (209.5 ± 15.06/cm2; n=3). Interestingly, at 65 dpi transgenic nerves had dramatically increased to WT levels of intact, myelinated axons. Importantly, at 65 dpi the quantity of intact, myelinated axons in both groups had increased to naïve levels, a finding indicative of successful regeneration. However, the size (Fig. 7C) and total quantity of visible axons (data not shown) were similar between groups, suggesting that fewer transgenic axons had regenerated to the whisker pad at 31 dpi, since there was less FG uptake by MNs (Fig. 5B). Taken together, these findings demonstrate an initial decrease followed by a significant increase in the quantity of regenerating MNs when NF-κB activation is inhibited in SCs.
Figure 5.

NF-κB activation in denervated SCs significantly influences axonal regeneration. (A) Fluorogold (FG) labeling of motor neuron (MN) cell bodies in the facial motor nucleus (FMN) of normal (WT) and transgenic (IκBα-dn) mice 12, 31 and 65 days post crush injury (dpi). Scale Bar: 50 μm (B) Stereological quantification of the number of FG+ MNs in the injured FMN. Results are expressed as the mean ± SEM of 5–9 animals/group. *p < .05, **p < .01, unpaired Student’s t-test (WT cpmared to transgenic). #p < .05, ###p < .001, one-way ANOVA.
Figure 6.

Glial NF-κB activation is required for efficient axonal regeneration but not motor neuron survival 31 days after facial nerve axotomy. (A) Retrogradely labeled facial motor neuron soma within the facial motor nucleus of the brainstem following FG administration. Co: uninjured side. Ax: injured side. Scale Bar: 100 μm. (B) Quantification of FG+ MNs. Results expressed as a ratio (axotomized vs. control). Quantification of vibrissae motor performance scores. (C) Quantification of the total number of Cresyl Violet MNs (ipsilateral/contralateral). Quantification of MN soma area (μm2). Data expressed as the mean ± SEM of 4–5 anmials/group; Student’s -test (*, p<.05).
Since NF-κB activation within Schwann cells has been implicated in SC survival and proper regeneration (Weinstein, 1999; Boyle et al., 2005), we were surprised to find no significant differences in the number of SC nuclei assessed in electron micrographs between groups, before or after injury (Fig. 7G). There was, however, a significant increase in the quantity of SC nuclei from naïve levels at 31 dpi in both groups, which likely reflects injury-induced SC proliferation.
Functional inhibition of NF-κB activation in denervated and unmyelinated Schwann cells delays sensory bundle formation
In naïve nerves, unmyelinated axons were clearly sorted and organized into Remak bundles in both groups (Fig. 7A). Each bundle was well defined and often contained more than 10 axons separated by the cytoplasm of an unmyelinated SC. At 31 dpi, Remak bundles in transgenic mice appeared unorganized (Fig. 7A), where the extracellular matrix was greatly compromised and loosely surrounding disrupted cytoplasmic space housing vacuoles. The total number of intact Remak bundles was significantly less in GFAP- IκBα-dn (15.11 ± 8.36/cm2; n=3) compared to WT (111.4 ± 25.71/cm2; n=3) mice 31 days following injury (Fig. 7E). Interestingly, the number of unmyelinated axons was unaltered between naïve and 31 dpi nerves in both groups, suggesting a reduction in the quantity of unmyelinated axons within Remak bundles. At 65 dpi, both groups had the same number of organized Remak bundles as well as unmyelinated axons (Fig. 7E,F). However, there were far fewer sorted axons within each bundle (usually 2–3), when compared to naïve conditions, which may necessitate the overall increase in the number of SCs seen in injured and restored facial nerves.
Inhibition of Schwann cell NF-κB activation delays compact re-myelination
Next, we sought to determine if there were any changes in re-myelination associated with NF-κB activation. Electron micrographs of cross sections distal to injury revealed a general lack of myelin compaction and adaxonal contact in GFAP-IκBα-dn nerves (Fig. 7A); although some compact myelin rings were present 31 dpi. Since both groups had the same average axonal diameter (Fig. 7C), a significant reduction in the gRatio (Fig. 7D) at 31 dpi supports the lack of ring compaction seen in transgenic nerves. There were no apparent differences in myelin ring size or compaction at 65 dpi between groups. These data suggest that NF-κB activation in SCs is required for timely re-myelination following injury.
Expression of myelin regulatory transcription factors and structural proteins
Following loss of axonal-SC contact, effective regeneration in the PNS is heavily reliant on SC dedifferentiation into an “immature-like” phenotype (Jessen and Mirsky, 2008). Developmental myelination initiated by immature SCs and re-myelination orchestrated by denervated SCs are thought to utilize similar genetic programs (Stoll and Muller, 1999; Jessen and Mirsky, 2008). SC de-differentiation is dependent on intracellular signaling molecules such as c-Jun and Krox-20 which serve as negative and positive regulators of myelination, respectively. To assess SC-mediated events following injury, we performed Western blot analysis on harvested distal nerve tissue. In both animal groups, uninjured nerves from naïve animals exhibited virtually no c-Jun protein expression (Fig. 8); however, by 4 dpi, a strong increase in c-Jun expression was found, indicative of SC de-differentiation. At later time points c-Jun was attenuated, as SCs presumably re-entered the promyelin state (Fig. 8).
Figure 8.

Myelin-associated protein expression is unaltered in Schwann cells lacking NF-κB activation following crush injury. (A) Western blots of myelin-associated transcription factor (cJun, Krox-20) and structural (MPZ, MBP) protein expression within the distal nerve of normal (WT) and transgenic mice before (N) and 4, 12, and 31 days after injury. Lamin A/C serves as a loading control. (B) Densitometric quantification of protein expression normalized to WT naïve (N). AU: arbitrary units. Results are expressed as the mean ± SEM of 3–7 animals/group. *p<.05, unpaired Student’s t-test.
Krox-20 is a zinc finger protein essential for myelin basic protein (MBP) and myelin protein zero (MPZ) production, two structural proteins involved in myelin sheath formation and maintenance (Topilko et al., 1994; Decker et al., 2006). We found a significant reduction in krox-20 expression in naïve GFAP-IκBα-dn nerves when compared to WT littermates, suggesting that NF-κB may positively regulate krox-20 expression under normal physiological conditions (Fig. 8). Krox-20 protein expression levels were greatly reduced by 4 dpi in distal nerves as SCs phenotypically revert to an immature/denervated state, and are steadily recapitulated at 12 and 31 dpi in both groups (Fig. 8).
Next, we explored the expression levels of myelin structural proteins utilized for lamallae compaction by SCs: MBP and MPZ (Fig. 8). We were surprised to find no differences between MBP and MPZ protein expression before or after injury in both groups (n=3/group/time point). Taken together, these data suggest that NF-κB activation in denervated SCs is not required for demyelination or the subsequent synthesis of key myelin structural proteins critical for re-myelination.
NF-κB activation in denervated SCs is not required for Wallerian degeneration
A delay in axonal degeneration could explain the delay in regeneration and re-myelination seen in transgenic mice. Reduced macrophage infiltration into the injured nerve, for example, impedes proper clearance of myelin and axonal debris which serves as an inhibitory substrate to fiber regeneration (Boivin et al., 2007). This is unlikely in our model, since MBP and MPZ expression levels decrease following injury (Fig. 8); however, we wanted to carefully assess Wallerian degeneration.
Since degeneration and regeneration occur somewhat simultaneously, following nerve crush, we completely transected the buccal branch and sutured the proximal stump to underlying muscle tissue to prevent reinnervation of the distal nerve segment (Fig. 9A). Transverse, semi-thin cross sections of distal nerves stained with p-phenylenediamine (PPD) and TB revealed no differences in the rate of degeneration between groups (Fig. 9B). At 4 dpi, nerves primarily contained demyelinating axons and myelin debris with few intact axons visible. By 12 dpi, both groups had virtually no intact axons or myelin debris, and fewer axons undergoing demyelination. Additionally, macrophage infiltration to the injured nerve, determined by quantification of CD11b+ cells, was equivalent between groups (Fig. 9C). MBP immunofluorescence on longitudinal, distal nerve segments demonstrated effective demyelination in both groups (Fig. 9D). Hence, there is little evidence to support the role of Schwann cell NF-κB activation in Wallerian degeneration. Furthermore, NF-κB inactivation in Schwann cells does not appear to regulate inflammation, as evidenced by macrophage infiltration, following injury; a surprising finding considering its frequent role in the immune response of many cell types.
Figure 9.

Wallerian degeneration is unaltered by transgenic inhibition of NF-κB in denervated Schwann cells. (A) Schematic representation of facial nerve transection model. (B) Transverse, semi-thin sections stained for myelin with paraphenylenediamine (PPD) and toluidine blue (TB). Sections were collected 4 mm distal to transection at 4 and 12 days post injury (dpi). Scale Bar: 10 μm. Bar graphs represent the total number of de-myelinating and intact axons. Data expressed as the mean ± SEM of 4–8 animals/group. WT: wild type. IκBα-dn: transgenic. (C) CD11b immunostain of macrophages in the distal facial nerve following injury and a naïve sciatic nerve. Bar graph represents stereological quantification of CD11b+ macrophages. Scale Bar: 20 μm. X-axis: days post injury. Results expressed as the mean ± SEM of 3–6 animals/group. (D) Longitudinal facial nerves distal to injury stained with myelin protein zero. Naïve: uninjured sciatic nerve from an adult WT mouse. Scale Bar: 20 μm.
DISCUSSION
Peripheral nerve regeneration is a highly dynamic process orchestrated by several types of cells in a spatiotemporal manner; impairment of certain cellular events associated with injury and recovery can result in unfavorable outcomes. Here, we show a novel function for NF-κB signaling in Schwann cells during peripheral nerve injury and repair: without NF-κB activation in Schwann cells, regeneration and compact re-myelination is transiently delayed 31 days following buccal crush injury. Later, this results in enhanced regeneration and normal myelin compaction 65 days after injury. These findings were not a result of impaired Wallerian degeneration, macrophage infiltration, Schwann cell de- and/or re-differentiation, or myelin protein synthesis.
We were surprised to find no notable differences in macrophage infiltration or Wallerian degeneration in the absence of NF-κB activation in denervated SCs (Fig. 9). Toll-like receptor signaling, known to activate NF-κB, regulates macrophage infiltration into injured nerves. Perturbations of Toll-like receptor signaling significantly delays macrophage recruitment following sciatic nerve injury, thereby impeding the proper clearance of myelin and axonal debris which serve as an inhibitory substrate to fiber regeneration (Boivin et al., 2007). Although we found no differences in Wallerian degeneration following transection, we cannot completely rule out the possibility of delayed degeneration after crush injury, since they are subtly different paradigms. However, several observations suggest that degeneration is unaltered following pinch crush injury: WT and IκBα-dn littermates exhibit similar axonal degeneration profiles 1 dpi, demonstrated by NF-H immunostains (Fig. 2), similar levels of regeneration 12 dpi (Fig. 5), and a similar Schwann cell de-/re-differentiation profile (Fig. 8).
Our findings are likely the result of an increase in axonal regeneration but a decrease in the rate of regeneration and, therefore, a decrease in the rate of re-myelination and myelin compaction. However, there are several, unexplored explanations for our findings, including an ablation or impairment in lipid biosynthesis and/or perturbed Krox-20 transcriptional regulation.
Myelin is highly enriched in glycosphingolipids and cholesterol; protein trafficking and myelin compaction require cholesterol in Schwann cells. Lipids constitute 70–80% of the dry weight of myelin, whereas plasma membranes exhibit a lipid to protein ratio of 1:1 (Norton and Cammer, 1984; Saher et al., 2009). Although cholesterol reutilization by SCs is not required for axonal regeneration, it has been shown to be necessary for proper myelination and compaction by regulating MPZ exit from the endoplasmic reticulum en route to myelin compartments (Saher et al., 2009). Perhaps a reduction in cholesterol, supplied by Schwann cells, could result in slower neurite and axonal membrane outgrowth following injury, resulting in a ~1:1 pairing of regenerating fibers and Schwann cell columns; in this scenario, axons may reach target tissues in a more effective ratio and require less pruning. Future studies on this process will better elucidate the mechanisms by which SC NF-κB governs regeneration and re-myelination of the PNS.
Krox-20 (Egr-2) was recently shown to regulate, at least in part, cholesterol/lipid biosynthetic genes during peripheral nerve myelination in the sciatic nerve (Leblanc et al., 2005). Krox-20 and sterol regulatory element binding protein (SREBP) transactivators synergistically activate promoters of many SREBP target genes to regulate myelination during development (Leblanc et al., 2005). Immunoblots showed a significant decrease in naïve, krox-20 protein expression levels in IκBα-dn mice, paired with normal myelin structure (Fig. 8). This suggests that naïve, transgenic animals require less cholesterol for myelin maintenance during adulthood; perinatal studies of transgenic animals may exhibit a delay in myelination that is compensated for by adulthood (2–4 months of age).
Our findings in adult, naïve transgenic mice demonstrate normal myelination at two months of age within the facial nerve. Following injury, transgene expression is robustly elevated and effectively inhibits p65 activation in denervated Schwann cells in transgenic mice. However, once denervated Schwann cells begin maturing into a myelinating phenotype they will forfeit GFAP expression; those pursuing a non-myelinating phenotype will retain GFAP expression. Although there are advantages to the conditional activation of our transgene in that we can study the population of denervated GFAP-expressing Schwann cells, as well as mature non-myelinating Schwann cells, GFAP is not expressed in mature, myelinating Schwann cells; therefore, we cannot directly understand the initial role played by NF-κB activation in these cells. Future studies using different genetic approaches will help elucidate these matters.
Acknowledgments
We would like to thank D. Meijer for the krox-20 antibody, P. Monje for constructive discussions. We would also like to thank the University of Miami Surgery, Histology and EM core facilities; in particular, M. Bates, R. Puzis, and A. Gomez for EM studies and B. Frydel for microscopy assistance. This work was supported by the National Institutes of Health training Grant [T32 NS007459 to P.D.M.], funded by the National Institute of Neurological Disorders and Stroke and National Institute of Health Predoctoral Training Program in central nervous system injury and repair; and the National Institute of Health RO1 Grant [NS51709 to J.R.B.].
References
- Ashwell KW. The adult mouse facial nerve nucleus: morphology and musculotopic organization. J Anat. 1982;135(Pt 3):531–8. [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Boivin A, Pineau I, Barrette B, Filali M, Vallieres N, Rivest S, Lacroix S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J Neurosci. 2007;27(46):12565–76. doi: 10.1523/JNEUROSCI.3027-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle K, Azari MF, Cheema SS, Petratos S. TNFalpha mediates Schwann cell death by upregulating p75NTR expression without sustained activation of NFkappaB. Neurobiol Dis. 2005;20(2):412–27. doi: 10.1016/j.nbd.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Bracchi-Ricard V, Brambilla R, Levenson J, Hu WH, Bramwell A, Sweatt JD, Green EJ, Bethea JR. Astroglial nuclear factor-kappaB regulates learning and memory and synaptic plasticity in female mice. J Neurochem. 2008;104(3):611–23. doi: 10.1111/j.1471-4159.2007.04993.x. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202(1):145–56. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Hurtado A, Persaud T, Esham K, Pearse DD, Oudega M, Bethea JR. Transgenic inhibition of astroglial NF-kappa B leads to increased axonal sparing and sprouting following spinal cord injury. J Neurochem. 2009;110(2):765–78. doi: 10.1111/j.1471-4159.2009.06190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang H, Yoon SO, Xu X, Hottiger MO, Svaren J, Nave KA, Kim HA, Olson EN, Lu QR. HDAC-mediated deacetylation of NF-kappaB is critical for Schwann cell myelination. Nat Neurosci. 2011;14(4):437–41. doi: 10.1038/nn.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Yu WM, Strickland S. Peripheral regeneration. Annu Rev Neurosci. 2007;30:209–33. doi: 10.1146/annurev.neuro.30.051606.094337. [DOI] [PubMed] [Google Scholar]
- Decker L, Desmarquet-Trin-Dinh C, Taillebourg E, Ghislain J, Vallat JM, Charnay P. Peripheral myelin maintenance is a dynamic process requiring constant Krox20 expression. J Neurosci. 2006;26(38):9771–9. doi: 10.1523/JNEUROSCI.0716-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernyhough P, Smith DR, Schapansky J, Van Der Ploeg R, Gardiner NJ, Tweed CW, Kontos A, Freeman L, Purves-Tyson TD, Glazner GW. Activation of nuclear factor-kappaB via endogenous tumor necrosis factor alpha regulates survival of axotomized adult sensory neurons. J Neurosci. 2005;25(7):1682–90. doi: 10.1523/JNEUROSCI.3127-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu ES, Zhang YP, Sagen J, Candiotti KA, Morton PD, Liebl DJ, Bethea JR, Brambilla R. Transgenic inhibition of glial NF-kappa B reduces pain behavior and inflammation after peripheral nerve injury. Pain. 2010;148(3):509–18. doi: 10.1016/j.pain.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6(9):671–82. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia. 2008;56(14):1552–65. doi: 10.1002/glia.20761. [DOI] [PubMed] [Google Scholar]
- Leblanc SE, Srinivasan R, Ferri C, Mager GM, Gillian-Daniel AL, Wrabetz L, Svaren J. Regulation of cholesterol/lipid biosynthetic genes by Egr2/Krox20 during peripheral nerve myelination. J Neurochem. 2005;93:737–748. doi: 10.1111/j.1471-4159.2005.03056.x. [DOI] [PubMed] [Google Scholar]
- Limpert AS, Carter BD. Axonal neuregulin 1 type III activates NF-kappaB in Schwann cells during myelin formation. J Biol Chem. 2010;285(22):16614–22. doi: 10.1074/jbc.M109.098780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Bisby MA. Increased activation of nuclear factor kappa B in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Brain Res. 1998;797(2):243–54. doi: 10.1016/s0006-8993(98)00380-1. [DOI] [PubMed] [Google Scholar]
- Miller SC, Huang R, Sakamuru S, Shukla SJ, Attene-Ramos MS, Shinn P, Van Leer D, Leister W, Austin CP, Xia M. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem Pharmacol. 2010;79(9):1272–80. doi: 10.1016/j.bcp.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje PV, Athauda G, Wood PM. Protein kinase A-mediated gating of neuregulin-dependent ErbB2-ErbB3 activation underlies the synergistic action of cAMP on Schwann cell proliferation. J Biol Chem. 2008;283(49):34087–100. doi: 10.1074/jbc.M802318200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols JC, Valentine W, Kanwal S, Carter BD. Activation of the transcription factor NF-kappaB in Schwann cells is required for peripheral myelin formation. Nat Neurosci. 2003;6(2):161–7. doi: 10.1038/nn995. [DOI] [PubMed] [Google Scholar]
- Norton WT, Cammer W. In: Isolation and characterization of myelin. Morell P, editor. Myelin. New York: Plenum Press; 1984. pp. 147–195. [Google Scholar]
- Pollock G, Pennypacker KR, Memet S, Israel A, Saporta S. Activation of NF-kappaB in the mouse spinal cord following sciatic nerve transection. Exp Brain Res. 2005;165(4):470–7. doi: 10.1007/s00221-005-2318-6. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, et al. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43(1):57–67. doi: 10.1016/j.neuron.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Saher G, Quintes S, Mobius W, Wehr MC, Kramer-Albers EM, Brugger B, Nave KA. Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J Neurosci. 2009;29(19):6094–104. doi: 10.1523/JNEUROSCI.0686-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Smith D, Tweed C, Fernyhough P, Glazner GW. Nuclear Factor-κB activation in axons and Schwann cells in experimental sciatic nerve injury and its role in modulating axon regeneration: studies with Etanercept. Exp Neurol. 2009;68:691–700. doi: 10.1097/NEN.0b013e3181a7c14e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Muller HW. Nerve injury, axonal degeneration and neural regeneration: basic insights. Brain Pathol. 1999;9:313–25. doi: 10.1111/j.1750-3639.1999.tb00229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svaren J, Meijer D. The molecular machinery of myelin gene transcription in Schwann cells. Glia. 2008;56:1541–51. doi: 10.1002/glia.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, Babinet C, Charnay P. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–99. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- Weinstein DE. Review: The role of Schwann cells in neural regeneration. Neuroscientist. 1999;5:208–216. [Google Scholar]
- Werner A, Willem M, Jones LL, Kreutzberg GW, Mayer U, Raivich G. Impaired axonal regeneration in alpha7 integrin-deficient mice. J Neurosci. 2000;20(5):1822–30. doi: 10.1523/JNEUROSCI.20-05-01822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XM, Guenard V, Kleitman N, Bunge MB. Axonal regeneration into Schwann cell-seeded guidance channels grafted into transected adult rat spinal cord. J Comp Neurol. 1995;351(1):145–60. doi: 10.1002/cne.903510113. [DOI] [PubMed] [Google Scholar]
- Yoon C, Korade Z, Carter BD. Protein kinase A-induced phosphorylation of the p65 subunit of nuclear factor-kappaB promotes Schwann cell differentiation into a myelinating phenotype. J Neurosci. 2008;28(14):3738–46. doi: 10.1523/JNEUROSCI.4439-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]