

Abstract
Guanidinium-rich scaffolds facilitate cellular translocation and delivery of bioactive cargos through biological barriers. Although impressive uptake has been demonstrated for nonoligomeric and nonpept(o)idic guanidinylated scaffolds in cell cultures and animal models, the fundamental understanding of these processes is lacking. Charge pairing and hydrogen bonding with cell surface counterparts have been proposed, but their exact role remains putative. The impact of the number and spatial relationships of the guanidinium groups on delivery and organelle/organ localization is yet to be established.
While it has been known for half a century that certain polybasic proteins can enhance the cellular uptake of biomolecules,1 the past two decades have seen tremendous progress in advancing the basic science, applications, and preclinical evaluation of such remarkable tools.2 The potential of protein transduction domains (PTD) was first realized using the homeodomain of antennapedia (Antp).3 This transcription factor was shown to mediate cellular uptake in numerous cell types.4 Its DNA-binding domain was demonstrated to be sufficient for mediating cell uptake5 and was named penetratin-1.6 In 1988, two groups independently reported that HIV Tat possesses unique cell uptake features.7 Conjugation of Tat fragments that included the RNA binding domain to large proteins was then shown to facilitate the cellular uptake of these proteins.8 An important step forward was the development of genetically engineered fusion proteins, which were reported to efficiently internalize and to exhibit the expected biological activity.9 A database search, inspired by the translocation properties of Tat, identified a number of membrane-permeable peptides that contain clustered arginine residues.10 Although numerous other naturally occurring and chimeric peptides exhibit effective translocation across cell membranes, efforts have focused on arginine-rich sequences.6,11 Further exploration of the impact of stereochemistry and composition on cell uptake has identified d-Tat and (Arg)9 as competent transporters.10 Additionally, significant cellular internalization was observed for branched arginine-rich oligomers.12 Taken together, the guanidinium moiety emerged as the responsible molecular feature, which has triggered the exploration of diverse per-guanidinylated scaffolds for cellular delivery. Before elaborating on diverse cellular transporters, we discuss the salient features of guanidine and its protonated form, guanidinium. We briefly highlight the fundamental entry pathways into mammalian cells and then elaborate on diverse delivery vehicles. We close with a short discussion of guanidinylating reagents.
Guanidine and Guanidinium
Guanidine, first isolated in 1861 by oxidizing guanine,13 is found in a wide variety of natural products, including the amino acid arginine.14 Although known for more than 150 years, the first solid-state structure was solved in 2007 by co-crystallization with 2-amino-4,6-dimethyl-1,3,5-triazine.15 Two years later, the structure of the free base was reported,16 and more recently, neutron diffraction studies accurately positioned the hydrogen atoms (Figure 1a).17 As a strong base (pKb ≈ 0.5), at physiological pH’s guanidine exists in its protonated form, the highly stabilized guanidinium cation.
Figure 1.
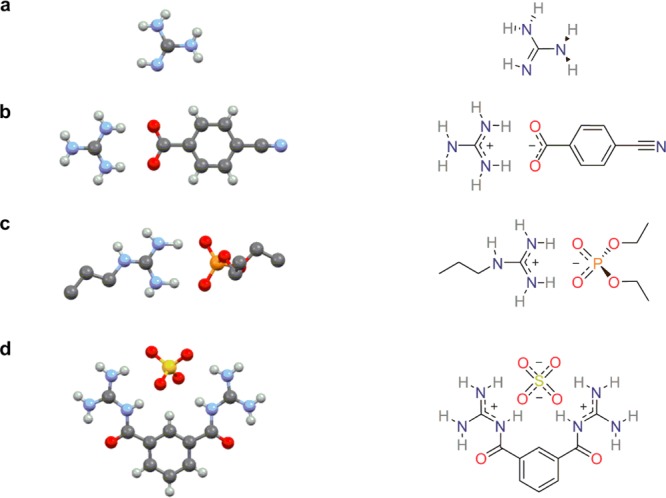
Crystal structures and 2D representations of (a) free base guanidine,16 (b) a guanidinium carboxylate salt,20 (c) a propylguanidinium phosphate salt,21 and (d) a sulfate salt of a synthetic bisguanidinium receptor.22
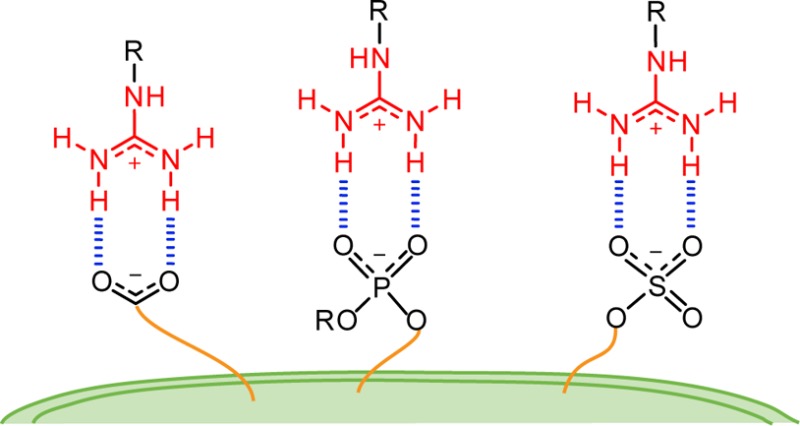
The Y-shaped guanidinium group is a highly symmetric planar functionality that can form two strong parallel hydrogen bonds with biologically relevant counterparts.18 Its geometry generates a more favorable hydrogen bond alignment compared to ammonium groups, which are also widely found in biomolecules. Additionally, binding can occur through both charge pairing and hydrogen bonding as the group maintains its protonated state over a wide range of pH. Moreover, unlike for the ammonium cations where the charge is localized (hard), the interaction with softer ions such as phosphates and sulfates is facilitated by delocalization of the positive charge in the guanidinium group (Figure 1).19
Comparing the binding energies of ammonium and guanidinium groups indicates that both have high affinities and selectivities for phosphates and arsenates over other anions, while higher binding constants are found for the guanidinium groups.23 Furthermore, while the formation of ammonium–phosphate complexes was found to be primarily entropy driven, favorable enthalpy and entropy changes were reported for the guanidinium-phosphate complexation.23 This distinct thermodynamic behavior was suggested to derive from differences in the solvation shell of the two groups.23 Isothermal titration calorimetry (ITC) measurements with a series of substituted bicyclic guanidiniums and different counterions assigned a decisive role to solvation in this enthalpy–entropy compensation and highlighted the significance of the coordinating ability of the counterion.24
Cellular Uptake: Mechanisms
Endocytosis, the energy-dependent vesicular uptake of extracellular substrates, has been established as the main mechanism for cellular uptake of nonviral vectors.25 The mechanisms of endocytosis have been extensively reviewed by Doherty and McMahon.26 Briefly, endocytosis pathways can be divided into clathrin-mediated endocytosis, the best characterized endocytic pathway;27 caveolin-mediated endocytosis;28 phagocytosis, typically restricted to specialized mammalian cells;29 and macropinocytosis, which refers to the formation of large endocytic vesicles.30 They differ in the composition of the vesicle coat (if any) and in the fate of the internalized particles.31 Although most receptors are internalized by clathrin-mediated endocytosis, other endocytic pathways are capable of selective receptor-mediated endocytosis events.32 After endocytosis, internalized cargo is trafficked into endosomes, from where it can either escape or be sorted back to the surface of the cell or into other compartments such as lysosomes for degradation.33 Nonendocytic delivery was initially suggested for the direct translocation of cationic peptides such as Tat across cell membranes.34 However, the actual pathway for their entry into cells has remained controversial.35 These possible pathways for cellular uptake are schematically depicted in Figure 2.
Figure 2.
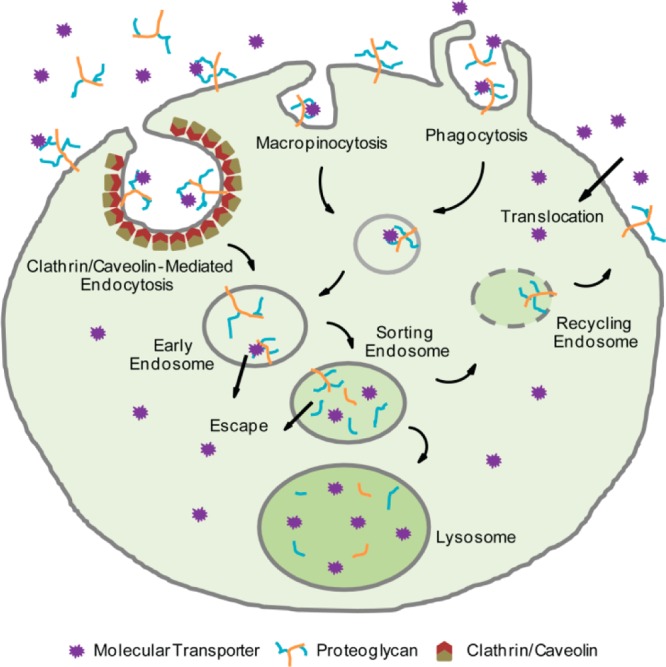
Potential mechanisms for cellular uptake including clathrin- and caveolin-mediated endocytosis, macropinocytosis, phagocytosis, and direct translocation across the plasma membrane.
Arginine-Rich Peptides: Tat as Inspiration for Synthetic Transporters
Following the discovery of HIV-Tat’s basic sequence (Tat49–57, RKKRRQRRR) as the module responsible for cellular entry,7a alanine scan indicated that the arginine residues are critical for cellular uptake and that the analogous (Arg9) displayed increased translocation efficiency.36 The nature of the counterion was also shown to play an important role in the translocation ability of oligo/polyarginines in vitro, with lipophilic anions altering the highly hydrophilic characteristic of the guanidinium-containing entities into lipophilic complexes, therefore facilitating the translocation through lipophilic membranes.37 The opposite effect is observed for hydrophilic anions, and while some amphiphilic counterparts were not shown to mediate such phase transfer, others could solubilize polyarginine (but not polylysine) in chloroform.37
In a similar context, Wender and co-workers investigated the importance of hydrogen bonding to membrane embedded constituents and correlated it with cellular uptake. They compared the uptake of Arg8 with that of octamers of monomethylated and asymmetrically dimethylated arginine. The results showed that increasing methylation decreased cellular uptake.38 This observation correlates with the ability of the octamers to form bidentate hydrogen bonds, further supported by molecular modeling of the possible isomers of the methylated guanidiniums and estimation of their energies.38
Given the ability of the guanidinium group to bind biologically abundant counterions and its involvement in translocation through mammalian cell membranes, researchers have focused on the design and development of guanidinium-containing oligomeric transporters, with the aim of delivering diverse cargo into mammalian cells. Wender and co-workers have recently reviewed oligomeric and polymeric guanidinium-rich molecular transporters where the guanidinium groups are linked to either peptidic or peptoidic backbones as well as to longer oligocarbamates and oligocarbonates.39 The use of guanidinylated dendritic molecular transporters in cell transfection, as reported by Goodman and others, has been presented by Gillies and co-workers.40 Moreover, the utility of cationic lipids for gene delivery has recently been discussed by Zhao and co-workers.41 This synopsis, therefore, focuses on the development of nonoligomeric nonpept(o)idic guanidinylated scaffolds designed and used to deliver low and high molecular weight cargo, from small drugs to quantum dots, across cellular membranes.
Guanidinoglycosides, Inositol, and Carbohydrate Scaffolds
Guanidinoglycosides are guanidinylated aminoglycoside antibiotics where all the ammonium groups are converted to guanidinium groups.42 BODIPY-tagged guanidinotobramycin and guanidinoneomycin were shown to translocate across the cellular membrane with considerably improved efficiency compared to their parent aminoglycosides (Figure 3).43 Furthermore, significantly higher translocation efficiency was observed for guanidinoneomycin (GNeo), containing six guanidinium moieties, compared to oligo-Arg peptide Arg9, suggesting that the semirigid and perhaps more globular organization of the guanidinium groups might play an important role in facilitating cellular uptake.43
Figure 3.
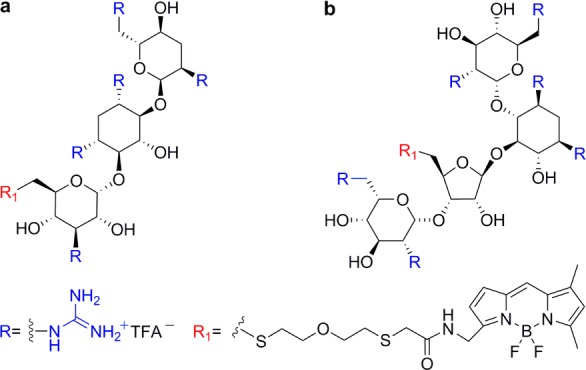
BODIPY-tagged (a) guanidinotobramycin and (b) guanidinoneomycin.43
The cellular binding and uptake of GNeo at nanomolar concentrations exclusively depends on cell surface heparan sulfate (HS) proteoglycans.44 The number of guanidinium groups and, to a lesser extent, their spatial distribution on the guanidinoglycoside core, significantly impact cellular uptake.45 Comparing the uptake of monomeric and dimeric guanidinoglycosides derived from tobramycin, paromomycin, and neomycin B containing a different number and 3D arrangement of guanidinium groups established a correlation between valency and uptake efficiency (Figure 4).45
Figure 4.
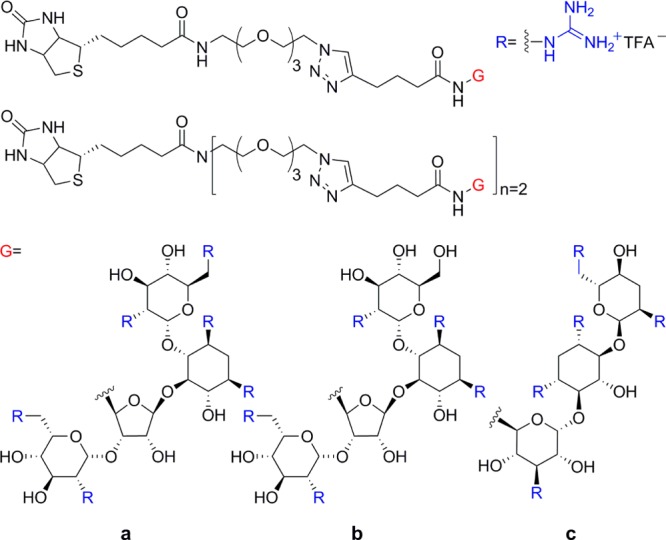
Monomeric and dimeric structures of (a) guanidinoneomycin, (b) guanidinoparomomycin, and (c) guanidinotobramycin.45
Specific mutant cell lines derived from Chinese hamster ovary (CHO) cells, differing in their expression of HS, were used to investigate the effect of sulfation patterns on the cellular recognition and uptake of guanidinoglycosides.45 HS-deficient cells showed very poor uptake (<5%, compared to wild type).45 Uptake of monomeric guanidinoglycoside constructs was reduced to <20% in undersulfated cell lines, when compared to wild-type cells.45 Unlike the monomeric carriers, the multivalent constructs were able to overcome lower sulfation levels and displayed higher uptake levels in such cell lines (between 50 and 75% compared to that observed in wild-type cells).45 These results identify the nature of cell surface HS as a key parameter affecting the cellular uptake and recognition of guanidinoglycosides, suggesting an additional variable to consider when evaluating the behavior of molecular transporters.
Guanidinoglycosides can translocate large bioactive molecules through cell membranes.46 When biotinylated GNeo is conjugated to streptavidin-coated quantum dots (QD525), approximately 90% of internalized nanoparticles colocalize with lysosomes after 3 h, suggesting that GNeo can deliver very high molecular weight cargo (>107 Da) to these organelles (Figure 5).46 To facilitate conjugation of diverse biomolecules, an N-hydroxysuccinimide activated ester of guanidinoneomycin was prepared (GNeo-NHS, Scheme 1).46 Two lysosomal enzymes, β-d-glucuronidase and α-l-iduronidase, were conjugated to GNeo without interfering with their enzymatic activity and delivered to patient cells lacking the corresponding lysosomal enzyme in sufficient amounts to restore normal turnover of glycosaminoglycans.46
Figure 5.
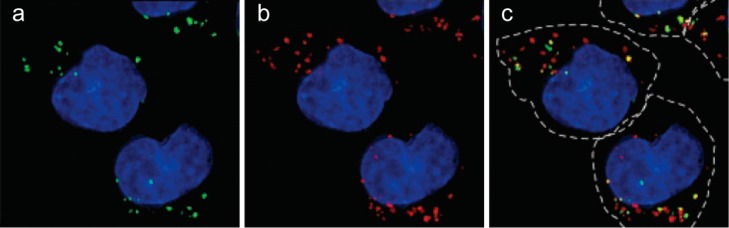
GNeo-QD525 conjugate colocalizes with lysosomes. Wild-type Chinese hamster ovary cells were incubated with 5 nmol/L GNeo-QD525 in growth medium for 30 min. After the cells were rinsed three times, fresh medium was added, and 2.5 h later, they were rinsed with Hank’s balanced salt solution and labeled with Hoechst dye and LysoTracker Red. Images were captured with a DeltaVision Restoration microscope system and were deconvolved to show the localization of (a) GNeo-QD525 and (b) lysosomes in a single Z-stack plane. The merged images from (a) and (b) are shown in (c) with the outline of cells (hatched line) drawn based on a phase contrast micrograph. Reprinted with permission from ref (46). Copyright 2010 Nature Publishing Group.
Scheme 1. Synthesis of a NHS Ester of Guanidinoneomycin46.

To shed light on the uptake mechanism of guanidinoglycosides, fluorescently tagged streptavidins [streptavidin–pycoerythrin-Cy5 (ST–PECy5), streptavidin–phycoerythrin (ST–PE), and streptavidin–Cy5 (ST–Cy5)] were used as model cargos. Evaluating the uptake and binding of streptavidin–GNeo conjugates by flow cytometry and cell-surface FRET analysis suggested that heparan sulfate proteoglycan aggregation is a pivotal step for endocytic translocation of guanidinoglycosides.47 This pathway can be altered by selective acylation of guanidinoneomycin-based transporters with long alkyl chains, which enhances the macromolecular cellular uptake with little or no heparan sulfate aggregation (Figure 6).48 These findings suggest an alternative and distinct pathway involving hydrophobic interactions impacting membrane curvature while assisting the uptake.
Figure 6.
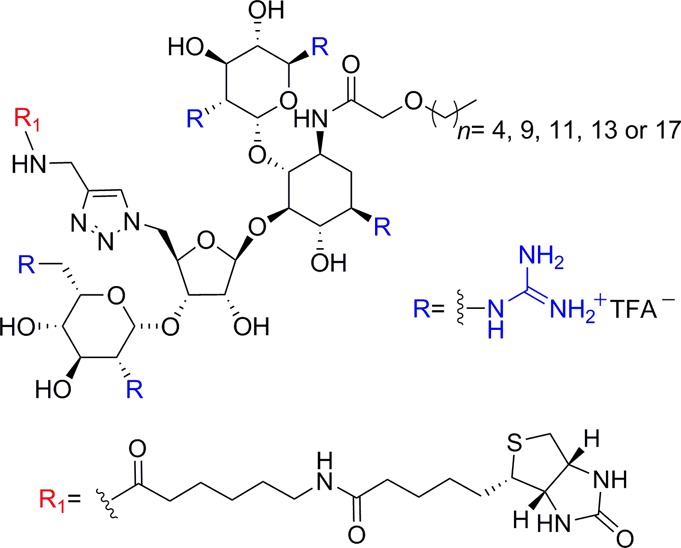
Alkyl-chain-modified guanidinoneomycin.48
It is worth noting that HS proteoglycans were also identified as cell surface receptors for Tat internalization.49 The ability of a carrier to bind to HS is not always sufficient, however, for efficient uptake, as suggested by the differences observed for arginine-rich cell penetrating peptides (CPPs) featuring d-amino acids compared to those with l-amino acids, both having comparable HS binding constants.50 The ability to cluster HS has been shown to contribute to the efficiency of endocytosis of CPPs, typically initiated through electrostatic interactions with cell-surface HS.51 The endocytic mechanism of internalization has been proposed to relate to the nature and distribution of proteoglycans expressed on the cell surface,52 and a recent report established structure–activity relationships for HS binding and uptake for a set of CPPs where stoichiometry was the decisive factor.53
Searching for naturally occurring scaffolds, Chung and co-workers focused on carbohydrates as polyfunctional cores for molecular transporters. The intracellular localization of myo- and scyllo-inositol dimers bearing eight guanidinium groups differed from that of the Tat and (Arg)8 peptides, suggesting a distinct clathrin-independent internalization pathway (Figure 7).54 Unlike Tat peptides, these compounds were found mainly along the heart, lung and brain tissues, displaying unique distribution both in vitro and in vivo. In addition, conjugation of doxorubicin to one of the transporters significantly increased drug uptake and its intracellular permeation in brain tissues.54
Figure 7.
Representative structures of dimeric (a) myo-inositol and (b) scyllo-inositol.54
Despite of the promising features of these dimeric inositol based transporters, Chung and co-workers explored a different scaffold, based on sorbitol, keeping the eight guanidinium units (Figure 8a,b).55 Similarly to the di-inositol transporters, the internalization mechanism was observed to be different from that of the Tat peptide. More significant was their high selectivity for mitochondria and higher distribution in the heart muscle and brain sections.55 Taking advantage of their ability to cross the blood–brain barrier (BBB), these octa-guanidinylated sorbitol-based molecular transporters were covalently linked to paclitaxel (Figure 8c). Good antitumor activity was observed in a mouse model of glioblastoma.56 Furthermore, a conjugate of 3′-azido-3′-deoxythymidine (AZT) showed effective cellular uptake in HeLa cells with preferred localization in mitochondria and nucleoli (Figure 8d).57 This conjugate also crossed the BBB.57 In a similar approach, 5-fluorouridine (5-FU) was covalently attached to the sorbitol-based molecular transporter through a succinate ester linker at position 5′ of the ribose ring (Figure 8e).58 Like the AZT derivative, 5-FU conjugates displayed good cellular uptake and mitochondrial localization. Although they showed different biodistribution in mouse tissue, these 5-FU conjugates crossed the BBB and showed more potent in vitro cytotoxicity than unconjugated 5-FU.58
Figure 8.
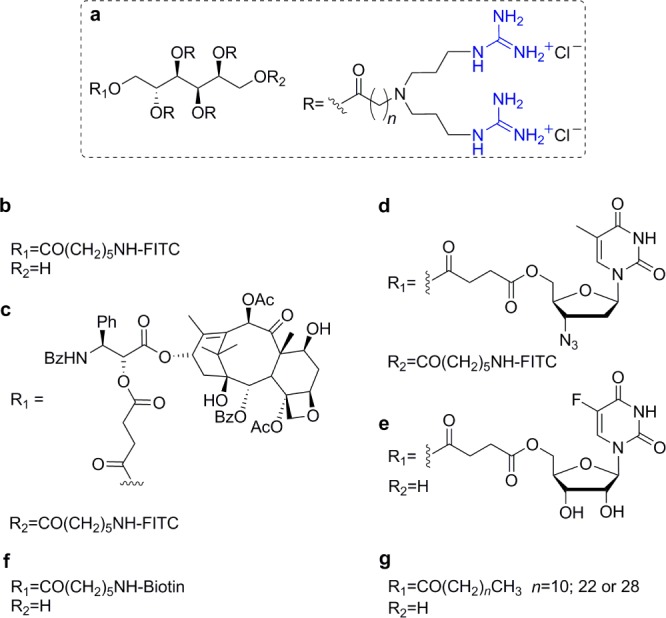
Sorbitol-based transporters: (a) general structure of the sorbitol scaffold, conjugated with (b) FITC,55 (c) paclitaxel,56 (d) AZT,57 (e) 5-FU,58 (f) biotin,59 and (g) fatty acids.60
Internalization of quantum dots (QD) into HeLa cells was facilitated using a biotinylated version of the sorbitol-based transporter bound to streptavidin–QD conjugates (Figure 8f).59 After long incubation times, these conjugates appeared mostly in the perinuclear region but did not enter the nucleus. Moreover, QD conjugates were observed to efficiently cross the BBB in mice when administered via tail vein.59 Finally, lipidated derivatives of the sorbitol-based transporters were evaluated for their ability to condense either DNA or siRNA and their applicability in nonviral gene delivery systems (Figure 8g).60 Compounds with a short lipid chain (C12) were proven particularly useful for nucleic acid condensation, whereas those with a long lipid chain (C30) were optimal for surface modification of nucleic acid containing lipid vesicles.60
Other scaffolds investigated by Chung and co-workers include the disaccharides lactose,61 sucrose,62 and trehalose (Figure 9),63 the monosaccharides glucose, mannose, allose, and galactose (Figure 10),64 and monomeric myo- and scyllo-inositols (Figure 11).65 Both the lactose- and sucrose-based transporters feature seven guanidinium groups tethered to the sugar through linkers of different lengths (Figure 9a,b). For both, the intracellular localization was influenced by the length, or lipophilicity, of the linker and in the case of sucrose scaffold also by the nature of the fluorescent dye attached to the transporter.61,62
Figure 9.
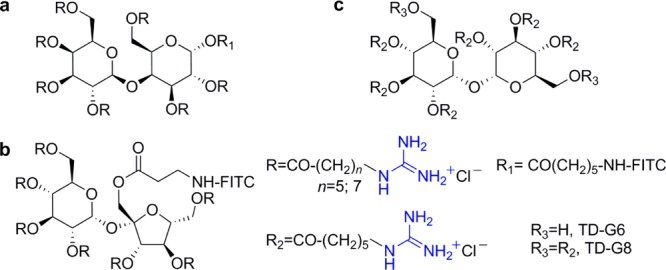
Representative structures of disaccharide-based transporters: (a) lactose,61 (b) sucrose,62 and (c) trehalose.63
Figure 10.
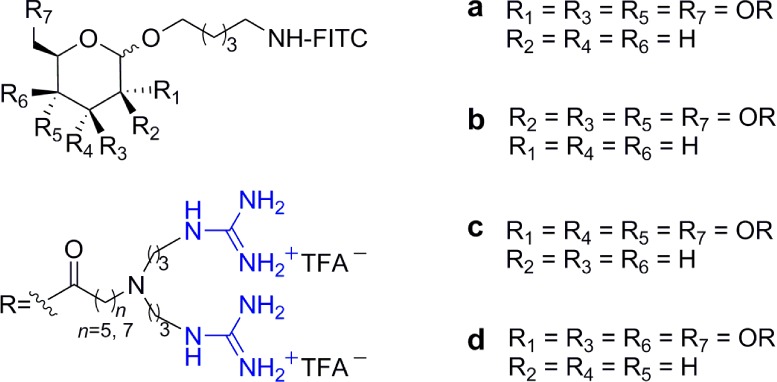
Representative structures of monosaccharide-based transporters: (a) glucose, (b) mannose, (c) allose, and (d) galactose.64
Figure 11.
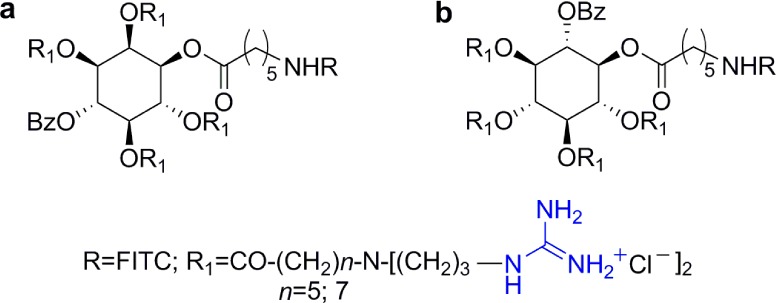
Representative structures of monomeric inositol-based transporters: (a) myo-inositol and (b) scyllo-inositol.65
Trehalose, a neuroprotective disaccharide with poor cellular uptake,66 was decorated with either six (TD-G6) or eight (TD-G8) guanidinium moieties and administered to Huntington disease model mice (Figure 9c).63 TD-G8 resulted to be more toxic than TD-G6, and the latter showed enhanced neuroprotective activity compared to trehalose itself. While all monosaccharide-based molecular transporters shown in Figure 10 displayed good permeability to brain tissues, the intracellular localization, particularly their mitochondrial affinity, was found to be related to their stereochemistry and to a lesser extent to the lipophilicity of the linker between the guanidinium moiety and the sugar scaffold.64
To better understand the correlation between stereochemistry and mitochondrial localization, a series of octa-guanidinylated molecular transporters based on two inositol stereoisomers, myo- and scyllo-inositol, was explored (Figure 11). Derivatives of myo-inositol were found to target the mitochondria, whereas the more symmetric scyllo-inositol derivatives did not show significant mitochondrial colocalization. In addition, while all the transporters showed good affinity for brain tissues, scyllo-inositol-based transporters, unlike their myo-inositol stereoisomers, were widely distributed in all organs.65 It seems that although the structures of the saccharide scaffolds are closely related, several parameters need to be tuned for optimal organellar selectivity and tissue distribution.
Miscellanea
As mentioned above, the use of high-order guanidinylated dendrimers was reviewed by Gillies et al.40 Related reports, include different platforms such as dendronized nanoparticles,67 “vivo-morpholinos”,68 and guanidinium dendron–carbon nanotubes (Figure 12).69 Jeong and co-workers encapsulated a hydrophobic peptide model drug into dendritic amine and guanidinium group-modified nanoparticles.67 In this system, up to four amine or guanidinium groups are connected to a hydrophobic stearyl tail through a short oligophenylalanine linker introduced to provide structural rigidity (Figure 12a).67 It was shown that the uptake efficiency increased with the number of the positively charged groups and that guanidinium-functionalized nanoparticles had better ability to cross membranes than the amino-functionalized ones. Moreover, no significant cytotoxicity was observed for the tetravalent carriers.67
Figure 12.
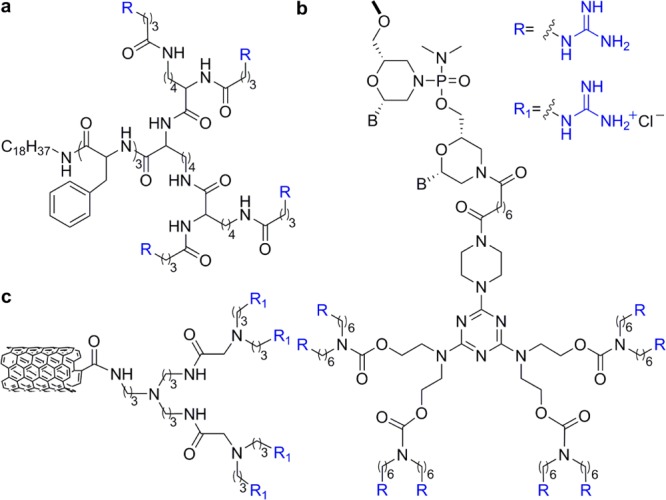
Dendrimer-like transporters: (a) dendronized nanoparticles,67 (b) “vivo morpholinos”,68 and (c) a representative structure of guanidinium carbon nanotubes.69
Morcos et al. developed a octaguanidinylated dendritic structure, built around a triazine core.68 This transporter structure was conjugated to a morpholino oligomer yielding a product referred to as a vivo-morpholino (Figure 12b).68 Vivo-morpholinos were shown to effectively silence genes within cultured cells. This structure also proved to effectively deliver morpholino antisense oligomers into a wide variety of tissues in living mice.68 Also aimed to deliver antisense oligonucleotides, Chi et al. designed and prepared multiwalled carbon nanotubes conjugated to positively charged dendrons bearing either ammonium or guanidinium groups (Figure 12c).69 Interestingly, ammonium-decorated nanotubes displayed notably better siRNA complexation, cellular uptake and gene silencing activity than the guanidinium-decorated counterparts.69
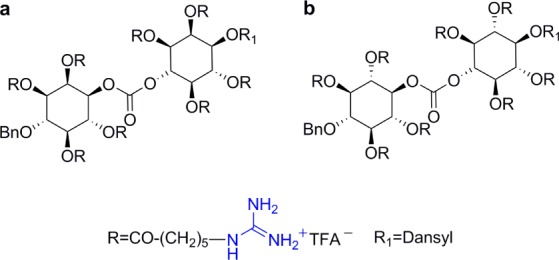
Ungaro and co-workers reported the synthesis of calix[n]arenes (n = 4, 6, and 8) bearing guanidinium groups in the aromatic rings (upper rim) showing good water solubility and proven ability to bind linear and plasmid DNA (Figure 13a).70 Their findings indicate that cell transfection, promoted by guanidinylated calix[n]arenes, is highly influenced by small changes in conformation, ring size, and the nature of alkyl substitutions in the lower rim. Subtle changes in these constituents notably affect their ability to bind to DNA and condensate it, which correlates with their ability to transfect cells and deliver DNA.71 These upper rim guanidinylated calix[n]arenes, however, showed low overall transfection efficiency and relatively high cytotoxicity. These drawbacks were overcome by attaching the guanidinium groups to the lower rim, through the phenolic moieties (Figure 13b).72 Calix[4]arenes, containing four guanidinium groups, showed significantly enhanced cell transfection efficiency and reduced cytotoxicity compared to the upper ring analogues.72 Moreover, one of the reported structures, linking the guanidinium moiety and the phenolic oxygen through a propylene bridge and lacking substitutions in the upper rim, had higher transfection efficiency than lipofectamine when formulated with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE). Again, like the upper rim modified platforms, subtle structural variations in these vectors can cause drastic changes in their biological properties.72 More recently, calix[4]arenes bearing four lysine or arginine units on either the upper or lower rim were disclosed (Figure 13c).73 These tetraargininocalix[4]arene constructs displayed higher efficiency in DNA transfection when compared to their lysine counterparts.73
Figure 13.
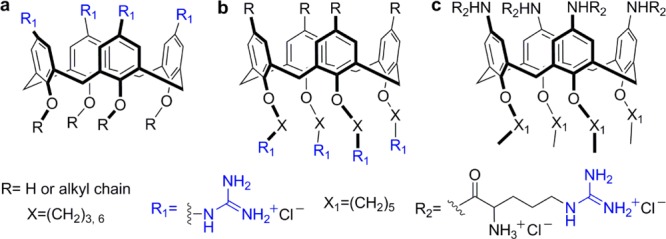
Representative structures of (a) upper rim guanidinocalix[4]arenes,70 (b) lower rim guanidinocalix[4]arenes,72 and (c) tetraargininocalix[4]arene.73
A family of tetraguanidinium vectors that efficiently internalized in human tumor cells was reported by Giralt and co-workers.74 These vectors consist of chiral bicyclic guanidinium subunits linked together through short thioether spacers (Figure 14). It was shown that these compounds translocated through HeLa membranes more efficiently than Antp or Tat peptides at very low concentrations.74 These compounds appear to specifically accumulate in mitochondria and showed no cytotoxicity at relevant concentrations. Following kinetic and temperature-dependent experiments showed that the internalization pathway involved both active energy-dependent transport and passive internalization.74
Figure 14.
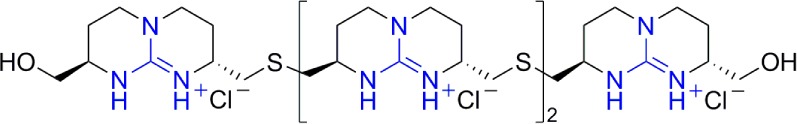
Bicyclic chiral guanidinium tetramer.74
Guanidinylating Agents
As apparent from the above discussion, guanidinium groups confer unique translocation features upon polyfunctional scaffolds. Their introduction into diverse cores frequently involves modification of the corresponding polyamines. Numerous strategies to convert a primary amine into a guanidinium group have been reported and comprehensively reviewed by Katritzky and Rogovoy.75 Here, we briefly describe the synthetic approaches used in the reports mentioned above (Figure 15). For the conversion of aminoglycosides into guanidinoglycosides, we typically use Boc-protected triflylguanidine, a reagent developed by Goodman and co-workers.76 The same reagent was used by Chung and co-workers.54−65 Ungaro and co-workers used either Boc-protected triflylguanidine or N,N′-di(tert-butoxycarbonyl)thiourea in the presence of mercuric chloride to guanidinylate the calix[n]arenes derivatives.70−72 The use of di-Boc-4-pyrazole-1-carboximidamide, described by Drake et al.,78 was the choice of Battigelli et al. to guanidinylate the carbon nanotube transporters,69 and Morcos and co-workers used O-methylisourea for the “vivo-morpholinos” conjugates.68
Figure 15.
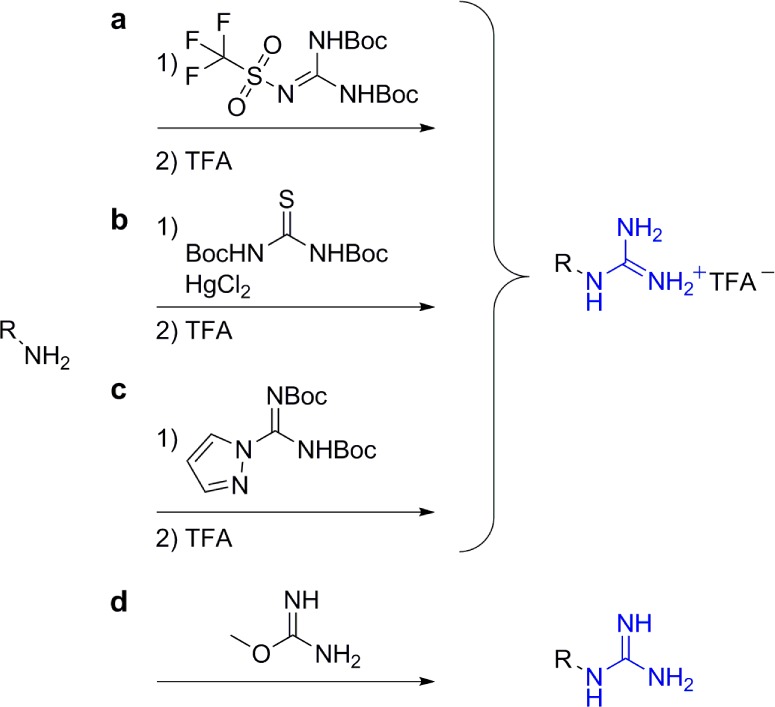
Guanidinylating strategies described in this Synopsis. Reaction conditions: (a) (i) DCM, TEA, −78 °C; (ii) TFA, rt;76 (b) (i) dimethylformamide (DMF), TEA, rt; (ii) TFA, rt;77 (c) (i) DMF, DIEA, rt; (ii) TFA, rt;78 (d) 1,3-Dimethyl-2-imidazolidinone, 1-hydroxybenzotriazole, TEA, ammonium hydroxide, 50 °C.68
Although high yields were reported for the guanidinylation of primary and secondary amines using protected thioureas, the need for mercury salts makes this procedure somewhat less atractive.77 The use of di-Boc-4-pyrazole-1-carboximidamide is often associated with moderate yields,78 while high to quantitative yields have been reported for the use of triflylguanidine.76 The inexpensive O-methylisourea is useful for guanidinylation in aqueous solutions without the need for a deprotection step, and high yields were also reported for its reactions.68 When it comes to the perguanidinylation of multifunctional scaffolds, triflylguanidine remains our guanidinylating agent of choice.
Conclusions and Perspectives
The introduction of guanidinium groups into multifunctional scaffolds has proven to be a very efficient strategy to generate cellular delivery vehicles. Such molecular transporters have been shown to facilitate the delivery of diverse cargos, ranging from low molecular weight small molecules to extraordinary high molecular weight quantum dots. Certain guanidinylated scaffolds have also been shown to overcome important biological barriers such as the BBB, allowing for the delivery of therapeutic agents to the brain.
Although extensive efforts have advanced the utility of diverse guanidinium-based molecular transporters, the fundamental understanding of their cell entry processes and the role of guanidinium groups in cellular delivery remain elusive. Subtle structural changes in the carriers and linkers often result in unexpectedly distinct celullar delivery profiles or in completely different biodistribution patterns and intracellular localization. Systematic correlations between structural features of the molecular transporters and both their cellular delivery efficiency and biodistribution at the organelle/organ level are therefore needed. A deeper molecular level understanding of these processes will likely further advance such guanidinium-rich molecular transporters as therapeutic and diagnostic tools.
Acknowledgments
We are grateful to the W. M. Keck Foundation, the National Science Foundation (Grant No. CHE-1303554) and the National Institutes of Health (Grant No. GM077471) for supporting research in our laboratories.
Biographies

Dr. Ezequiel Wexselblatt completed his Ph.D. in 2010 at the Hebrew University of Jerusalem under the supervision of Prof. Katzhendler and Dr. Yavin. After a short postdoctoral fellowship in Prof. Gibson’s group, in 2012 he moved to the University of California, San Diego, where he currently is a postdoctoral fellow in Prof. Yitzhak Tor’s group.

Professor Jeffrey D. Esko received his Ph.D. in Biochemistry at the University of Wisconsin, Madison. After an independent fellowship at the Molecular Biology Institute at the University of California, Los Angeles, he moved to the University of Alabama at Birmingham (1983) and then to the Department of Cellular and Molecular Medicine at the University of California, San Diego, in 1996 to help build a program in Glycobiology. He is currently the Co-Director of the Glycobiology Research and Training Center. Work in his laboratory focuses on the structure, biosynthesis, and function of proteoglycans.

Professor Yitzhak Tor received his undergraduate education at Tel Aviv University and his Ph.D. at the Weizmann Institute of Science (1990). Following a postdoctoral stay at the California Institute of Technology, he began his independent career at the University of Chicago. A year later, in 1994, he moved to the University of California, San Diego, where he is currently a Professor of Chemistry and Biochemistry and the George W. and Carol A. Lattimer Professor. His research interests include chemistry and biology of nucleosides, nucleotides, and nucleic acids, as well as the development of cellular delivery agents and fluorescent probes.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Ryser H. J. P. Science 1968, 159, 390. [DOI] [PubMed] [Google Scholar]
- Dietz G. P. H.; Bahr M. Mol. Cell. Neurosci. 2004, 27, 85. [DOI] [PubMed] [Google Scholar]
- Perez F.; Joliot A.; Blochgallego E.; Zahraoui A.; Triller A.; Prochiantz A. J. Cell Sci. 1992, 102, 717. [DOI] [PubMed] [Google Scholar]
- Leroux I.; Joliot A. H.; Blochgallego E.; Prochiantz A.; Volovitch M. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derossi D.; Joliot A. H.; Chassaing G.; Prochiantz A. J. Biol. Chem. 1994, 269, 10444. [PubMed] [Google Scholar]
- Derossi D.; Chassaing G.; Prochiantz A. Trends Cell Biol. 1998, 8, 84. [PubMed] [Google Scholar]
- a Frankel A. D.; Pabo C. O. Cell 1988, 55, 1189–93. [DOI] [PubMed] [Google Scholar]; b Green M.; Loewenstein P. M. Cell 1988, 55, 1179. [DOI] [PubMed] [Google Scholar]
- Fawell S.; Seery J.; Daikh Y.; Moore C.; Chen L. L.; Pepinsky B.; Barsoum J. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia J. S.; Dowdy S. F. Adv. Drug Deliver. Rev. 2005, 57, 579. [DOI] [PubMed] [Google Scholar]
- Futaki S.; Suzuki T.; Ohashi W.; Yagami T.; Tanaka S.; Ueda K.; Sugiura Y. J. Biol. Chem. 2001, 276, 5836. [DOI] [PubMed] [Google Scholar]
- Other approaches have recently been reviewed; see:Matile S.; Fyles T. Acc. Chem. Res. 2013, 46, 2741. [DOI] [PubMed] [Google Scholar]
- Futaki S.; Nakase I.; Suzuki T.; Zhang Y. J.; Sugiura Y. Biochemistry 2002, 41, 7925. [DOI] [PubMed] [Google Scholar]
- Strecker A. Liebigs Ann. Chem. 1861, 118, 151. [Google Scholar]
- Berlinck R. G. S.; Trindade-Silva A. E.; Santos M. F. C. Nat. Prod. Rep. 2012, 29, 1382. [DOI] [PubMed] [Google Scholar]
- Gobel M.; Klapotke T. M. Chem. Commun. 2007, 3180. [DOI] [PubMed] [Google Scholar]
- Yamada T.; Liu X. H.; Englert U.; Yamane H.; Dronskowski R. Chem.—Eur. J. 2009, 15, 5651. [DOI] [PubMed] [Google Scholar]
- Sawinski P. K.; Meven M.; Englert U.; Dronskowski R. Cryst. Growth Des. 2013, 13, 1730. [Google Scholar]
- Yokomori Y.; Hodgson D. J. Int. J. Pept. Prot. Res. 1988, 31, 289. [Google Scholar]
- a Houk R. J. T.; Tobey S. L.; Anslyn E. V. Top. Curr. Chem. 2005, 255, 199. [Google Scholar]; b Fromm J. R.; Hileman R. E.; Caldwell E. E. O.; Weiler J. M.; Linhardt R. J. Arch. Biochem. Biophys. 1995, 323, 279. [DOI] [PubMed] [Google Scholar]
- Han J.; Yau C. W.; Chan C. W.; Mak T. C. W. Cryst. Growth Des. 2012, 12, 4457. [Google Scholar]
- Furberg S. Acta Chem. Scand. 1972, 26, 3699. [DOI] [PubMed] [Google Scholar]
- Grossel M. C.; Merckel D. A. S.; Hutchings M. G. CrystEngComm 2003, 5, 77. [Google Scholar]
- Tobey S. L.; Anslyn E. V. J. Am. Chem. Soc. 2003, 125, 14807. [DOI] [PubMed] [Google Scholar]
- Haj-Zaroubi M.; Mitzel N. W.; Schmidtchen F. P. Angew. Chem., Int. Ed. 2002, 41, 104. [DOI] [PubMed] [Google Scholar]
- Friend D. S.; Papahadjopoulos D.; Debs R. J. Biochim. Biophys. Acta 1996, 1278, 41. [DOI] [PubMed] [Google Scholar]
- Doherty G. J.; McMahon H. T. Annu. Rev. Biochem. 2009, 78, 857. [DOI] [PubMed] [Google Scholar]
- Takei K.; Haucke V. Trends Cell Biol. 2001, 11, 385. [DOI] [PubMed] [Google Scholar]
- Matveev S.; Li X.; Everson W.; Smart E. J. Adv. Drug Deliver. Rev. 2001, 49, 237. [DOI] [PubMed] [Google Scholar]
- Conner S. D.; Schmid S. L. Nature 2003, 422, 37. [DOI] [PubMed] [Google Scholar]
- Swanson J. A.; Watts C. Trends Cell Biol. 1995, 5, 424. [DOI] [PubMed] [Google Scholar]
- Khalil I. A.; Kogure K.; Akita H.; Harashima H. Pharmacol. Rev. 2006, 58, 32. [DOI] [PubMed] [Google Scholar]
- Subtil A.; Hemar A.; Dautryvarsat A. J. Cell Sci. 1994, 107, 3461. [DOI] [PubMed] [Google Scholar]
- Varkouhi A. K.; Scholte M.; Storm G.; Haisma H. J. J. Controlled Release 2011, 151, 220. [DOI] [PubMed] [Google Scholar]
- Vives E.; Brodin P.; Lebleu B. J. Biol. Chem. 1997, 272, 16010. [DOI] [PubMed] [Google Scholar]
- Trehin R.; Merkle H. P. Eur. J. Pharm. Biopharm. 2004, 58, 209. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Mitchell D. J.; Pattabiraman K.; Pelkey E. T.; Steinman L.; Rothbard J. B. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N.; Matile S. J. Am. Chem. Soc. 2003, 125, 14348. [DOI] [PubMed] [Google Scholar]
- Rothbard J. B.; Jessop T. C.; Lewis R. S.; Murray B. A.; Wender P. A. J. Am. Chem. Soc. 2004, 126, 9506. [DOI] [PubMed] [Google Scholar]
- Stanzl E. G.; Trantow B. M.; Vargas J. R.; Wender P. A. Acc. Chem. Res. 2013, 46, 2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin V.; Bonduelle C. V.; Gillies E. R. Pharmaceuticals 2010, 3, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi D.; Zhang S.; Cui S.; Zhao Y.; Wang Y.; Zhao D. Bioconjugate Chem. 2013, 24, 487. [DOI] [PubMed] [Google Scholar]
- Luedtke N. W.; Baker T. J.; Goodman M.; Tor Y. J. Am. Chem. Soc. 2000, 122, 12035. [DOI] [PubMed] [Google Scholar]
- Luedtke N. W.; Carmichael P.; Tor Y. J. Am. Chem. Soc. 2003, 125, 12374. [DOI] [PubMed] [Google Scholar]
- Elson-Schwab L.; Garner O. B.; Schuksz M.; Crawford B. E.; Esko J. D.; Tor Y. J. Biol. Chem. 2007, 282, 13585. [DOI] [PubMed] [Google Scholar]
- Dix A. V.; Fischer L.; Sarrazin S.; Redgate C. P.; Esko J. D.; Tor Y. ChemBioChem 2010, 11, 2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin S.; Wilson B.; Sly W. S.; Tor Y.; Esko J. D. Mol. Ther. 2010, 18, 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M.; Tong W.; Esko J. D.; Tor Y. ACS Chem. Biol. 2013, 8, 1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M.; Wexselblatt E.; Esko J. D.; Tor Y. ChemBioChem. 2014, 15, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M.; Rusnati M.; Presta M.; Giacca M. J. Biol. Chem. 2001, 276, 3254. [DOI] [PubMed] [Google Scholar]
- Verdurmen W. P.; Bovee-Geurts P. H.; Wadhwani P.; Ulrich A. S.; Hallbrink M.; van Kuppevelt T. H.; Brock R. Chem. Biol. 2011, 18, 1000. [DOI] [PubMed] [Google Scholar]
- Ziegler A.; Seelig J. Biophys. J. 2008, 94, 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon G. M. K.; Gariepy J. Biochem. Soc. T. 2007, 35, 788. [DOI] [PubMed] [Google Scholar]
- Wallbrecher R.; Verdurmen W. P.; Schmidt S.; Bovee-Geurts P. H.; Broecker F.; Reinhardt A.; van Kuppevelt T. H.; Seeberger P. H.; Brock R.. Cell. Mol. Life Sci. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti K. K.; Jeon O.-Y.; Lee W. S.; Kim D.-C.; Kim K.-T.; Takeuchi T.; Futaki S.; Chung S.-K. Angew. Chem., Int. Ed. 2006, 45, 2907. [DOI] [PubMed] [Google Scholar]
- Maiti K. K.; Lee W. S.; Takeuchi T.; Watkins C.; Fretz M.; Kim D.-C.; Futaki S.; Jones A.; Kim K.-T.; Chung S.-K. Angew. Chem., Int. Ed. 2007, 46, 5880. [DOI] [PubMed] [Google Scholar]
- Jin J.; Lee W. S.; Joo K. M.; Maiti K. K.; Biswas G.; Kim W.; Kim K.-T.; Lee S. J.; Kim K.-H.; Nam D.-H.; Chung S.-K. MedChemComm 2011, 2, 270. [Google Scholar]
- Im J.; Kim W.; Kim K.-T.; Chung S.-K. Chem. Commun. 2009, 4669. [DOI] [PubMed] [Google Scholar]
- Im J.; Biswas G.; Kim W.; Kim K.-T.; Chung S.-K. Bull. Korean Chem. Soc. 2011, 32, 873. [Google Scholar]
- Im J.; Maiti K. K.; Kim W.; Kim K.-T.; Chung S.-K. Bull. Korean Chem. Soc. 2011, 32, 1282. [Google Scholar]
- Higashi T.; Khalil I. A.; Maiti K. K.; Lee W. S.; Akita H.; Harashima H.; Chung S.-K. J. Controlled Release 2009, 136, 140. [DOI] [PubMed] [Google Scholar]
- Biswas G.; Jeon O.-Y.; Lee W. S.; Kim D.-C.; Kim K.-T.; Lee S.; Chang S.; Chung S.-K. Chem.—Eur. J. 2008, 14, 9161. [DOI] [PubMed] [Google Scholar]
- Lee W. S.; Im C.-N.; Teng Q. Y.; Chang Y.-T.; Kim D.-C.; Kim K.-T.; Chung S.-K. Mol. BioSyst. 2009, 5, 822. [DOI] [PubMed] [Google Scholar]
- Im J.; Kim S.; Jeong Y.-H.; Kim W.; Lee D.; Lee W. S.; Chang Y.-T.; Kim K.-T.; Chung S.-K. MedChemComm 2013, 4, 310. [Google Scholar]
- Lee W. S.; Kim W.; Kim K.-T.; Chung S.-K. Bull. Korean Chem. Soc. 2011, 32, 2286. [Google Scholar]
- Ghosh S. C.; Kim B.; Im J.; Lee W. S.; Im C.-N.; Chang Y.-T.; Kim W.; Kim K.-T.; Chung S.-K. Bull. Korean Chem. Soc. 2010, 31, 3623. [Google Scholar]
- Tanaka M.; Machida Y.; Niu S.; Ikeda T.; Jana N. R.; Doi H.; Kurosawa M.; Nekooki M.; Nukina N. Nat. Med. 2004, 10, 148. [DOI] [PubMed] [Google Scholar]
- Chi B.; Park S. J.; Park M. H.; Lee S. Y.; Jeong B. Bioconjugate Chem. 2010, 21, 1473. [DOI] [PubMed] [Google Scholar]
- Li Y.-F.; Morcos P. A. Bioconjugate Chem. 2008, 19, 1464. [DOI] [PubMed] [Google Scholar]
- Battigelli A.; Wang J. T.-W.; Russier J.; Da Ros T.; Kostarelos K.; Al-Jamal K. T.; Prato M.; Bianco A. Small 2013, 9, 3610. [DOI] [PubMed] [Google Scholar]
- Dudic M.; Colombo A.; Sansone F.; Casnati A.; Donofrio G.; Ungaro R. Tetrahedron 2004, 60, 11613. [Google Scholar]
- Sansone F.; Dudic M.; Donofrio G.; Rivetti C.; Baldini L.; Casnati A.; Cellai S.; Ungaro R. J. Am. Chem. Soc. 2006, 128, 14528. [DOI] [PubMed] [Google Scholar]
- Bagnacani V.; Franceschi V.; Fantuzzi L.; Casnati A.; Donofrio G.; Sansone F.; Ungaro R. Bioconjugate Chem. 2012, 23, 993. [DOI] [PubMed] [Google Scholar]
- Bagnacani V.; Franceschi V.; Bassi M.; Lomazzi M.; Donofrio G.; Sansone F.; Casnati A.; Ungaro R. Nat. Commun. 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Carneado J.; Van Gool M.; Martos V.; Castel S.; Prados P.; De Mendoza J.; Giralt E. J. Am. Chem. Soc. 2005, 127, 869. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Rogovoy B. V. ARKIVOC 2005, iv, 49. [Google Scholar]
- Feichtinger K.; Zapf C.; Sings H. L.; Goodman M. J. Org. Chem. 1998, 63, 3804. [Google Scholar]
- Kim K. S.; Qian L. G. Tetrahedron Lett. 1993, 34, 7677. [Google Scholar]
- Drake B.; Patek M.; Lebl M. Synthesis—Stuttgart 1994, 579. [Google Scholar]