

Abstract
Recent evidence has shown a role for the serine/threonine protein kinase D (PKD) in the regulation of acute aldosterone secretion upon angiotensin II (AngII) stimulation. However, the mechanism by which AngII activates PKD remains unclear. In this study, using both pharmacological and molecular approaches, we demonstrate that AngII-induced PKD activation is mediated by protein kinase C (PKC) and Src family kinases in primary bovine adrenal glomerulosa cells and leads to increased aldosterone production. The pan PKC inhibitor Ro 31-8220 and the Src family kinase inhibitors PP2 and Src-1 inhibited both PKD activation and acute aldosterone production. Additionally, like the dominant-negative serine-738/742-to-alanine PKD mutant that cannot be phosphorylated by PKC, the dominant-negative tyrosine-463-tophenylalanine PKD mutant, which is not phosphorylatable by the Src/Abl pathway, inhibited acute AngII-induced aldosterone production. Taken together, our results demonstrate that AngII activates PKD via a mechanism involving Src family kinases and PKC, to underlie increased aldosterone production.
Keywords: adrenal glomerulosa, aldosterone, protein kinase D (PKD), angiotensin II (AngII), protein kinase C (PKC), Src family kinases
1. INTRODUCTION
Aldosterone is produced by the glomerulosa cells of the adrenal cortex to regulate salt and water balance. However, excessive aldosterone production can result in primary aldosteronism (PA) which affects 5-10% of hypertensives (Funder, et al. 2009). Patients with primary aldosteronism have an increased prevalence of cardiac fibrosis and congestive heart failure (Funder et al. 2009; Milliez, et al. 2005), and aldosterone increases collagen synthesis in rat cardiac fibroblasts after myocardial infarction (Mill, et al. 2003). About 35% of PA cases are caused by an aldosterone-producing adenoma, a condition also known as Conn’s syndrome, whereas approximately 60% of PA results from bilateral idiopathic hyperaldosteronism (IHA) [reviewed in (Young 2007)]. Although patients with Conn’s syndrome tend to have higher blood pressures than those with IPA (Young 2007), the mechanisms underlying these two disorders are still poorly understood.
In addition to angiotensin-converting enzyme inhibitors, calcium channel blockers, low-dose diuretics, and beta blockers have been used as treatment options for primary hypertension. Nevertheless, patients with hypertension still require combination therapy to achieve optimal blood pressure goals. Thus, there is great interest in the development of medical interventions to reduce the incidence of hypertension and its associated complications.
We have previously shown that the serine/threonine protein kinase D (PKD) mediates AngII-induced aldosterone synthesis acutely (Shapiro, et al. 2010), and there is also evidence for its role in chronic aldosterone secretion (Romero, et al. 2006). Thus, work from the laboratory of Gomez-Sanchez demonstrated that AngII-induced PKD activation is able to increase the expression of the aldosterone-synthesizing enzyme, CYP11B2 (Romero et al. 2006). Additionally this group suggested the importance of novel PKCs in PKD activation and chronic AngII-mediated aldosterone production. Our previous data also suggest a role for phospholipase D in AngII-induced PKD activation and aldosterone production (Olala, et al. 2013). However, the complete mechanisms underlying this PKD activation are not entirely clear.
PKD belongs to a family of three isozymes within the calcium/calmodulin-dependent protein kinase (CaMK) family (Hanks 2003): PKD1/PKCu (Valverde, et al. 1994), PKD2 (Sturany, et al. 2001) and PKD3/PKCv (Rey, et al. 2003), which are activated by phorbol esters, diacylglycerol, growth factors and hormones (Zugaza, et al. 1997). Novel PKCs activate mouse PKD by phosphorylating serines 744 and 748 in the activation loop (serines 738 and 742 in human) (Waldron and Rozengurt 2003), with inhibition of PKC activity abrogating PKD transphosphorylation and thus its activation (Waldron, et al. 1999). In other systems, PKD has also been shown to be activated by tyrosine kinases of the Src family via an Abl-mediated phosphorylation of tyrosine 463 (tyrosine 469 in mouse) (Storz, et al. 2003). A role for Src family kinases in aldosterone production was suggested by studies in rat adrenal glomerulosa cells treated with the Src kinase inhibitor tyrphostin 23 (Kapas, et al. 1995), as well as in the adrenocortical tumor cell line NCI-H295R cells treated with the Src kinase inhibitor PP2 (Sirianni, et al. 2001). Activity of PKD leads to autophosphorylation at serine 916 (910 in human), which can be used as a marker of PKD activation status (Matthews, et al. 1999). In this study, we investigated the mechanisms by which AngII activates PKD to underlie acute aldosterone production.
2. MATERIALS AND METHODS
2.1. Adrenal glomerulosa cell culture and treatment
Glomerulosa cell slices were prepared from near-term fetal bovine adrenal glands obtained from a local meat-packing plant, and the cells were dispersed from collagenase-digested slices by mechanical agitation. Freshly isolated cells were cultured overnight in Falcon Primaria dishes in a Dulbecco’s modified Eagles’s medium–Ham’s F-12 medium (1:1) containing 10% horse serum (v/v), 2% fetal bovine serum (v/v), ascorbate (100 μM), α-tocopherol (1.2 μM), Na2SeO3 (0.05 μM), butylated hydroxyanisole (50 μM), metyrapone (5 μM), penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin-B (0.25 μg/ml). After replacement of the serum-containing medium with serum-free medium plus 0.2% bovine serum albumin (BSA), the cells were incubated for an additional 20–24 hours before treatment. Prior to stimulation, the medium was removed and the cells washed three times with Krebs-Ringer bicarbonate-buffered (KRB+) solution containing sodium acetate (120 mM NaCl, 24.9 mM NaHCO3, 3.5 mM KCl, 1.2 mM MgSO4, 1.2 mM NaH2PO4, 1.25 mM CaCl2, 0.1% dextrose, 0.2% BSA and 2.5mM sodium acetate) and incubated at 37°C in KRB+ equilibrated with 5% CO2 for 30-60 minutes (eqKRB+). AngII and 22(R)-hydroxycholesterol were obtained from Sigma (St. Louis, MO) as were the inhibitors PP2 and Src-1. Ro 31-8220 was obtained from EMD Biosciences (San Diego, CA).
2.2 Measurement of aldosterone production
Cultured adrenal glomerulosa cells were incubated with eqKRB+ containing the appropriate agents for the indicated times, and the supernatants collected and stored frozen until aldosterone was assayed using a solid-phase radioimmunoassay kit (Siemens, Los Angeles, CA).
2.3. Plasmid constructs
Recombinant DNA techniques were performed as described in (Luo, et al. 2007). The human tyrosine-463-to-phenylalanine PKD mutant was kindly provided to us by Dr. Alex Toker (Harvard Medical School, Boston, MA), and has been previously described (Storz et al. 2003). The recombinant adenovirus construct was generated by homologous recombination between the pAdTrack-CMV shuttle plasmid and an adenoviral backbone vector, pAdEasy-1, in electrocompetent BJ5183 cells. Recombinant plasmids were then transfected into Ad-293 cells to generate the replication-deficient adenovirus. Large-scale virus production was achieved by amplification in Ad-293 cells and purification using cesium chloride gradient banding followed by dialysis with storage buffer [10% glycerol, 10 mM Tris, 0.9% NaCl (pH 8.1)] for an hour, with multiple changes of buffer. Titers were determined by measuring OD260. Primary cultures were infected with the adenoviral constructs at a multiplicity of infection (MOI) of 25 in serum-free medium. After 4-hours of incubation at 37°C, GFP expression was observed using confocal microscopy to confirm infection, and media were replaced with serum-free media for 20 hours.
2.4. Western blot analysis
Cultured adrenal glomerulosa cells were incubated with eqKRB+ containing the appropriate agents for the indicated times. Cells were solubilized with warm lysis buffer containing Tris–HCl (0.175 M, pH 8.5), SDS (3%, v/v) and EGTA (1.5 mM) and scraped, and buffer containing 30% (v/v) glycerol, 15% 2-mercaptoethanol (v/v) and 0.1% bromophenol blue (v/v) was added to the lysates to constitute Laemmli buffer. Since the protein concentrations measured were within 5% in individual experiments, equal volumes of sample were subjected to (8%) SDS-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to Immobilon-P or Immobilon-FL membranes and incubated with the appropriate primary and secondary antibodies. Antibodies used were anti-PKD and anti-phosphoserine 916 (serine 910 in human) PKD antibodies from Cell Signaling (Danvers, MA), anti-phosphotyrosine from Millipore (Billerica, MA), anti-GFP from Epitomics (Burlingame, CA), and anti-actin antibody from Sigma (St. Louis, MO). Immunoreactivity was visualized using the ECF system (Amersham, Piscataway, NJ) and imaged on a Typhoon imager (Molecular Dynamics, Sunnyvale, CA). Alternatively, we performed infrared imaging on an Odyssey imaging system (LI-COR Biosciences, Lincoln, NE).
2.5. Immunoprecipitation (IP)
Cultured adrenal glomerulosa cells on Falcon Primaria dishes were incubated with eqKRB+ in the presence or absence of AngII for 30 min. The supernatants were collected for aldosterone assay, and the cells were washed at least three times with ice cold 0.9% NaCl, after which the cells were collected on ice in 500 μl of IP lysis buffer. The IP lysis buffer was composed of the following: 50 mM Tris (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM MgCl2, 5 mM NaF, 1 mM Na3VO3, and 1 Complete Protease Inhibitor Cocktail Tablet (Roche Pharmaceuticals, San Francisco, CA) per 7 mLs of buffer. The cells were incubated with either anti-PKCμ/PKD (Santa Cruz) or anti-phosphotyrosine antibody (Millipore) for 2 hours at 4°C, after which the samples were incubated with Tru-blot anti-rabbit IgG beads (eBioscience) for 1 hour. The samples were then subjected to SDS-PAGE as described above.
2.6. Immunohistochemistry (IHC)
Adrenal tissue was fixed in 10% formalin. The tissues were then dehydrated and paraffin embedded. 10-μm thick sections of paraffin-embedded tissues were dewaxed, and antigen retrieval was performed in boiling 10 mM citrate buffer (pH 6). Endogenous peroxidases were quenched in 0.3% hydrogen peroxide (Sigma, UK) before blocking in normal serum (VectorLabs, UK). Sections were incubated with the PKD primary antibody [a mouse monoclonal that cross-reacts with human and bovine (Abcam, catalog no. ab57114)] for 45 min at room temperature followed by two changes of 1X phosphate-buffered saline. Sections were incubated with a peroxidase-conjugated AffiniPure F(ab’) fragment donkey anti-mouse (Jackson ImmunoResearch Laboraties, Inc., West Grove, PA) and the bound antibody was detected with a 3,3′-diaminobenzidine (DAB) substrate kit (Dako North America Inc., Carpinteria, CA). Sections incubated in the absence of primary antibody served as negative controls. For assessment of staining intensity, three regions corresponding to the capsule, the zona glomerulosa and the zona fasciculata were marked (with the letters A, B and C) on photomicrographs of stained human adrenal glands. The regions were assigned a score of 0 (no staining), 1 (minimal staining), 2 (moderate staining) or 3 (intense staining) by three uninvolved observers. Values were then averaged for the three observers and presented as the means ± SEM of the average staining of three glands. Deidentified cadaver adrenal glands were obtained as discarded tissue during kidney transplantation procedures; consent for the use of tissue for research was obtained from the organ donor’s family by the organ procurement team.
2.7. Statistical analysis
All experiments were repeated a minimum of three times, and within each experiment, variables were performed at least in duplicate. Statistically significant differences were determined by ANOVA with a Newman-Keuls post-hoc test using the computer program Prism (GraphPad Software, San Diego, CA).
3. RESULTS
3.1. PKD was expressed in the zonae glomerulosa and fasciculata of near-term fetal bovine and in adult human adrenal glands
Previous work in our laboratory has demonstrated that both AngII and the PKC activator phorbol 12-myristate 13-acetate (PMA) time- and dose-dependently increase PKD activation and acute aldosterone secretion (within 60 minutes) (Shapiro et al. 2010) in bovine adrenal glomerulosa cells in vitro. Gomez-Sanchez and colleagues have also shown that AngII promotes PKD activation, as well as chronic aldosterone and cortisol production, in the human adrenocortical carcinoma cell line NCI H295R (Romero et al. 2006). PKD may thus play a role in both aldosterone and cortisol production. To determine PKD’s distribution in the intact gland in situ, we used immunohistochemical analysis and found that PKD was expressed not only in the zona glomerulosa, but also in the zona fasciculata in the adult human adrenal (Figure 1). PKD was also expressed in near-term fetal bovine adrenals in areas consistent with localization in zonae glomerulosa and fasiculata; however, the adrenal cortex of the near-term fetal adrenal may not be fully differentiated and the cells stained may be progenitor cells that express phenotypic characteristics of the fully differentiated zones.
Figure 1. PKD distribution in human and bovine adrenal glands.

(A) PKD distribution was determined in the positive control, human tonsillar tissue (a) and in human [(b) through (d)] and bovine [(e) through (d)] adrenal gland as described in Methods. PKD is present both in the zona glomerulosa and zona fasciculata regions. Sections incubated in the absence of primary antibody served as negative controls [(b) and (e)]. Panels (c) and (f) show PKD immunoreactivity, noted as the brown staining in these tissue sections, photographed at a 10x magnification and panels (d) and (g) show a 40x magnification. (B) The intensity of PKD staining in different regions of human adrenal gland sections was assessed as described in the Methods and presented as the means ± SEM of the values obtained in three glands.
3.2. AngII-induced PKD activation and aldosterone production is mediated, in part, by PKC
Several reports have demonstrated a role for PKC in the expression of different steroidogenic enzymes (Manna, et al. 2009, 2011; Romero et al. 2006). In addition, we have also demonstrated that PMA not only time- and dose-dependently promotes PKD activation, but also acute aldosterone secretion (Shapiro et al. 2010). Therefore, we tested whether inhibition of AngII-induced PKC activation decreased PKD phosphorylation/activation and acute aldosterone production in primary bovine adrenal glomerulosa cells. Primary bovine adrenal glomerulosa cells were treated with or without AngII (10 nM) in the presence or absence of the pan-PKC inhibitor Ro 31-8220 (3 μM) for 30 minutes. Cell lysates were analyzed by western blotting for PKD autophosphorylation, as measured by increased immunoreactivity of phosphoserine 910, a marker for PKD activation (Matthews et al. 1999). The supernatants were assayed for aldosterone production as previously described (Betancourt-Calle, et al. 1999). The PKC inhibitor, Ro 31-8220 alone had no significant effect on basal PKD autophosphorylation but reduced AngII-induced PKD activation (Figure 2A and B), suggesting that PKC modulates PKD activity in primary bovine adrenal glomerulosa cells. Ro 31-8220 also inhibited AngII-stimulated aldosterone production (Figure 2C). No inhibition by Ro 31-8220 was observed with 22(R)-hydroxycholesterol-mediated aldosterone production (Figure 2D). 22R-hydroxycholesterol is a synthetic cholesterol analog that readily permeates into the mitochondria in the absence of steroidogenic acute regulatory (StAR) protein (Pezzi, et al. 1996), thus bypassing signaling mechanisms, and can be used to determine the non-specific effects of inhibitors. Thus, these results indicated that Ro 31-8220 exhibited neither cytotoxicity to the cells nor non-specific effects on steroidogenic enzyme activities.
Figure 2. The PKC inhibitor Ro 31-8220 inhibited AngII-induced PKD activation and aldosterone secretion.

Primary cultures of bovine adrenal glomerulosa cells were pretreated for 30 minutes with 3 μM Ro 31-8220 prior to a 30-minute stimulation with or without 10 nM AngII. Panel (A) illustrates a representative blot. Note that the lower band in this blot appears to be non-specific, as it is only observed with certain lot numbers of the polyclonal antibody. (B) Band intensities from multiple experiments were quantified and normalized to actin. (C) Supernatants from the PKD activation experiments were assayed for aldosterone content. Values are expressed relative to the maximal response (in the presence of AngII) and represent means ± SEM from three experiments performed in duplicate; ***p<0.001 vs. the control, †p<0.05 vs. AngII. (D) Cells were pretreated for 30 minutes with 3 μM Ro 31-8220 prior to a 30-minute incubation with or without 25 μM 22(R)-hydroxycholesterol after which supernatants were assayed for aldosterone content. Values represent the means ± SEM of three separate experiments performed in duplicate.
3.3. Tyrosine phosphorylation of PKD is increased by AngII
Results from Toker’s group have demonstrated that phosphorylation by a Src/Abl tyrosine kinase pathway at tyrosine 463 is important for PKD activation in HeLa cells (Storz et al. 2003). Additionally, work from our laboratory has demonstrated that Src family kinase pathway-mediated phosphorylation at the tyrosine 463 residue is involved in ultraviolet light-induced PKD activation in keratinocytes (Arun, et al. 2011). To determine whether AngII increased tyrosine phosphorylation of PKD, glomerulosa cells were treated with or without 10nM AngII for 30 minutes, immunoprecipitated with a PKD antibody, and the immunoprecipitates analyzed by western blotting with an anti-phosphotyrosine antibody. Tyrosine phosphorylation of PKD was upregulated in AngII-treated cells (Figure 3A and B), confirming our hypothesis that PKD is tyrosine phosphorylated in primary bovine AG cells in response to AngII treatment. Thus, we next sought to determine if tyrosine-phosphorylating Src family kinases mediated this AngII-stimulated PKD activation and subsequent aldosterone production.
Figure 3. AngII increased tyrosine phosphorylation of PKD.

Primary cultures of bovine adrenal glomerulosa cells were stimulated with or without 10 nM AngII and immunoprecipitated with total PKD antibody (Santa Cruz). Immunoblot analysis was performed using an antibody recognizing phosphotyrosine (Millipore). Panel (A) illustrates a representative blot. Immunoblot analysis with total PKD antibody was performed on one-tenth of the immunoprecipitated protein, which served as loading control (input). Panel (B) illustrates band intensities from multiple experiments quantified and normalized to the loading control and are expressed as fold over control values. Values represent means ± SEM from four experiments performed in duplicate; *p<0.05 vs. control. Note that similar results were observed when lysates were immunoprecipitated with anti-phosphotyrosine antibody and the immunoprecipitate probed with anti-PKD antibody (data not shown).
3.4. Src family kinases mediate AngII-induced PKD activation and aldosterone production
AngII has been shown to activate Src family kinases in vascular smooth muscle cells (Ushio-Fukai, et al. 1999) as well as in the adrenocortical cell line NCI-H295R cells (Sirianni et al. 2001). In addition, Src/Abl family kinases induce PKD activation by phosphorylating the enzyme at tyrosine 463 (tyrosine 469 in mouse), as has been demonstrated in several cell types including Swiss 3T3 fibroblasts and HeLa cells (Storz et al. 2004; Waldron et al. 2004). Therefore, we sought to determine whether inhibition of the Src/Abl pathway, using the selective Src family kinase inhibitors Src-1 and PP2, attenuates AngII-stimulated PKD activation and aldosterone production. Primary bovine adrenal glomerulosa cells were pretreated with or without 10 μM PP2, a Src family kinase inhibitor, followed by stimulation with or without 10 nM AngII. PP2 inhibited AngII-elicited PKD activation (Figure 4A and B) by about 30% and aldosterone production by approximately 25% (Figure 4C). To rule out non-specific effects of the inhibitor, we incubated the cells with 25 μM 22(R)-hydroxycholesterol and observed that the resulting aldosterone production was unaffected by 10 μM PP2 (Figure 4D). Additionally, another Src family kinase inhibitor, Src-1, was used to verify the role of Src family kinases in AngII-elicited PKD activation and aldosterone production. Cells were pretreated with 0.75 μM Src-1, and then incubated in the presence or absence of 10 nM AngII. Src-1 also attenuated AngII-induced PKD activation (Figure 5A and B) and blunted AngII-stimulated aldosterone production by approximately 70% (Figure 5C). To determine if the concentration of Src-1 used exerted non-specific cytotoxic effects on the cells, primary bovine adrenal glomerulosa cells were incubated with 25 μM 22R-hydroxycholesterol in the presence of the inhibitor. Src-1 had no effect on 22(R)-hydroxycholesterol-mediated aldosterone secretion (Figure 5D).
Figure 4. PP2 inhibited AngII-induced PKD activation and aldosterone secretion.

Primary cultures of bovine adrenal glomerulosa cells were pretreated for 30 minutes with 10 μM PP2 prior to a 30-minute stimulation with or without 10 nM AngII. Panel (A) illustrates a representative blot. (B) Band intensities from multiple experiments were quantified and normalized to actin. (C) Supernatants from the PKD activation experiments were assayed for aldosterone content. Values represent means ± SEM from four experiments performed in duplicate and are expressed relative to the maximal response (in the presence of AngII); **p<0.01 vs. the control, ***p<0.001 vs. the control, †p<0.05 vs. AngII. (D) Cells were pretreated for 30 minutes with 10 μM PP2 prior to a 30-minute incubation with or without 25 μM 22(R)-hydroxycholesterol, after which supernatants were assayed for aldosterone content. Values represent the means ± SEM of four separate experiments performed in duplicate.
Figure 5. Src-1 decreased AngII-mediated PKD activation and aldosterone secretion.

Primary cultures of bovine adrenal glomerulosa cells were pretreated for 30 minutes with 0.75 μM Src-1 prior to a 30-minute stimulation with or without 10 nM AngII. Panel (A) illustrates a representative blot. (B) Band intensities from multiple experiments were quantified and normalized to actin. (C) Supernatants from the PKD activation experiments were assayed for aldosterone content. Values are expressed relative to the maximal response (in the presence of AngII) and represent means ± SEM from three experiments performed in duplicate; *p<0.05 vs. the control, ***p<0.001 vs. the control, ††p<0.01 vs. AngII. (D) Cells were pretreated for 30 minutes with 0.75 μM Src-1 prior to a 30-minute incubation with or without 25 μM 22(R)-hydroxycholesterol after which supernatants were assayed for aldosterone content. Values represent the means ± SEM of four separate experiments performed in duplicate.
3.5. Overexpression of the tyrosine-463-to-phenylalanine PKD mutant inhibited AngII-induced aldosterone production
Since tyrosine 463 is critical for Src family kinase-mediated PKD activation in other cell types, we anticipated that mutation of this residue would inhibit the activation of PKD by AngII and decrease AngII-induced aldosterone production. Primary bovine adrenal glomerulosa cells were mock-infected, or infected with empty vector (pAdtrackCMV) or the tyrosine-463-tophenylalanine PKD mutant for 4 hours, after which time the media was replaced with serum-free media for an additional 16-20 hours and the cells stimulated with or without AngII. Western blot analysis of cell lysates confirmed overexpression of the PKD construct and demonstrated expression of GFP as a marker of adenoviral infection (Figure 6A). Supernatants were collected and assayed for aldosterone content. Overexpression of the tyrosine-463-to-phenlyalanine PKD mutant decreased AngII-stimulated aldosterone production (Figure 6B). Our results demonstrate that phosphorylation of tyrosine 463 residue of PKD was important for PKD activation, suggesting the involvement of Src family tyrosine kinases in AngII-mediated acute aldosterone production. To determine if the concentration of the tyrosine-463-to-phenylalanine PKD adenovirus used exerted non-specific cytotoxic effects on the cells, mutant PKD-infected primary bovine adrenal glomerulosa cells were incubated with 22(R)-hydroxycholesterol. The tyrosine-463-to-phenylalanine PKD adenovirus did not inhibit 22(R)-hydroxycholesterol-mediated aldosterone secretion compared to vector-infected cells (Figure 7).
Figure 6. The tyrosine-463-to-phenylalanine PKD mutant decreased aldosterone production.
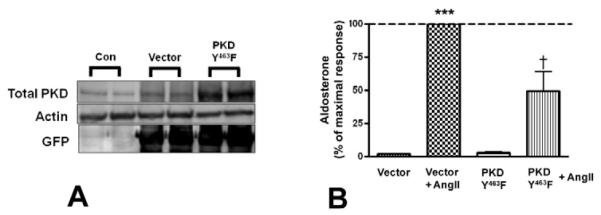
Primary cultures of bovine adrenal glomerulosa cells were infected with pAdtrackCMV (empty vector), or the tyrosine 463-to-phenylalanine PKD mutant (PKDY463F) for 4 hours, and media was replaced with serum-free media for an additional 16-20 hours before treatment with or without 10nM AngII. Panel (A) illustrates a representative experiment showing PKDY463F (and GFP) overexpression in adenovirus-infected cells 20-24 hours post-infection. (B) Supernatants from the PKD overexpression experiments were assayed for aldosterone content. Values represent means ± SEM from three experiments performed in duplicate and are expressed relative to the maximal response (in the presence of AngII); *p<0.05 vs. the control, ***p<0.001 vs. the control (vector), †p<0.05 vs. vector + AngII.
Figure 7. The tyrosine-463-to-phenylalanine PKD mutant did not inhibit 22(R)-hydroxycholesterol-mediated aldosterone production.
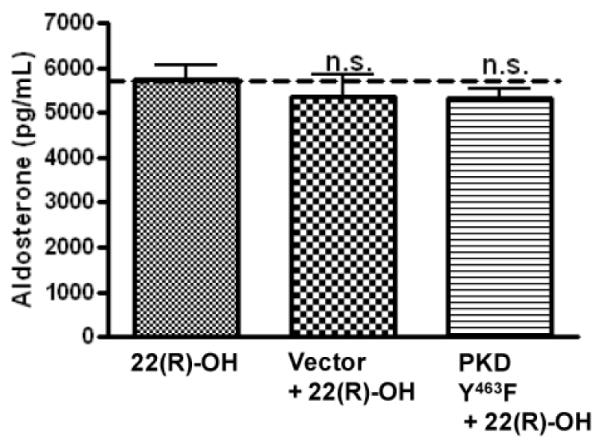
Primary cultures of bovine adrenal glomerulosa cells were incubated for 4 hours with adenovirus expressing pAdtrackCMV (empty vector) or tyrosine-463-to-phenylalanine PKD (PKDY463F) and media was replaced with serum-free media for an additional 16-20 hours before treatment with or without 25 μM 22(R)-hydroxycholesterol for one hour and assay of aldosterone content in the media. 22(R)-Hydroxycholesterol-mediated aldosterone secretory rates were not stat istically different. Data represent means ± SEM from three experiments performed in duplicate.
3.6. Inhibition of both PKC and Src family kinases additively attenuated AngII-induced PKD activation and aldosterone production
To further understand the regulation of AngII-induced PKD activation and aldosterone secretion by PKC and Src family kinase pathways, we sought to determine if the combination of both the PKC inhibitor, Ro 31-8220 (3 μM), and the Src family kinase inhibitor, PP2 (10 μM), would enhance the inhibitory response observed with either inhibitor alone. Indeed, Ro 31-8220 and PP2 had an additive inhibitory effect on AngII-induced PKD activation and aldosterone production (Figure 8A-C), and did not elicit any cytotoxicity (Figure 8D), suggesting the importance of both PKC and Src family kinases to AngII-induced PKD activation and aldosterone production.
Figure 8. Ro 31-8820 and PP2 additively decreased AngII-mediated PKD activation and aldosterone secretion.

Primary cultures of bovine adrenal glomerulosa cells were pretreated for 30 minutes with 3 μM Ro 31-8220, 10 μM PP2, or a combination of Ro 31-8220 and PP2 prior to a 30-minute stimulation with or without 10 nM AngII. Panel (A) illustrates a representative blot. (B) Band intensities from multiple experiments were quantified and normalized to actin. (C) Supernatants were assayed for aldosterone content. Values represent means ± SEM from six experiments performed in duplicate and are expressed relative to the maximal response (in the presence of AngII); *p<0.05 vs. the control, ***p<0.001 vs. the control, †p<0.05 vs. AngII, †††p<0.001 vs. AngII §§p<0.01 vs. PP2 + AngII, #p<0.05 vs. Ro 31-8220 + AngII. (D) Cells were pretreated for 30 minutes with 3 μM Ro 31-8220 and 10 μM PP2 prior to a 30-minute incubation with or without 25 μM 22(R)-hydroxycholesterol, after which supernatants were assayed for aldosterone content. Values represent the means ± SEM of three separate experiments performed in duplicate.
4. DISCUSSION
Several studies have demonstrated that PKD, a novel diacyglycerol/phorbol ester-responsive enzyme, is regulated by PKC-mediated transphosphorylation, and that this regulation is important for its role in modulating several cellular processes (Abedi, et al. 1998; Iglesias, et al. 1998; Rozengurt, et al. 2005; Waldron et al. 2004; Zugaza, et al. 1996). PKD possesses a pleckstrin homology domain that regulates its activity, as well as a highly hydrophobic stretch of amino acids in its N-terminal region (Lint, et al. 2002; Storz et al. 2003; Van Lint, et al. 2002). PKD has been implicated in the regulation of a variety of cellular functions, including signal transduction, Golgi trafficking, protein transport, cell survival, migration, differentiation, and proliferation (Arun et al. 2011; Guha, et al. 2010; Haworth, et al. 1999; Jamora, et al. 1999). We have recently demonstrated a role for PKD in AngII-induced acute (within 30 minutes) aldosterone production in primary bovine adrenal glomerulosa cells (Shapiro et al. 2010). Overexpression of the constitutively active serine 738/742-to-glutamate PKD mutant increased acute AngII-elicited aldosterone production, whereas the effect of AngII on aldosterone production was inhibited by overexpression of a dominant-negative serine-toalanine PKD mutant in which serines 738 and 742 were mutated to alanines, and thus could not be phosphorylated by PKC (Shapiro et al. 2010). Additionally, others have reported that PKCε-mediated PKD transphosphorylation in response to AngII is involved in AngII-induced chronic (within 24 hours) aldosterone production (Romero et al. 2006). In this study, overexpression of PKCε increased PKD activation as well as CYP11B2 mRNA expression. Thus, data in the literature support a role for PKC and PKD in aldosterone production in glomerulosa cells.
The ability of PKC/PKD to modulate steroidogenesis has also been demonstrated in MA-10 mouse Leydig tumor cells, in which the PKC-stimulating diacylglycerol analog PMA increases the phosphorylation of (i.e., activated) novel PKCs as well as PKD. In this report an increase in the levels of steroidogenic acute regulatory protein and progesterone synthesis was also observed (Manna et al. 2011). The StAR protein shuttles cholesterol from the outer to the inner mitochondrial membrane and is the rate-limiting step in steroid production. StAR protein expression and phosphorylation regulate its cholesterol-transporting activity (Clark, et al. 1994; Stocco and Clark 1993, 1996). Our recent studies also support a role for AngII-activated PKD-mediated StAR expression, induced through activation of transcription factors of the activating transcription factor/cAMP response element binding protein family, in regulating aldosterone secretion (Olala, et al. 2014). Nevertheless, the role of PKC/PKD signaling in aldosterone production remains controversial. Rasmussen and colleagues have previously shown a synergistic effect of the PKC/PKD-activating PMA and agents that increase calcium to trigger steroidogenesis (Barrett, et al. 1989; Rasmussen, et al. 1995). As discussed above, chronic AngII-elicited aldosterone production has also been shown to be mediated by PKD in the NCIH295R cells (Chang, et al. 2007; Romero et al. 2006). On the other hand, it has been reported that aldosterone production is enhanced in diacylglycerol-stimulated rat glomerulosa cells pre-treated with the non-selective PKC/PKD inhibitor staurosporine (Hajnoczky, et al. 1992), suggesting an aldosterone inhibitory role for this pathway. Likewise, another study has shown that CYP11B2 promoter activity is increased in hamster adrenal glomerulosa cells treated with the selective PKC inhibitor bisindolymaleimide I as well as the selective PKC/PKD inhibitor Gödecke 6976 (LeHoux, et al. 2001). These authors also showed that overexpression of some isoforms of PKC inhibited CYP11B2 promoter activity (Lehoux and Lefebvre 2007). Thus, additional studies must be performed to fully understand the mechanism through which PKC and PKD modulate aldosterone production. However, in our hands, AngII-mediated PKD activation increases aldosterone production, as we have demonstrated here and elsewhere (Shapiro et al. 2010), using adenoviral-mediated downregulation and upregulation of PKD activity as well as selective PKC inhibitors (Figure 2).
The PKC signaling pathway has also been recently implicated in regulating adrenocortical zonation (Hofland, et al. 2013). These authors demonstrated that in human adrenocortical carcinoma and primary adrenocortical cells PMA and AngII-induced PKC signaling results in increased activin A expression and levels, thereby attenuating CYP17A1 expression and function. These authors also demonstrated, in contrast, that ACTH- or forskolin-activated PKA increases inhibin expression. PKA signaling also blocks the downregulation of CYP17A1 expression, presumably because inhibin inhibits activin A action. Indeed, CYP17A1 is differentially expressed in the adrenal, with more expression detected in the zonae fasciculata and reticularis (Briere, et al. 1997). P450c17 catalyzes two distinct steroid biosynthetic activities, 17alpha-hydroxylase and 17,20-lyase activities (leading to cortisol synthesis in the fasciculata and androgen production in the reticularis) (Miller 2008). Thus, PKC may help to promote aldosterone synthesis in the glomerulosa by decreasing the expression and activity of CYP17A1, which would otherwise direct steroidogenesis away from aldosterone production and towards the synthesis of cortisol and adrenal androgens.
We were also interested in determining the role of Src family kinases in AngII-mediated PKD activation and aldosterone production, since Toker and colleagues have demonstrated that Src/Abl can activate PKD in HeLa cells (Storz et al. 2003). These authors reported that PKD is tyrosine-phosphorylated within the PH domain at tyrosine 463 (tyrosine 469 in mouse) via a pathway consisting of the Src/Abl tyrosine kinases, leading to activation in response to pervanadate stimulation and oxidative stress. Studies in the adrenocortical cell line NCI-H295R cells also demonstrated the involvement of the Src family kinases in aldosterone production (Sirianni et al. 2001). Here, we describe for the first time that Src family kinases activate PKD in primary bovine adrenal glomerulosa cells to underlie acute aldosterone production. This involvement was demonstrated using the Src family kinase inhibitors, PP2 and Src-1 to inhibit AngII-induced PKD activation (Figures 4 and 5), as well as the tyrosine-463-to-phenylalanine PKD mutant that cannot be phosphorylated by the Src family kinase cascade (Figure 7). Importantly, all of these manipulations inhibited AngII-stimulated acute aldosterone secretion in bovine adrenal glomerulosa cells (Figures 4-7). Given the studies by Rainey and colleagues suggesting that Src family kinases inhibit CYP17A1 expression (Sirianni et al. 2001) and the work by Hofland et al. (Hofland et al. 2013) demonstrating similar effects for PKC, we would hypothesize that AngII-mediated PKD activation, through Src family kinases and PKC, may regulate the activin-inhibin signaling cascade, which appears to play a role in AngII-mediated adrenal zonation (Hofland et al. 2013).
In summary, our results demonstrate that PKD is activated through two mechanisms involving PKC and Src family kinases to mediate acute AngII-induced aldosterone production. Thus, PKD, or its upstream activators or downstream effectors, may be potential pharmaceutical targets for the development of drugs to decrease the production of aldosterone, which is implicated in the development or exacerbation of several cardiovascular diseases, such as congestive heart failure. Clearly, additional studies are necessary to fully understand the entire pathway regulating aldosterone production in the adrenal gland as well as the role and mechanisms by which this steroid hormone contributes to cardiovascular diseases.
Highlights.
Angiotensin II activates protein kinase D via protein kinase C in glomerulosa cells
Angiotensin II activates protein kinase D via Src family kinases in glomerulosa cells
Protein kinase D activation via these two pathways mediates aldosterone secretion
ACKNOWLEDGEMENTS
We would like to acknowledge the excellent technical assistance of Mr. Peter Parker and Ms. Mariya V. George for preparation of the glomerulosa cells. This project was supported in part by award #HL70046 from the National Institutes of Health/National Heart, Lung and Blood Institute, a Grant-in-Aid award #0350166N from the American Heart Association and VA Merit Award #I01BX001344. WBB is supported by a VA Research Career Scientist Award.
Abbreviations
- AngII
angiotensin II
- DAG
diacylglycerol
- PKD
protein kinase D
- ZG
zona glomerulosa
- PKC
protein kinase C
- KRB+
Bicarbonate-buffered Kreb’s Ringer containing 2.5 mM sodium acetate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abedi H, Rozengurt E, Zachary I. Rapid activation of the novel serine/threonine protein kinase, protein kinase D by phorbol esters, angiotensin II and PDGF-BB in vascular smooth muscle cells. FEBS Lett. 1998;427:209–212. doi: 10.1016/s0014-5793(98)00427-x. [DOI] [PubMed] [Google Scholar]
- Arun SN, Kaddour-Djebbar I, Shapiro BA, Bollag WB. Ultraviolet B irradiation and activation of protein kinase D in primary mouse epidermal keratinocytes. Oncogene. 2011;30:1586–1596. doi: 10.1038/onc.2010.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett PQ, Bollag WB, Isales CM, McCarthy RT, Rasmussen H. Role of calcium in angiotensin II-mediated aldosterone secretion. Endocr Rev. 1989;10:1–22. doi: 10.1210/edrv-10-4-496. [DOI] [PubMed] [Google Scholar]
- Betancourt-Calle S, Bollag WB, Jung EM, Calle RA, Rasmussen H. Effects of angiotensin II and adrenocorticotropic hormone on myristoylated alanine-rich C-kinase substrate phosphorylation in glomerulosa cells. Mol Cell Endocrinol. 1999;154:1–9. doi: 10.1016/s0303-7207(99)00111-2. [DOI] [PubMed] [Google Scholar]
- Bollag WB, Dodd ME, Shapiro BA. Protein kinase D and keratinocyte proliferation. Drug News Perspect. 2004;17:117–126. doi: 10.1358/dnp.2004.17.2.829045. [DOI] [PubMed] [Google Scholar]
- Briere N, Martel D, Cloutier M, LeHoux JG. Immunolocalization and biochemical determination of cytochrome P450C17 in adrenals of hamsters treated with ACTH. J Histochem Cytochem. 1997;45:1409–1416. doi: 10.1177/002215549704501009. [DOI] [PubMed] [Google Scholar]
- Chang HW, Chu TS, Huang HY, Chueh SC, Wu VC, Chen YM, Hsieh BS, Wu KD. Down-regulation of D2 dopamine receptor and increased protein kinase Cmu phosphorylation in aldosterone-producing adenoma play roles in aldosterone overproduction. J Clin Endocrinol Metab. 2007;92:1863–1870. doi: 10.1210/jc.2006-2338. [DOI] [PubMed] [Google Scholar]
- Clark BJ, Wells J, King SR, Stocco DM. The purification, cloning, and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR) J Biol Chem. 1994;269:28314–28322. [PubMed] [Google Scholar]
- Funder J, Carey R, Fardella C, Gomez-Sanchez C, Mantero F, Stowasser M, Young W, Montori VM. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an Endocrine Society clinical practice guideline. Eur J Endocrinol. 2009 doi: 10.1530/EJE-09-0870. [DOI] [PubMed] [Google Scholar]
- Guha S, Tanasanvimon S, Sinnett-Smith J, Rozengurt E. Role of protein kinase D signaling in pancreatic cancer. Biochem. Pharmacol. 2010;80:1946–1954. doi: 10.1016/j.bcp.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Varnai P, Buday L, Farago A, Spat A. The role of protein kinase-C in control of aldosterone production by rat adrenal glomerulosa cells: activation of protein kinase-C by stimulation with potassium. Endocrinology. 1992;130:2230–2236. doi: 10.1210/endo.130.4.1547736. [DOI] [PubMed] [Google Scholar]
- Hanks SK. Genomic analysis of the eukaryotic protein kinase superfamily: a perspective. Genome Biol. 2003;4:111. doi: 10.1186/gb-2003-4-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth RS, Sinnett-Smith J, Rozengurt E, Avkiran M. Protein kinase D inhibits plasma membrane Na+/H+ exchanger activity. Am. J. Physiol. 1999;277:C1202–C1209. doi: 10.1152/ajpcell.1999.277.6.C1202. [DOI] [PubMed] [Google Scholar]
- Hofland J, Steenbergen J, Hofland LJ, van Koetsveld PM, Eijken M, van Nederveen FH, Kazemier G, de Herder WW, Feelders RA, de Jong FH. Protein kinase C-induced activin A switches adrenocortical steroidogenesis to aldosterone by suppressing CYP17A1 expression. Am J Physiol Endocrinol Metab. 2013;305:E736–744. doi: 10.1152/ajpendo.00034.2013. [DOI] [PubMed] [Google Scholar]
- Iglesias T, Waldron RT, Rozengurt E. Identification of in vivo phosphorylation sites required for protein kinase D activation. J. Biol. Chem. 1998;273:27662–27667. doi: 10.1074/jbc.273.42.27662. [DOI] [PubMed] [Google Scholar]
- Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, Malhotra V. Gbg-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98:59–68. doi: 10.1016/S0092-8674(00)80606-6. [DOI] [PubMed] [Google Scholar]
- Kapas S, Purbrick A, Hinson JP. Role of tyrosine kinase and protein kinase C in the steroidogenic actions of angiotensin II, a-melanocyte-stimulating hormone and corticotropin in the rat adrenal cortex. Biochem J. 1995;305:433–438. doi: 10.1042/bj3050433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeHoux JG, Dupuis G, Lefebvre A. Control of CYP11B2 gene expression through differential regulation of its promoter by atypical and conventional protein kinase C isoforms. J Biol Chem. 2001;276:8021–8028. doi: 10.1074/jbc.M009495200. [DOI] [PubMed] [Google Scholar]
- Lehoux JG, Lefebvre A. Angiotensin II activates p44/42 MAP kinase partly through PKCepsilon in H295R cells. Mol Cell Endocrinol. 2007;265-266:121–125. doi: 10.1016/j.mce.2006.12.027. [DOI] [PubMed] [Google Scholar]
- Lint JV, Rykx A, Vantus T, Vandenheede JR. Getting to know protein kinase D. Int J Biochem Cell Biol. 2002;34:577–581. doi: 10.1016/s1357-2725(01)00163-7. [DOI] [PubMed] [Google Scholar]
- Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2:1236–1247. doi: 10.1038/nprot.2007.135. [DOI] [PubMed] [Google Scholar]
- Manna PR, Soh JW, Stocco DM. The Involvement of Specific PKC Isoenzymes in Phorbol Ester-Mediated Regulation of Steroidogenic Acute Regulatory Protein Expression and Steroid Synthesis in Mouse Leydig Cells. Endocrinology. 2009;152:313–325. doi: 10.1210/en.2010-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna PR, Soh JW, Stocco DM. The involvement of specific PKC isoenzymes in phorbol ester-mediated regulation of steroidogenic acute regulatory protein expression and steroid synthesis in mouse Leydig cells. Endocrinology. 2011;152:313–325. doi: 10.1210/en.2010-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem. 1999;274:26543–26549. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- Mill JG, Milanez Mda C, de Resende MM, Gomes Mda G, Leite CM. Spironolactone prevents cardiac collagen proliferation after myocardial infarction in rats. Clin Exp Pharmacol Physiol. 2003;30:739–744. doi: 10.1046/j.1440-1681.2003.03906.x. [DOI] [PubMed] [Google Scholar]
- Miller WL. Steroidogenic enzymes. Endocr Dev. 2008;13:1–18. doi: 10.1159/000134751. [DOI] [PubMed] [Google Scholar]
- Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005;45:1243–1248. doi: 10.1016/j.jacc.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Olala LO, Choudhary V, Johnson M, Bollag WB. Angiotensin II-Induced Protein Kinase D Activates the ATF/CREB Family of Transcription Factors and Promotes StAR mRNA Expression. Endocrinology. 2014 doi: 10.1210/en.2013-1485. en20131485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olala LO, Seremwe M, Tsai YY, Bollag WB. A role for phospholipase D in angiotensin II-induced protein kinase D activation in adrenal glomerulosa cell models. Mol Cell Endocrinol. 2013 doi: 10.1016/j.mce.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzi V, Clark BJ, Ando S, Stocco DM, Rainey WE. Role of calmodulin-dependent protein kinase II in the acute stimulation of aldosterone production. J Steroid Biochem Mol Biol. 1996;58:417–424. doi: 10.1016/0960-0760(96)00052-0. [DOI] [PubMed] [Google Scholar]
- Rainey WE, Bird IM, Mason JI. Angiotensin-II-directed glomerulosa cell function in fetal adrenal cells. J Steroid Biochem Mol Biol. 1992;43:847–854. doi: 10.1016/0960-0760(92)90311-6. [DOI] [PubMed] [Google Scholar]
- Rasmussen H, Isales CM, Calle R, Throckmorton D, Anderson M, Gasalla-Herraiz J, McCarthy R. Diacylglycerol production, Ca2+ influx, and protein kinase C activation in sustained cellular responses. Endocr. Rev. 1995;16:649–681. doi: 10.1210/edrv-16-5-649. [DOI] [PubMed] [Google Scholar]
- Rey O, Yuan J, Young SH, Rozengurt E. Protein kinase C nu/protein kinase D3 nuclear localization, catalytic activation, and intracellular redistribution in response to G protein-coupled receptor agonists. J Biol Chem. 2003;278:23773–23785. doi: 10.1074/jbc.M300226200. [DOI] [PubMed] [Google Scholar]
- Romero DG, Welsh BL, Gomez-Sanchez EP, Yanes LL, Rilli S, Gomez-Sanchez CE. Angiotensin II-mediated protein kinase D activation stimulates aldosterone and cortisol secretion in H295R human adrenocortical cells. Endocrinology. 2006;147:6046–6055. doi: 10.1210/en.2006-0794. [DOI] [PubMed] [Google Scholar]
- Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280:13205–13208. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- Shapiro BA, Olala L, Arun SN, Parker PM, George MV, Bollag WB. Angiotensin II-activated protein kinase D mediates acute aldosterone secretion. Mol Cell Endocrinol. 2010;317:99–105. doi: 10.1016/j.mce.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirianni R, Sirianni R, Carr BR, Pezzi V, Rainey WE. A role for src tyrosine kinase in regulating adrenal aldosterone production. J Mol Endocrinol. 2001;26:207–215. doi: 10.1677/jme.0.0260207. [DOI] [PubMed] [Google Scholar]
- Stocco DM, Clark BJ. The requirement of phosphorylation on a threonine residue in the acute regulation of steroidogenesis in MA-10 mouse leydig cells. J. Steroid Biochem. Mol. Biol. 1993;46:337–347. doi: 10.1016/0960-0760(93)90223-j. [DOI] [PubMed] [Google Scholar]
- Stocco DM, Clark BJ. Role of the steroidogenic acute regulatory protein (StAR) in steroidogenesis. Biochem. Pharmacol. 1996;51:197–205. doi: 10.1016/0006-2952(95)02093-4. [DOI] [PubMed] [Google Scholar]
- Storz P, Doppler H, Johannes FJ, Toker A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J Biol Chem. 2003;278:17969–17976. doi: 10.1074/jbc.M213224200. [DOI] [PubMed] [Google Scholar]
- Storz P, Döppler H, Toker A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol. Cell Biol. 2004;24:2614–2626. doi: 10.1128/MCB.24.7.2614-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturany S, Van Lint J, Muller F, Wilda M, Hameister H, Hocker M, Brey A, Gern U, Vandenheede J, Gress T, et al. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem. 2001;276:3310–3318. doi: 10.1074/jbc.M008719200. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Alexander RW, Akers M, Lyons PR, Lassegue B, Griendling KK. Angiotensin II receptor coupling to phospholipase D is mediated by the betagamma subunits of heterotrimeric G proteins in vascular smooth muscle cells. Mol Pharmacol. 1999;55:142–149. doi: 10.1124/mol.55.1.142. [DOI] [PubMed] [Google Scholar]
- Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: A target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc. Natl. Acad. Sci. USA. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint J, Rykx A, Vantus T, Vandenheede JR. Getting to know protein kinase D. Int. J. Biochem. Cell Biol. 2002;34:577–581. doi: 10.1016/s1357-2725(01)00163-7. [DOI] [PubMed] [Google Scholar]
- Waldron RT, Iglesias T, Rozengurt E. Phosphorylation-dependent protein kinase D activation. Electrophoresis. 1999;20:382–390. doi: 10.1002/(SICI)1522-2683(19990201)20:2<382::AID-ELPS382>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Waldron RT, Rey O, Zhukova E, Rozengurt E. Oxidative stress induces protein kinase C-mediated activation loop phosphorylation and nuclear redistribution of protein kinase D. J Biol Chem. 2004;279:27482–27493. doi: 10.1074/jbc.M402875200. [DOI] [PubMed] [Google Scholar]
- Waldron RT, Rozengurt E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J Biol Chem. 2003;278:154–163. doi: 10.1074/jbc.M208075200. [DOI] [PubMed] [Google Scholar]
- Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf) 2007;66:607–618. doi: 10.1111/j.1365-2265.2007.02775.x. [DOI] [PubMed] [Google Scholar]
- Yuan J, Bae D, Cantrell D, Nel AE, Rozengurt E. Protein kinase D is a downstream target of protein kinase Cq. Biochem. Biophys. Res. Comm. 2002;291:444–452. doi: 10.1006/bbrc.2002.6469. [DOI] [PubMed] [Google Scholar]
- Zugaza JL, Sinnett-Smith J, Van Lint J, Rozengurt E. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J. 1996;15:6220–6230. [PMC free article] [PubMed] [Google Scholar]
- Zugaza JL, Waldron RT, Sinnett-Smith J, Rozengurt E. Bombesin, vasopressin, endothelin, bradykinin, and platelet-derived growth factor rapidly activate protein kinase D through a protein kinase C-dependent signal transduction pathway. J Biol Chem. 1997;272:23952–23960. doi: 10.1074/jbc.272.38.23952. [DOI] [PubMed] [Google Scholar]