

Abstract
A peptide-based hydrogel has been designed that directs the formation of hydroxyapatite. MDG1, a twenty-seven residue peptide, undergoes triggered folding to form an unsymmetrical β-hairpin that self-assembles in response to an increase in solution ionic strength to yield a mechanically rigid, self supporting hydrogel. The C-terminal portion of MDG1 contains a heptapeptide (MLPHHGA) capable of directing the mineralization process. Circular dichroism spectroscopy indicates that the peptide folds and assembles to form a hydrogel network rich in β-sheet secondary structure. Oscillatory rheology indicates that the hydrogel is mechanical rigid (G′ ∼ 2500 Pa) before mineralization. In separate experiments, mineralization was induced both biochemically and with cementoblast cells. Mineralization-domain had little effect on the mechanical rigidity of the gel. SEM and EDS show that MDG1 gels are capable of directing the formation of hydroxapatite. Control hydrogels, prepared by peptides either lacking the mineral-directing portion or reversing its sequence, indicated that the heptapeptide is necessary and its actions are sequence specific.
Keywords: Biomineralization, Biomimetic Material, Peptide, Hydrogel, Scaffold, Hydroxyapatite
Introduction
A major ongoing challenge in the field of reconstructive and regenerative medicine is the successful repair or replacement of hard tissue, such as cartilage, bone, and dental tissues, which has been lost due to disease, congenital defects or trauma. Autograft tissue is considered as one of the best sources for the replacement of lost hard tissue. Its use, however, is limited due to difficulties in harvesting substantial amounts of autologous grafts with the appropriate shapes.[1] Allografts and xenografts of freeze-dried or de-mineralized hard tissue have also been utilized for repair; however major limitations exist for these materials, such as the risk of host immune response and, more importantly, the transfer of pathogens such as HIV and hepatitis viruses. [2] To overcome the inadequacies of current tissue repair and reconstruction strategies, a variety of tissue engineering approaches has been developed and expanded with a focus on utilizing synthetic tissue scaffolds to replace or regenerate the lost tissue.
Some of the most commonly investigated materials for possible hard tissue scaffolds include natural materials, such as cornstarch-based polymers [3], chitosan [4], collagen [5], and natural calcium phosphates [6-8], and synthetic materials, such as calcium phosphates of inorganic origin [9-10] and synthetic inorganic polymers.[11-12] Many of these scaffolds have been designed to be mimics of bone, e.g., possessing similar mechanical properties of the hard tissue. The complexity and highly hierarchical structure of hard tissues, however, often require tissue scaffolds that provide an architectural support for the cells, as well as influence and guide the structural properties of the newly formed mineral phase. Soluble extracellular matrix (ECM) proteins play an important role in this process.[13-15] Therefore, many of the key proteins thought to regulate bone development have been incorporated within tissue scaffolds. For example, bone morphogenetic proteins (BMPs), Arginine-Glycine-Aspartic acid (RGD) peptides, and P15 peptides [13, 16-17] are amongst the most widely used bioactive molecules to develop biomimetic tissue scaffolds. Although there are many exciting examples, the lack of knowledge on both the function and the temporal and spatial distribution of the proteins involved in biological mineralization have largely limited the successful use of these proteins in tissue engineering. An alternate approach is to incorporate small peptides within a material matrix that can guide biomineralization. Our labs are developing genetically engineered peptides that selectively bind to inorganics (GEPIs). The GEPIs are selected by exposing a pool of random amino acid sequences, displayed on the surface of a host organism (e.g. bacteriophage or bacteria) [18-19] to a target inorganic solid substrate and identifying the strongly binding sequences. [20] It has been shown that GEPIs can bind to solid substrates specifically and significantly influence the inorganic material formation processes. [21-26]
For many implantable hard materials, regardless of how similar the mechanical and physiochemical properties of designed scaffold are to the living bone, when clinical application is considered, the many advantages offered by these materials can be lost if the scaffold cannot be implanted correctly at the defect site. Poor tissue integration due to improper apposition at the site of implantation is a major failure mechanism of many tissue engineering materials. Thus, an in situ forming scaffold, where the three-dimensional matrix is formed at the defect site, would be advantageous. These materials eliminate the complications originating from the prefabrication process and improve the physical contact between the scaffold and the surrounding tissue. These materials can be introduced to the wound bed in a minimally invasive manner via a syringe. For example, the synthetic polymer, poly(methyl methacrylate) (PMMA), is a widely used in situ forming material in clinical orthopedics. Although PMMA has been shown to be very successful in many applications, its use, however, is limited due to the exothermic polymerization reaction that occurs during material formation, usually leading to the necrosis of the surrounding tissue. [27] Moreover, the non-degradable nature of PMMA poses a risk for foreign body response and may slow down the healing of the hard tissue. Synthetic polymers, such as poly(propylene fumarate) [28] and polyanhydride [29], have also been utilized as in situ forming scaffolds. These polymers are cyto-compatible and, hence, partially eliminate the risk of foreign body response, but their utility is curtailed due to the highly cross-linked nature of the network formed, posing limitations in maintaining cell viability. An attractive alternative for in situ forming tissue scaffolds would be hydrogels formed from self assembling materials such as amphiphilic peptides or proteins. [30-43] Peptide hydrogels enable formation of scaffold materials that mechanically resemble the native ECM and can be formed at the site of implantation without the use of harsh chemical cross-linking agents or the presence of adverse assembling reactions that can affect the surrounding tissues.
In this work, we describe the development of an in situ forming, self-assembling peptide hydrogel that is capable of directing the mineralization of calcium phosphate. The peptide, MDG1 (Mineral Directing Gelator), undergoes triggered intra-molecular folding into a conformation that subsequently self-assembles to form a fibrillar network where a mineral directing GEPI sequence is displayed from the fibrils that constitute the gel, Figure 1. The ability to form three-dimensional networks that carry an inherent functionality makes these hybrid peptides attractive candidates for use in tissue engineering applications and suitable models for developing, multifunctional structures for nano-technological applications.
Figure 1.
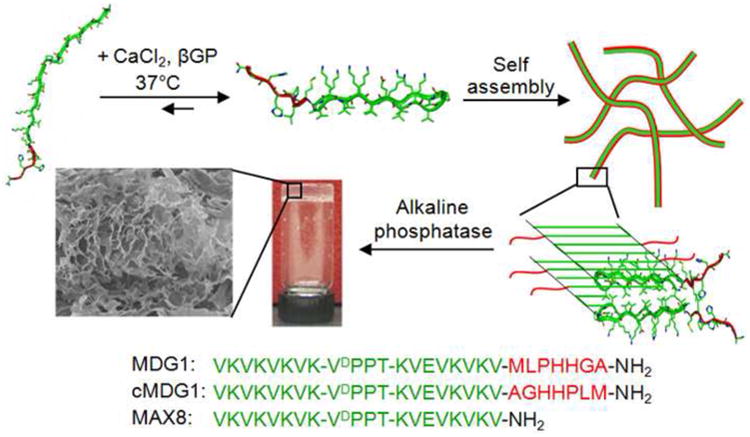
Schematic representation of the folding, self assembly, and resultant hydrogelation of MDG1, cMDG1, and MAX8 peptides. Sequences of the three peptides are shown.
Materials and Methods
Peptide Synthesis and Purification
Peptides were synthesized on RINK amide resin via an automated ABI 433A peptide synthesizer employing standard Fmoc-protocol and HCTU activation. The resulting resin-bound peptides were cleaved and side-chain deprotected for 2 hours using a TFA: thioanisole: ethanedithiol: anisole (90:5:3:2) cocktail. Filtration followed by ether precipitation yielded crude peptides that were purified by RP-HPLC using a Vydac C18 peptide/protein column.[44] Analytical HPLC profiles and ESI (+) mass spectroscopy spectrum of the pure peptides are provided in the Supporting Information.
Circular Dichroism
Circular dichroism experiments were performed on a Jasco J-810 spectropolarimeter. Stock 300 μM solutions of each peptide were prepared in chilled pH 7.4, 25 mM Tris buffer. 150 μL of the peptide stock was placed in a 1 mm quartz cell. To the same cell, 150 μL of chilled pH 7.4, 25 mM Tris buffer containing 48 mM CaCl2 and 28.8 mM β-glycerophosphate (β-GP) was added. The quartz cell was gently mixed by cell inversion to yield the final peptide solution (150 μM peptide, 25 mM Tris, 24 mM CaCl2, and 14.4 mM β-GP). The cell was immediately placed in the CD cell holder pre-equilibrated at 5°C. Temperature dependent wavelength spectra were collected every 5°C up to a maximum temperature of 80°C with a 10 min equilibration time at each temperature. The concentration of the peptide solutions were determined by UV-Vis absorbance at 220 nm (ε = 21263 cm−1 M−1 for MDG1 and cMDG1, ε = 15,750 cm−1 M−1 for MAX8). Mean residue ellipticity [θ] was calculated from the equation, [θ]= θobs/(10*l*c*r), where θobs is the measured ellipticity (millidegrees), l is the length of the cell (centimeters), c is the concentration (molar), and r is the number of residues.
Dynamic Oscillatory Rheology
Oscillatory rheology experiments were performed on a Paar Physica MCR 500 rheometer using a 25 mm diameter stainless steel parallel plate geometry. For the one hour rheological measurements, gels were prepared directly on the rheometer plate. To prepare 1 wt% hydrogels, peptide stock solutions were first prepared in glass vials by dissolving 3.5 mg of each peptide in 175 μL of chilled pH 7.4, 25 mM Tris buffer. To the stock solution, 175 μL of chilled Tris buffer (25 mM, pH 7.4) containing 48 mM CaCl2 and 28.8 mM β-GP was added. Then, 300 μL of the stock solution was quickly added to the rheometer plate, which was pre-equilibrated at 5°C. The parallel plate tool was then lowered to a gap height of 0.5 mm and the temperature was ramped linearly to 37°C to initiate gelation. Initially, a dynamic time sweep was performed to measure the storage (G′) and loss (G″) modulus at a frequency of 6 rad/sec and 0.2% strain as a function of time for an hour. A dynamic frequency sweep (0.1 to 100 rad/sec at constant 0.2% strain) was then performed, followed by a dynamic strain sweep (0.1 to 1000% strain at constant 6 rad/sec) for all samples to ensure the hydrogels were within the linear viscoelastic regime.
For the 24 hour rheological measurements, gels were made in a 3.5 K molecular weight cut-off dialysis cassette (Pierce, USA) and then transferred to the rheometer plate for assessment. Peptide stock solutions were first prepared in glass vials by fully dissolving 6 mg of each peptide in 300 μL of sterile, chilled pH 7.4, 25 mM Tris buffer. To this solution, 300 μL of chilled Tris buffer (25 mM, pH 7.4) containing 48 mM CaCl2 and 1.4×10−6 mg/mL alkaline phosphatase was then added. Then, 500 μL of the stock solution was degassed with continuous nitrogen flow and then was quickly added to the dialysis cassette. Control stock solutions were prepared with Tris CaCl2 buffer containing no alkaline phosphatase. The dialysis cassettes containing the hydrogels were placed in an incubator equilibrated at 37°C. Two hours after gel preparation, the dialysis cassettes were immersed in a 500 mL solution of pH 7.4, 25 mM Tris buffer containing 24 mM CaCl2 and 14.4 mM β-GP. Hydrogels were placed in an incubator equilibrated at 37°C overnight. After 24 hrs, the gels were removed from the dialysis cassettes and the gels were transferred to the rheometer plate, which was equilibrated at 37°C. The parallel plate tool was then lowered to a gap height of 0.5 mm. A dynamic time sweep was performed for 15 min to ensure G′ and G″ reached a plateau, followed by a dynamic frequency sweep and a dynamic strain sweep for all samples, with the same parameters as the one hour rheology studies.
Hydrogelation and Solution Mineralization
Mineralization experiments were performed in a 3.5 K molecular weight cut-off dialysis cassette (Pierce, USA) using an Alkaline Phosphatase (AP) mediated mineralization model that mimics matrix vesicle mineralization. A mineralization solution was prepared by diluting stock solutions of CaCl2 and AP to final concentrations of 24 mM and 1.4×10−6 mg/mL respectively, in 200 μL of pH 7.4 Tris buffer. 2 mg of peptide was dissolved in this mineralization solution. The solutions were degassed with continuous nitrogen flow and injected into the dialysis cassette. The solution was allowed to gelate at 37°C in a humidified chamber for 2 hours. After gelation was complete, the cassette was immersed into a 14.4 mM β-glycerophosphate solution in the same buffer and allowed to mineralize for an additional 22 hours. The mineralization took place through the hydrolysis of β-glycerophosphate by the AP entrapped in the gel and the reaction of the released inorganic phosphate group with the calcium.
Cell Culture
An immortalized mouse cementoblast cell line, (OCCM-30), was maintained in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were encapsulated within the hydrogel in the following manner. First, MDG1 and MAX8 were dissolved separately at a concentration of 1wt% in water. Then an equal volume of serum free DMEM, containing 2×104 OCCM-30 cells, 100μg/mL ascorbic acid and 40 mM β-GP was added to the peptide solution, resulting in a 0.5wt% hydrogel with 50μg/mL ascorbic acid and 20 mM β-GP. The mixture was allowed to gel in 96 well plates for 10 minutes at 37°C. DMEM containing 1% FBS was then added on top of the gels. The cells were cultured in the gels for 9 days and fresh media containing 1%FBS, 50μg/mL ascorbic acid and 20 mM β-GP was added every other day to the top of the gels.
A SYTO 9/Propidium iodide based live dead assay (Invitrogen, USA), where only the dead cells are stained with propidium iodide and gave red fluorescence, was used to test the viability of the cells in the hydrogel. The cells were encapsulated as described below and at the end of 3 days, a 1:1 mixture of SYTO 9/Propidium iodide was added on top of the gels and incubated for 30 minutes. The gels were then washed three times with serum free DMEM and imaged using a TE 300L microscope (Nikon, Japan) with the appropriate filters. A z-series of 10 images with 35μm step sizes were taken and the images were reconstructed using MetaMorph Imaging System version 6.2 (Photometrics UK Ltd.,UK, formerly Universal Imaging Co., USA). The viable and the dead cells were counted using ImageJ image processing software (NIH, USA).
Electron Microscopy Analysis
At the conclusion of the mineralization studies (24 hours for solution mineralization and 9 days for cell culture mineralization), the hydrogels were immersed into liquid nitrogen and lyophilized. The lyophilized gels were kept desiccated at −20°C until analyzed by microscopy. For scanning electron microscopy (SEM), the lyophilized gels were placed on a carbon tape and coated with platinum prior to microscopy. SEM imaging and energy dispersive spectroscopy (EDS) were performed using a JSM 7000F (JEOL) SEM at 10 kV in secondary electron imaging mode. For transmission electron microscopy (TEM), the lyophilized gels were mounted on double folding copper TEM grids. TEM imaging and electron diffraction were performed using an EMS 420T TEM at 120 kV (FEI, Inc., USA; formerly Philips, The Netherlands).
X-ray Diffraction Analysis
X-ray diffraction (XRD) measurements were performed to identify the final phase of the mineral deposited on the gels. The gel samples were collected by re-dissolving the lyophilized gel-mineral composite in distilled water to solubilize the peptide matrix. The mineral component was isolated by centrifugation. Collected mineral was dried in vacuum at room temperature prior to the measurements. The measurements were performed on a D8 Focus model diffractometer employing CuKα radiation, 40kV and 40mA. (Bruker, USA). Data were collected in the range of 2θ = 3° to 60° using a 0.05° step size and 1 sec/step scan speed. The XRD spectra were analyzed using Jade software version 8.5 (Materials Data Inc, USA)
Results and Discussion
Peptide Material Design
MDG1 is a twenty-seven residue peptide designed to undergo triggered intramolecular folding and subsequent self-assembly to form a fibillar network affording a mechanically rigid gel, Figure 1. When dissolved in pH buffered water at low ionic strength, the peptide remains unfolded due to unfavorable electrostatic interactions between residue side chains. When a solution containing CaCl2 and β-GP is added, these interactions are screened and the peptide is able to fold. The N-terminal twenty residues of MDG1 are designed to adopt an amphiphilic β-hairpin when the peptide folds. This N-terminal portion contains two β-strands connected by a four residue sequence (-VDPPT-) know to adopt a type II′ β-turn. [45] The β-strands are composed of alternating hydrophobic and hydrophilic residues that give the hairpin its amphiphilic character in the folded state. The N-terminal portion of MDG1 has been reported in the literature and studied extensively (e.g. MAX8: VKVKVKVKVDPPTKVEVKVKV-CONH2). [46-47] MAX8 hairpins readily self-assemble affording a fibrillar network where each fibril is composed of a bilayer of hairpins that have self-assembled laterally via intermolecular hydrogen bond formation along the long axis of a given fibril, Figure 1. [47-49] Hydrogels formed from MAX8 are mechanically rigid, viscoelastic materials that are cytocompatible. The C-terminal seven residues of MDG1 (MLPHHGA) were incorporated at the hairpin terminus so that this sequence could be displayed from the fibril surface and direct mineralization. This small peptide had been reported in the literature and named HABP1 (MLPHHGA). [50] It was identified via phage display and had been shown to control calcium phosphate mineral formation in solution. Specifically, HABP1 slows the mineralization rate and accelerates the transformation of amorphous calcium phosphate (ACP) into crystalline octacalcium phosphate (OCP) during mineralization. This results in the formation of elongated, plate-like particles. The full length peptide, MDG1 should fold and assemble, forming hydrogels capable of directing calcium phosphate mineralization.
In addition to the MDG1 hydrogel, two control hydrogels were prepared and studied. The first control gel is formed by the self-assembling peptide, cMDG1. This peptide is identical to the parent peptide except that the sequence of the seven C-terminal residues is reversed. cMDG1 examines the role that sequence specificity and amino acid composition play in directing mineral deposition. If sequence specific interactions are made between the C-terminal sequence of MDG1 and inorganic species during mineralization, distinct mineral morphology and type should be observed as compared to mineral deposits formed in hydrogels comprised of the control peptide (cMDG1). If there is little difference in the type of mineral deposited, this would suggest that sequence-specific interactions are largely not at play and that non-specific interactions, such as gross electrostatics, may be more important. This would suggest that amino acid composition, rather than sequence specificity, is the more relevant design parameter. The second control hydrogel is formed by the peptide MAX8, which lacks the mineral-directing portion. This control gel was used to determine if the fibrillar scaffold alone could direct mineralization. In addition, the MAX8 control was used to assess what effect adding a C-terminal sequence to an otherwise symmetrical β-hairpin had on peptide folding, self-assembly and the rheological properties of the resultant hydrogel.
Triggered Folding, Self-Assembly and Hydrogelation
The peptide construct MDG1 was designed to undergo triggered folding and self-assembly resulting in the formation of a mechanically rigid hydrogel in the presence of CaCl2 and β-GP. Mineralization within the gel is possible when alkaline phosphatase is added to cleave β-GP, liberating phosphate, which then paryicipates in the formation of calcium phosphate mineral. Circular dichroism (CD) spectroscopy was used to follow the folding and subsequent self-assembly of MDG1 at pH 7.4, 37°C in response to the addition of CaCl2 and β-GP. These salts screen unfavorable electrostatic side chain interactions that normally inhibit peptide folding. Figure 2a shows wavelength spectra for MDG1, cMDG1, and MAX8 peptide solutions after folding had been initiated. Low concentrations (150 μM) of peptide were used for the CD studies to limit scattering and aid data acquisition; at this concentration, the peptides do not form gels but still fold and assemble into soluble β-sheet-rich fibrils whose secondary structure can be easily monitored. All three peptides display spectra characteristic of β-sheet structure with minima at ∼217 nm. The spectra of MDG1 and cMDG1 are nearly identical indicating that the manner in which the GEPI, mineral-directing sequence is attached is inconsequential with respect to the ability of the hairpin portion to fold and assemble. Also evident is that the absolute value of the mean residue ellipticity at 217 nm for the MAX8 control peptide is greater. This indicates that the seven C-terminal residues of MDG1 and cMDG1 do not contribute to the β-sheet signal. Previous CD studies of the heptapeptide (MLPHHGA) alone indicate that this peptide exists in an equilibrium of polyproline and random coil conformations.[50] Taken together, the CD data suggests that MDG1 and cMDG1 fold, affording hairpins that assemble into β-sheet rich fibrils that display structurally flexible mineral-directing motifs as depicted in Figure 1.
Figure 2.
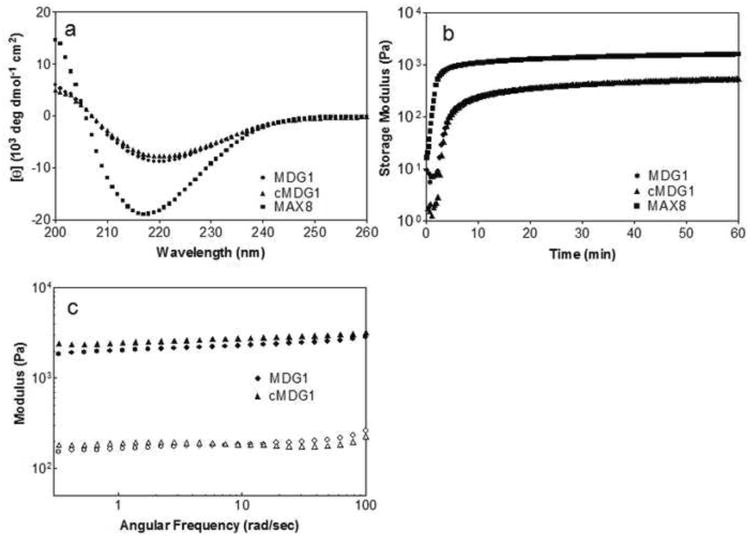
(a) CD wavelength spectra of 150 μM of MDG1, cMDG1, and MAX8 in pH 7.4 Tris-HCl buffer, 24 mM CaCl2, and 14.4 mM β-glycerophosphate at 37°C. (b) Dynamic oscillatory rheology of 1 wt% peptides in pH 7.4 Tris-HCl buffer, 24 mM CaCl2, and 14.4 mM β-glycerophosphate formed at 37°C. (c) Dynamic frequency sweep of 1 wt% peptides after 24 hr incubation at 37°C. Hydrogels are prepared with pH 7.4 Tris-HCl buffer containing 24 mM CaCl2 directly in a dialysis cassette that is then incubated for 2 hr at 37°C. The cassette is then immersed in a pH 7.4 Tris-HCl buffer containing 24 mM CaCl2 and 14.4 mM β-glycerophosphate for an additional 22 hr at 37°C.
Figure 2b shows dynamic oscillatory shear rheology time sweep experiments that monitor the evolution of the storage modulus (G′, a measure of a material's elastic response to applied stain) after peptide folding, self-assembly and subsequent hydrogelation has been triggered. Rheology of the three peptides was performed at a peptide concentration of 1 wt % at pH 7.4 in 25 mM Tris buffer containing 24 mM CaCl2 and 14.4 mM β-GP at 37°C. Both MDG1 and cMDG1 form viscoelastic hydrogels within the first ten minutes of the experiment ultimately yielding gels with storage moduli of 510 ± 10 Pa and 590 ± 100 Pa, respectively after one hour. In comparison, the control peptide MAX8 lacking any C-terminal extension forms a much more rigid gel, more quickly, with a modulus of 1650 ± 150 Pa after one hour. Taken together, this data suggests that the C-terminal extensions of MDG1 and cMDG1 decrease the rate of hydrogelation and reduce the mechanical rigidity of their respective hydrogels. The experiments in Figure 2b involve forming the gels in situ directly in the rheometer and the gels are allowed to cure for only one hour. Interestingly, when MDG1 and cMDG1 gels are incubated at 37°C for 24 h, their rigidities increase to nearly 2500 Pa. Figure 2c shows frequency sweep data taken at 24 h; G′ values for MDG1 and cMDG1 have increased to 2400 ± 300 Pa and 2700 ± 100 Pa at a frequency of 6 rad/sec, respectively and are an order of magnitude greater than the loss modulus (G″).
Biomineralization
The ability of the MDG1 hydrogel to promote and direct mineralization was probed by rheology, quantitative mineral load assessment, spectroscopy, microscopy and diffraction experiments. In the biomineralization experiments we used an enzyme-mediated mineralization model to mimic vertebrate biomineralization, where AP controls calcium phosphate formation by hydrolyzing organophosphate groups releasing inorganic phosphate. Here, therefore, our model consists of AP entrapped within the hydrogel in the presence of β-GP. Hydrogels used in the mineralization studies were prepared employing a dialysis cassette. Hydrogels were formed by initiating gelation with a solution of CaCl2 containing alkaline phosphatase directly in the cassette. Gelation was allowed to proceed over two hours and then the cassette was immersed in a bath that contained a buffered (pH 7.4) solution of β-GP and CaCl2. Mineralization occurred as the β-GP diffused into the cassette and was cleaved by the enzyme. For all the peptide gels (MDG1, cMDG1 and MAX8), as mineralization takes place, the initially optically clear gels become opaque. No mineralization was observed in the immersion buffer indicating that all the calcium phosphate deposition took place within the hydrogel. Figure 3a shows rheological data comparing mineralized and non-mineralized gels formed under identical conditions. The mechanical rigidity of MDG1 gels increased slightly after 24 h of mineralization. In comparison, the rigidities of both control peptides were affected little by the mineralization process. The measured mineral loads within each of the gels (Figure 3b) show a similar trend. The MDG1 gel accommodates a slightly higher load than the control gels. The data in Figure 3 indicate that all three peptide gels are capable of supporting mineralization and that the C-terminal, mineral-directing attachment has no effect on gross mineralization.
Figure 3.
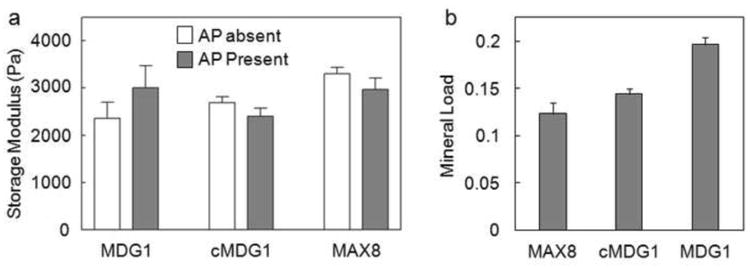
(a) Average storage modulus values (6 rad/sec) of 1 wt% gels after 24 hr incubation at 37°C in the absence or presence of alkaline phosphatase, which was used to promote mineraliztion. (b) Mineral load of the gels after 24 hours of mineralization.
Next, studies were performed to assess whether or not the mineral-directing peptide attachment could control the crystal morphology and the phase of the deposited calcium phosphate mineral. Scanning electron microscopy (SEM) images of lyophilized peptide hydrogels after mineralization are displayed in Figure 4a-f. With the understanding that the lyophilization treatment can influence material morphology, a highly porous structure with 10-15 μm pore sizes was the governing morphology observed for the MDG1, cMDG1 and MAX8 hydrogel networks. Energy dispersive spectroscopy (EDS) analysis reveals that calcium phosphate mineralization took place in all three gels (Figure 4a,c,e - insets). As expected, no calcium or phosphate was observed in the non-mineralized gels (data not shown). At higher magnification, morphological differences in the deposited mineral on MDG1 become evident, appearing more crystalline (Figure 4b), suggesting that the C-terminal appendage (MLPHHGA) may be influencing mineral morphology. In contrast, a poorly crystalline mineral film was observed on the control cMDG1 and MAX8 hydrogels (Figure 4d and f). Transmission electron microscopy (TEM) analysis further supports this assertion and shows distinct differences in the crystallinity of the deposited minerals within each of the peptide hydrogels, Figure 5. Mineral deposited within the MDG1 gel was highly crystalline and elongated, resembling biological apatite. The average length of these crystals was 100 nm with an approximate aspect ratio of between 1:10 – 1:20. Selected ara electron diffraction analysis confirmed the crystallinity of the particles. The mineral deposited within MDG1 showed a sharp 002 diffraction peak, indicating the thin plate-like nature of the crystals, and a broad diffraction band containing the 211, 112, and 300 triple diffraction peaks, which are characteristic to biological apatite. (Figure 5a - inset). Few individual particles were observed on the cMDG1 gel, which includes the reversed mineral-directing sequence, (Figure 5b). In addition, the poor quality of these crystals resulted in highly diffused electron scattering (Figure 5b – inset). Lastly, no individual particles were observed on the MAX8 gels, consistent with the the fact that the mineral-directing sequence was not present.
Figure 4.
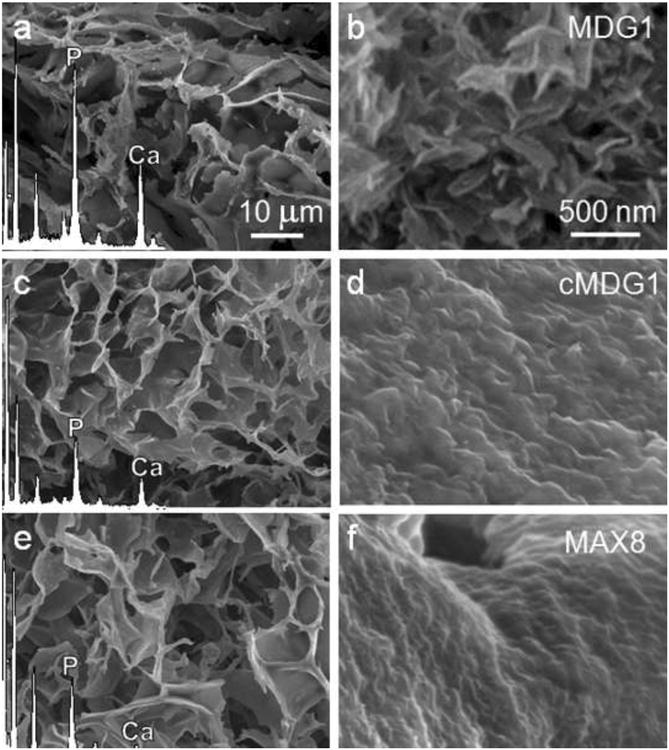
SEM micrographs and corresponding EDS spectra of lyophilized MDG1 (a), cMDG1 (c), and MAX8 (e) gels after 24 hr mineralization. The morphological differences among the deposited minerals are observable at higher magnifications in panels b, d and f, respectively.
Figure 5.

TEM micrographs and the corresponding electron diffraction pattern (insets) of lyophilized MDG1 (a), cMDG1 (b), and MAX8 (c) gels after 24 hr mineralization.
Identification of the final calcium phosphate phase is often difficult by electron diffraction analysis alone because of the similar crystal structure of octacalcium phosphate (OCP) and hydroxyapatite (HA). One of the key diffraction effect, i.e., the characteristic OCP ring that corresponds to (100) (d = 18.6Å), is usually masked by the transmitted beam in the electron diffraction pattern. Therefore, X-ray diffraction (XRD) measurements were performed in addition to electron diffraction analysis to eliminate these uncertainties in pahe identification. The results obtained from the XRD analyses were consistent with the morphological observations and the electron diffraction analyses obtained by TEM. The XRD pattern of the mineral deposited on MDG1 consisted of numerous sharp peaks indicative of crystalline HA, Figure 6. The major peaks were observed at 2θ = 31.8° (d = 2.81Å) and 32.1° (d = 2.78Å) corresponding to (211) and (300) planes, respectively. In comparison, the mineral deposited on the MAX8 control yielded only a weak and broad peak at 2θ = 31.8° (d = 2.81Å) indicating a small amount of poorly crystalline HA. For both MDG1 and MAX8 gels, diffraction peaks corresponding to other crystalline phases were not observed. Taken together, the microscopy and the diffraction studies suggest that the fibril network alone can accommodate mineralization. However, the mineral-directing attachment can influence the morphology of the deposited mineral. In addition, the data also indicates that the peptide attachment must be displayed from the fibrils with sequence fidelity, suggesting that, here, sequence-specific interactions with mineral components are being made to direct mineralization.
Figure 6.
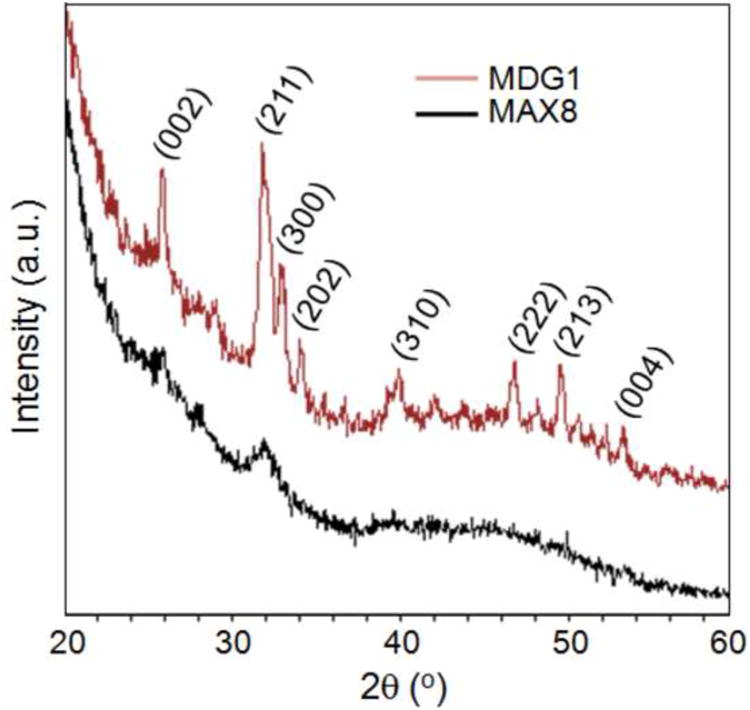
XRD patterns of mineral deposited on MDG1 and MAX8 hydrogels.
Cell Viability and Mineralization
The response of cells to the MDG1 matrix was assessed by monitoring mineralization induced by encapsulated cementoblasts. Cells were directly embedded in a 0.5 wt% MDG1 hydrogel by initiating peptide folding and self-assembly in the presence of cells (see Experimental Section). In this experiment, the DMEM media contained calcium and β-GP; viable cells should produce and secrete alkaline phosphatase to drive mineralization if the MDG1 hydrogel is cytocompatible. Upon 3 days of post encapsulation, cell viability assay shows that encapsulated cementoblasts were viable in the MDG1 matrix with 597 ± 25 live cells versus 157 ± 12 dead cells per 7.5 × 10−5cm3 (Figure 7a). After 9 days of culture in the mineralizing conditions, SEM analysis indicates that the cells were capable of mineralizing the MDG1 fibril network. The energy dispersive x-ray spectroscopy (EDS) results show clearly observable calcium-phosphate peaks. The mineral deposited on the MDG1 fibrils appeared homogenous in its consistency in contrast to the characteristic large, isolated mineral nodules observed when cementoblasts are cultured on a 2D tissue culture plate in the absence of gel, Figure S1a. The mineral morphology, observed in the MDG1 hydrogels, is consistent with that produced by cementoblast cultures in 2D in the presence of the mineral directing sequence (MLPHHGA) alone, Figure S1b. Previous studies show that this small sequence decreases the rate of mineralization resulting in a slow nucleation favored regime resulting in a homogeneous deposition of mineral instead of the rapid growth of isolated mineral nodules. [50] The data indicate the mineral directing sequence can also function when directly appended to a material scaffold and does not affect the ability of cementoblasts to produce alkaline phosphatase which, in turn, directs mineralization.
Figure 7.
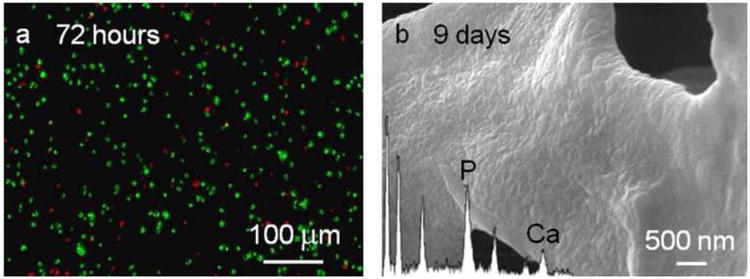
(a) Live/dead cytotoxicity assay on cementoblast cells 3 days after encapsulation into the MDG1 matrix. Viable cells fluoresce green and the dead cells fluoresce red. (b) SEM micrographs of the MDG1 matrix mineralized by cementoblasts after 9 days. The inset shows the corresponding EDS.
Conclusion
In this study, we have designed, synthesized and studied the mineralization potential of a peptide-based hydrogel. The peptide, MDG1, undergoes triggered folding to form an unsymmetrical β-hairpin that self-assembles in response to an increase in solution ionic strength to yield a mechanically rigid, self supporting hydrogel. The C-terminal portion of MDG1 contains a heptapeptide capable of directing the mineralization process of calcium and phosphate to produce hydroxyapatite. The ability to form well defined three-dimensional networks that carry an inherent functionality makes these hybrid peptides attractive candidates for use in tissue engineering applications and ideal models for developing, multifunctional structures for nano-technological applications.
Supplementary Material
Alizarin Red staining of the mineral deposited by cementoblasts in 2D culture containing (a) no peptide and (b) 100μg/ml of MLPHHGA.
(a) HPLC chromatogram and (b) ESI mass spectra of MDG1.
(a) HPLC chromatogram and (b) ESI mass spectra of cMDG1.
(a) HPLC chromatogram and (b) ESI mass spectra of MAX8.
Acknowledgments
This work was supported (MG, HF, CT, MS) mainly by the National Science Foundation through the Genetically Engineered Materials Science & Engineering Center (GEMSEC), an MRSEC at UW. This work was also supported (MB, JPS) by funding from the NIDCR-NIH grant, R01 DE01638601
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Mustafa Gungormus, Email: musgun@u.washington.edu.
Monica Branco, Email: monica.branco@nih.gov.
Hanson Fong, Email: hfong@u.washington.edu.
Joel P. Schneider, Email: Joel.Schneider@nih.gov.
Candan Tamerler, Email: tamerler@u.washington.edu.
Mehmet Sarikaya, Email: sarikaya@u.washington.edu.
References
- 1.Yaszemski MJ, Payne RG, Hayes WC, Langer R, Mikos AG. Evolution of bone transplantation: Molecular, cellular and tissue strategies to engineer human bone. Biomaterials. 1996;17:175–85. doi: 10.1016/0142-9612(96)85762-0. [DOI] [PubMed] [Google Scholar]
- 2.Buck BE, Malinin TI, Brown MD. Bone Transplantation and Human Immunodeficiency Virus: An Estimate of Risk of Acquired Immunodeficiency Syndrome (AIDS) Clin Orth Relat Res. 1989;240:129–36. [PubMed] [Google Scholar]
- 3.Stevenson S. Enhancement of Fracture Healing With Autogenous and Allogeneic Bone Grafts. Clin Orth Relat Res. 1998;355:S239–S46. doi: 10.1097/00003086-199810001-00024. [DOI] [PubMed] [Google Scholar]
- 4.Madihally SV, Matthew HWT. Porous chitosan scaffolds for tissue engineering. Biomaterials. 1999;20:1133–42. doi: 10.1016/s0142-9612(99)00011-3. [DOI] [PubMed] [Google Scholar]
- 5.Bet MR, Goissis G, Vargas S, Selistre-de-Araujo HS. Cell adhesion and cytotoxicity studies over polyanionic collagen surfaces with variable negative charge and wettability. Biomaterials. 2003;24:131–7. doi: 10.1016/s0142-9612(02)00270-3. [DOI] [PubMed] [Google Scholar]
- 6.Holmes RE. Bone Regeneration Within a Coralline Hydroxyapatite Implant. Plast Reconstr Surg. 1979;63:626–33. doi: 10.1097/00006534-197905000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Vacanti CA, Bonassar LJ, Vacanti MP, Shufflebarger J. Replacement of an Avulsed Phalanx with Tissue-Engineered Bone. N Engl J Med. 2001;344:1511–4. doi: 10.1056/NEJM200105173442004. [DOI] [PubMed] [Google Scholar]
- 8.Yuan HP, Kurashina K, de Bruijn JD, Li YB, de Groot K, Zhang XD. A preliminary study on osteoinduction of two kinds of calcium phosphate ceramics. Biomaterials. 1999;20:1799–806. doi: 10.1016/s0142-9612(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 9.Flatley TJ, Lynch KL, Benson M. Tissue Response to Implants of Calcium Phosphate Ceramic in the Rabbit Spine. Clin Orth Relat Res. 1983;179:246–52. [PubMed] [Google Scholar]
- 10.Graves GA, Noyes FR, Villanueva AR. The influence of compositional variations on bone ingrowth of implanted porous calcium aluminate ceramics. J Biomed Mater Res. 1975;9:17–22. doi: 10.1002/jbm.820090405. [DOI] [PubMed] [Google Scholar]
- 11.Cato TL, Saadiq FEA, Sobrasua EI, Darryl AW, Mohamed A, Harry RA, et al. A highly porous 3-dimensional polyphosphazene polymer matrix for skeletal tissue regeneration. J Biomed Mater Res. 1996;30:133–8. doi: 10.1002/(SICI)1097-4636(199602)30:2<133::AID-JBM1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 12.Dietmar WH, Thorsten S, Iwan Z, Kee Woei N, Swee Hin T, Kim Cheng T. Mechanical properties and cell cultural response of polycaprolactone scaffolds designed and fabricated via fused deposition modeling. J Biomed Mater Res. 2001;55:203–16. doi: 10.1002/1097-4636(200105)55:2<203::aid-jbm1007>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Addadi L, Weiner S. Interactions between acidic proteins and crystals: stereochemical requirements in biomineralization. Proc Natl Acad Sci U S A. 1985;82:4110–4. doi: 10.1073/pnas.82.12.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HD, Smith JG, Valentini RF. Bone Morphogenetic Protein 2-Coated Porous Poly-Llactic Acid Scaffolds: Release Kinetics and Induction of Pluripotent C3H10T1/2 Cells. Tissue Eng. 1998;4:35–51. [Google Scholar]
- 15.Lowenstam HA. Minerals formed by organisms. Science. 1981;211:1126–31. doi: 10.1126/science.7008198. [DOI] [PubMed] [Google Scholar]
- 16.Bhatnagar RS, Qian JJ, Wedrychowska A, Sadeghi M, Wu YM, Smith N. Design of Biomimetic Habitats for Tissue Engineering with P-15, a Synthetic Peptide Analogue of Collagen. Tissue Eng. 1999;5:53–65. doi: 10.1089/ten.1999.5.53. [DOI] [PubMed] [Google Scholar]
- 17.Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23:4315–23. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 18.Lee SY, Choi JH, Xu ZH. Microbial cell-surface display. Trends Biotechnol. 2003;21:45–52. doi: 10.1016/s0167-7799(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 19.Smith GP, Petrenko VA. Phage display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 20.Sarikaya M, Tamerler C, Jen AKY, Schulten K, Baneyx F. Molecular biomimetics: nanotechnology through biology. Nat Mater. 2003;2:577–85. doi: 10.1038/nmat964. [DOI] [PubMed] [Google Scholar]
- 21.Naik RR, Stringer SJ, Agarwal G, Jones SE, Stone MO. Biomimetic synthesis and patterning of silver nanoparticles. Nat Mater. 2002;1:169–72. doi: 10.1038/nmat758. [DOI] [PubMed] [Google Scholar]
- 22.Nam KT, Kim DW, Yoo PJ, Chiang CY, Meethong N, Hammond PT, et al. Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes. Science. 2006;312:885–8. doi: 10.1126/science.1122716. [DOI] [PubMed] [Google Scholar]
- 23.Sarikaya M. Biomimetics: Materials fabrication through biology. Proc Natl Acad Sci U S A. 1999;96:14183–5. doi: 10.1073/pnas.96.25.14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarikaya M, Tamerler C, Schwartz DT, Baneyx FO. Materials assembly and formation using engineered polypeptides. Annu Rev Mater Res. 2004;34:373–408. [Google Scholar]
- 25.Tamerler C, Khatayevich D, Gungormus M, Kacar T, Oren EE, Hnilova M, et al. Molecular Biomimetics: GEPI-Based Biological Routes to Technology. Biopolymers. 2010;94:78–94. doi: 10.1002/bip.21368. [DOI] [PubMed] [Google Scholar]
- 26.Sano KI, Sasaki H, Shiba K. Specificity and biomineralization activities of Ti-binding peptide-1 (TBP-1) Langmuir. 2005;21:3090–5. doi: 10.1021/la047428m. [DOI] [PubMed] [Google Scholar]
- 27.Jefferiss CD, Lee AJC, Ling RSM. Thermal Aspects of Self-Curing Polymethylmethacrylate. J Bone Joint Surg Br. 1975;57-B:511–8. [PubMed] [Google Scholar]
- 28.Yaszemski MJ, Payne RG, Hayes WC, Langer R, Mikos AG. In vitro degradation of a poly(propylene fumarate)-based composite material. Biomaterials. 1996;17:2127–30. doi: 10.1016/0142-9612(96)00008-7. [DOI] [PubMed] [Google Scholar]
- 29.Muggli DS, Burkoth AK, Anseth KS. Crosslinked polyanhydrides for use in orthopedic applications: Degradation behavior and mechanics. J Biomed Mater Res. 1999;46:271–8. doi: 10.1002/(sici)1097-4636(199908)46:2<271::aid-jbm17>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 30.Alireza SS, Xuezhong H, Esmaiel J. Effect of osteonectin-derived peptide on the viscoelasticity of hydrogel/apatite nanocomposite scaffolds. Biopolymers. 2007;85:370–8. doi: 10.1002/bip.20659. [DOI] [PubMed] [Google Scholar]
- 31.Aulisa L, Dong H, Hartgerink JD. Self-Assembly of Multidomain Peptides: Sequence Variation Allows Control over Cross-Linking and Viscoelasticity. Biomacromolecules. 2009;10:2694–8. doi: 10.1021/bm900634x. [DOI] [PubMed] [Google Scholar]
- 32.Cui HG, Webber MJ, Stupp SI. Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials. Biopolymers. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Y, Yang ZM, Kuang Y, Ma ML, Li JY, Zhao F, et al. Enzyme-Instructed Self-Assembly of Peptide Derivatives to Form Nanofibers and Hydrogels. Biopolymers. 2010;94:19–31. doi: 10.1002/bip.21321. [DOI] [PubMed] [Google Scholar]
- 34.Hosseinkhani H, Hosseinkhani M, Tian F, Kobayashi H, Tabata Y. Bone Regeneration on a Collagen Sponge Self-Assembled Peptide-Amphiphile Nanofiber Hybrid Scaffold. Tissue Engineering. 2007;13:11–9. doi: 10.1089/ten.2006.0120. [DOI] [PubMed] [Google Scholar]
- 35.Jung JP, Gasiorowski JZ, Collier JH. Fibrillar Peptide Gels in Biotechnology and Biomedicine. Biopolymers. 2010;94:49–59. doi: 10.1002/bip.21326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kazunori H, Motohiro H, Toshihiko Y, Hajime O. Spatial distribution of mineralized bone matrix produced by marrow mesenchymal stem cells in self-assembling peptide hydrogel scaffold. J Biomed Mater Res, Part A. 2008;84A:128–36. doi: 10.1002/jbm.a.31439. [DOI] [PubMed] [Google Scholar]
- 37.Kisiday J, Jin M, Kurz B, Hung H, Semino C, Zhang S, et al. Self-assembling peptide hydrogel fosters chondrocyte extracellular matrix production and cell division: Implications for cartilage tissue repair. Proc Natl Acad Sci U S A. 2002;99:9996–10001. doi: 10.1073/pnas.142309999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kretsinger JK, Haines LA, Ozbas B, Pochan DJ, Schneider JP. Cytocompatibility of self-assembled β-hairpin peptide hydrogel surfaces. Biomaterials. 2005;26:5177–86. doi: 10.1016/j.biomaterials.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 39.Krishna OD, Kiick KL. Protein- and Peptide-Modified Synthetic Polymeric Biomaterials. Biopolymers. 2010;94:32–48. doi: 10.1002/bip.21333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacEwan SR, Chilkoti A. Elastin-Like Polypeptides: Biomedical Applications of Tunable Biopolymers. Biopolymers. 2010;94:60–77. doi: 10.1002/bip.21327. [DOI] [PubMed] [Google Scholar]
- 41.Rapaport H, Grisaru H, Silberstein T. Hydrogel Scaffolds of Amphiphilic and Acidic β-Sheet Peptides. Adv Funct Mater. 2008;18:2889–96. [Google Scholar]
- 42.Woolfson DN. Building Fibrous Biomaterials From alpha-Helical and Collagen-Like Coiled-Coil Peptides. Biopolymers. 2010;94:118–27. doi: 10.1002/bip.21345. [DOI] [PubMed] [Google Scholar]
- 43.Zhan H, Timothy DS, James FH, Alvaro M, Pablo B, Jr, Chung-Yan K, et al. Bioactive Nanofibers Instruct Cells to Proliferate and Differentiate During Enamel Regeneration. J Bone Miner Res. 2008;23:1995–2006. doi: 10.1359/JBMR.080705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagarkar RP, Schneider JP. Synthesis and primary characterization of self-assembled peptide-based hydrogels. Methods Mol Biol. 2008;474:61–77. doi: 10.1007/978-1-59745-480-3_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pantoja-Uceda D, Santiveri CM, Jiménez MA. De novo Design of Monomeric β-Hairpin and β-Sheet Peptides. Methods Mol Biol. 2006;340:27–51. doi: 10.1385/1-59745-116-9:27. [DOI] [PubMed] [Google Scholar]
- 46.Branco MC, Nettesheim F, Pochan DJ, Schneider JP, Wagner NJ. Fast Dynamics of Semiflexible Chain Networks of Self-Assembled Peptides. Biomacromolecules. 2009;10:1374–80. doi: 10.1021/bm801396e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, et al. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc Natl Acad Sci. 2007;104:7791–6. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altunbas A, Sharma N, Lamm MS, Yan CQ, Nagarkar RP, Schneider JP, et al. Peptide-Silica Hybrid Networks: Biomimetic Control of Network Mechanical Behavior. ACS Nano. 2009;4:181–8. doi: 10.1021/nn901226h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hule RA, Nagarkar RP, Altunbas A, Ramay HR, Branco MC, Schneider JP, et al. Correlations between structure, material properties and bioproperties in self-assembled beta-hairpin peptide hydrogels. Faraday Discuss. 2008;139:251–64. doi: 10.1039/b717616c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gungormus M, Fong H, Kim IW, Evans JS, Tamerler C, Sarikaya M. Regulation of in vitro Calcium Phosphate Mineralization by Combinatorially Selected Hydroxyapatite-Binding Peptides. Biomacromolecules. 2008;9:966–73. doi: 10.1021/bm701037x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alizarin Red staining of the mineral deposited by cementoblasts in 2D culture containing (a) no peptide and (b) 100μg/ml of MLPHHGA.
(a) HPLC chromatogram and (b) ESI mass spectra of MDG1.
(a) HPLC chromatogram and (b) ESI mass spectra of cMDG1.
(a) HPLC chromatogram and (b) ESI mass spectra of MAX8.