

Abstract
The full details of a synthesis of the hetidine framework of the C20-diterpenoid alkaloids and its conversion to the atisine core structure are reported. The application of the hetidine framework to the synthesis of dihydronavirine, which is the formal reduction product of the natural product navirine, is also described. Key to the success of these studies is the use of a Ga(III)-catalyzed cycloisomerization reaction of alkynylindenes to prepare a [6–7–6] framework that was advanced to the hetidine skeleton. Furthermore, a Michael/aldol sequence was developed for the construction of the bicyclo[2.2.2] framework that is characteristic of the hetidines and atisines.
Introduction
The diterpenoid alkaloids (see representative examples in Figure 1) are a collection of complex alkaloids isolated from the Ranunculaceae and Rosaceae family of plants.1 These molecules are among the most architecturally complex natural products isolated to date. In addition to their imposing skeletal intricacy, they possess varied, yet dense, arrays of hydroxylation. To date, over 1100 natural products have been isolated and classified as diterpenoid alkaloids. On the basis of the number of constituent carbons, these natural products are described as C18-, C19-, or C20-diterpenoid alkaloids. The majority of these metabolites have been isolated from the Aconitum, Consolida, Delphinium, Rumex, and Spiraea genera, and extracts from these plants have been used in traditional Eastern herbal medicine for centuries as sedatives and fever reducers. Other pharmacological effects that have been discovered in these plants, including analgesic effects and anti-inflammatory, myorelaxant, and antiarrhythmic properties, are also attributed to their diterpenoid alkaloid constituents. Overall, this broad spectrum of biological activity likely arises because of the potent interactions of these alkaloids with voltage-dependent sodium, potassium, and calcium ion channels. The pharmaceutical potential of the diterpenoid alkaloids is continuing to emerge, and already, lappaconitine (allapinin, 1) and guan-fu base A (13, Figure 2; also known as acehytisine hydrochloride)2 are marketed as antiarrhythmia agents.34
Figure 1.

Representative C18-, C19-, and C20-diterpenoid alkaloids.
Figure 2.

Representative C20-diterpenoid alkaloids and derivatives in the atisine, hetidine, and hetisine subclass.
The classification of the C20-diterpenoid alkaloids by Wang and Liang on the basis of their carbon skeletons into the atisane, kaurane, rearranged, and bisditerpenoid classes is the most useful classification from a synthetic strategy perspective.5 In many ways, this delineation unveils a general pattern recognition6 that should facilitate the synthesis of not only a single member of this family of natural products but many members from a common intermediate. Our synthetic studies on these alkaloids began with an interest in the atisane class, specifically the atisine, hetidine, and hetisine-type skeletons. In this paper, we report the details of a strategy that led to the synthesis of an intermediate that provides access to the atisine and hetidine skeletons (A and B, respectively, Figure 2) and has culminated in a synthesis of dihydronavirine (8).
Background
Despite the isolation of a wide range of diterpenoid alkaloids over the last half-century across the various skeletal subclasses, there remains a relatively small number of total syntheses of these natural products. Early important contributions to the syntheses of the diterpenoid alkaloids (including a synthesis of talatisamine, 3, and atisine, 9) were made by Wiesner and co-workers,7 and recently, syntheses of nominine (12, by the groups of Gin8 and Muratake and Natsume9), neofinaconitine (2, by Gin and co-workers),10 and lepenine (4, by Fukuyama, Yokoshima, and co-workers)11 have appeared.12
In our approach to the C20-diterpenoid alkaloids, we envisioned the hetidine framework (see B, Figure 2) as a versatile carbon skeleton that could be converted at a late stage to the hetisine (i.e., C) and atisine (i.e., A) skeletons. Key to these conversions would be a late-stage C6–N bond formation (B → C; by employing a Hoffman–Löffler–Freytag-type reaction)13 or a C14–C20 bond cleavage (B → A; using an oxidative Grob-like fragmentation)14 to convert the hetidine skeleton to the hetisine and atisine skeletons, respectively. Importantly, Okamoto et al. have already demonstrated the feasibility of converting the hetidine core to a mixture of hetisine and atisine frameworks from the corresponding N-chloroamine,15 and so our studies have focused on the synthesis of the hetidine framework and its exclusive conversion to the atisine skeleton.
Results and Discussion
Previously, we reported the successful synthesis of 14 (Figure 3), which possesses key hydroxy groups at C11 and C14 that are critical for the synthesis of alkaloids such as spiramine H (10), kobusine (11), navirine (5), and guan-fu base A (13).16 In particular, we viewed the hydroxyl at C14 of 14 as an important functional handle that would facilitate the desired Grob-like fragmentation to arrive at the atisine skeleton and natural products such as 9 and 10. Herein, we report the full details of our studies using 14 as a key late-stage synthetic intermediate, which have culminated in the preparation of dihydronavirine (8) as well as our realization of the conversion of the hetidine framework to the atisine skeleton by employing a Grob-like fragmentation that is achieved under oxidation conditions. Our efforts to prepare 15, which represents the simplest synthetic rendition of the hetidine core, is also discussed.
Figure 3.

Potential synthetic equivalents to the hetidine core.
Initial Synthetic Studies on the Hetidine Core: Attempts To Prepare 15
From our synthetic analysis of the C20–diterpenoid alkaloids in the atisine, hetidine and hetisine skeletal subclasses, two key challenges have emerged. These are the installation of the [2.2.2] bicycle and the position-selective introduction of oxygenation, which for many of these molecules is required at sites distal from other functional groups. Our initial forays into the synthesis of the hetidine core sought to address the first of these challenges by preparing 15. From our perspective, the installation of the [2.2.2] bicycle has been most elegantly addressed by Gin and Peese, who employed an intramolecular Diels–Alder (IMDA) reaction to forge the [2.2.2] bicycle in their synthesis of nominine.8 Inspired by this powerful complexity building transformation, we envisioned the hetidine core (see 15, Scheme 1) arising from methoxyarene 16, where in the forward sense, the requisite diene for the IMDA reaction would be fashioned from the arene using a Birch reduction as previously demonstrated by Gin and Peese. Piperidine polycycle 16 could arise from amino alcohol 17 through a C20–N bond formation. In turn, tricycle 17 was envisioned from benzocycloheptadiene 18, which would be prepared from alkynylindene 19 using a Ga(III)-catalyzed cycloisomerization reaction developed in our laboratories.17
Scheme 1. Retrosynthetic Analysis of Hetidine Skeleton Equivalent 15.

Our planned synthesis of hexacycle 15 commenced with the preparation of alkynylindene 19 (Scheme 2) as previously described.16 Alkylation of commercially available β-ketoester 20(18) with alkyl iodide 21 (accessed in racemic form from 3-bromo-1-propanol in nine steps)19 proceeds in 78% yield to provide the alkylated β-ketoester adduct (22). Of note, we have also demonstrated the enantioselective synthesis of alkyl iodide 21 (in 89% ee) using a method developed by Sawamura and Ito.20 As such, the foundation for an enantioselective synthesis has been set. However, for the purposes of these studies, the racemic material was advanced. Saponification and decarboxylation of 22 using LiOH provides indanone 23 in 98% yield. Chemoselective reduction of the indanone carbonyl group is achieved in the presence of the nitrile group using NaBH4 as the reducing agent to provide an indanol (not shown; as a mixture of diastereomers) that was dehydrated using pyridinium p-toluenesulfonate (PPTS) to afford alkynylindene 19.
Scheme 2. Synthesis of Alkynylindene 19.

Following a comprehensive optimization study, it was found that subjecting rigorously dried alkynylindene 19 to a substoichiometric amount of GaI3 (25 mol %) in the presence of 4 Å powdered molecular sieves in toluene at 100 °C for 48 h gives benzannulated cycloheptadiene 18 in 89% yield (Scheme 3). The temperature and reaction time for the formation of 18 are higher and longer, respectively, compared to conditions employed previously for the cycloisomerization of other alkynylindene substrates. The higher temperature and longer reaction time for the conversion of 19 to 18 is attributed to several factors including the presence of the cyano group, which likely interacts with GaI3 and mitigates its Lewis acidity. In addition, the cyano group, by virtue of its inductive electron-withdrawing effect, likely decreases the inherent nucleophilicity of the alkyne group. Finally, in previous alkynylindene cycloisomerization reactions that we have conducted, highly electron-rich indenes were employed. As such, it seems reasonable that the less electron-rich indene moiety in 19 would require more forcing conditions for the desired transformation. Consistent with this line of reasoning is the observation that the use of other Lewis acid catalysts such as GaCl3 that had previously been efficient for the syntheses of 24–26, only resulted in the incomplete conversion of 19 to 18 and low isolated yields of the desired cycloheptadiene.
Scheme 3. Cycloisomerization of Indenylalkynes.

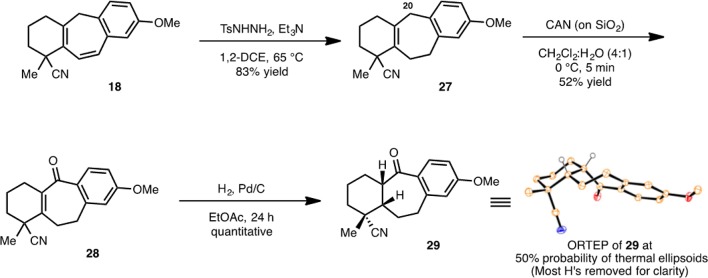
With benzannulated cycloheptadiene 18 in hand, selective reduction of the disubstituted double bond proceeds in good yield using diimide (Scheme 4), which is generated in situ from p-toluenesulfonyl hydrazide and triethylamine.21 Other conditions for diimide generation (e.g., hydrazine, CuSO4·H2O, EtOH)22 were less effective and led only to recovered starting material. Next, oxidation to install a carbonyl group at C20 (hetidine numbering) of 27 was pursued. This oxygenation proved to be one of the most capricious steps of our synthesis campaign. An extensive investigation of various oxidants including DDQ, CrO2, CrO3, Ba(MnO4)2, KMnO4, and MnO2 resulted in little to no oxidation, overoxidation, or nonspecific decomposition. Ultimately, ceric ammonium nitrate (CAN) adsorbed on silica gel proved to be the most effective oxidant and provided enone 28 in a workable 52% yield (0.75 mmol scale). Diastereoselective reduction of the tetrasubstituted enone double bond is achieved using H2 and Pd/C to provide a single diastereomer of ketone 29. The structure and relative stereochemistry of 29 was unambiguously confirmed by an X-ray crystallographic study of a single crystal (see ORTEP representation in Scheme 4). A possible rationale for the excellent diastereoselectivity that is observed in the hydrogenation of 28 is that hydrogen addition occurs from the face bearing the methyl group (A = 1.7),23 which adopts a pseudoequatorial position in 28, whereas the cyano group (A = 0.17) is axially disposed. As a result, approach from the face opposite the cyano group is more likely. While this “contra-steric” rationale is appealing,24 an electronic argument may also be advanced. Thus, Woodward has proposed that the facial selectivity of a reacting olefin group may be determined by the extent to which that face is electron-depleted.25 In our case, this would suggest that the face bearing the cyano group is electron-depleted (by virtue of an interaction between the π orbital of the tetrasubstituted double bond and the lower-lying π* orbital of the axially disposed cyano group), and as such, hydrogenation occurs from the face bearing the methyl substituent.26
Scheme 4. Functionalization of Cyloheptadiene 18 to Tricycle 29.
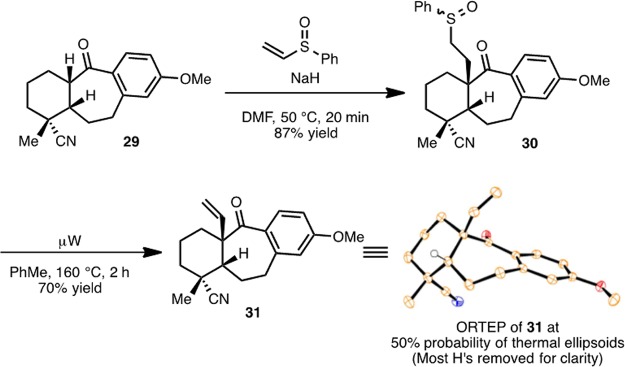
With ketone 29 in hand, the next task was to install a vinyl group α to the ketone group (Scheme 5). Deprotonation α to the carbonyl group in 29 is readily effected with sodium hydride and if conducted in the presence of phenyl vinylsulfoxide gives sulfoxide 30 in 87% yield. Sulfoxide extrusion at this stage installs the vinyl group in 31, which was obtained as a single diastereomer as confirmed by X-ray analysis of a single crystal of 31 (see ORTEP in Scheme 5). Surprisingly, every attempt to reduce both the nitrile and carbonyl groups of 31 (in preparation for the formation of a piperidine ring; see 17 to 16, Scheme 1) with LiAlH4 was frustrated by the formation of a complex mixture, which included the expected amino alcohol (i.e., 17, as a mixture of diastereomers) along with unidentifiable side products. Furthermore, attempts to form the piperidine ring by activation of the alcohol group of 17 (with SOCl2, oxalyl chloride, or MsCl) led to a complex mixture where it appeared the vinyl group was engaged by presumed carbocation intermediates formed by the activation of the C20 hydroxy group.
Scheme 5. Installation of the Vinyl Group.
We hypothesized that an allyl group in place of the vinyl group in 31 would mitigate the unwanted reactivity of the double bond of the angular substituent upon activation of the hydroxy group. If this was the case, then the allyl group could be converted to the vinyl group at a later stage following the formation of the piperidine ring. As shown in Scheme 6, with an allyl group in place of the vinyl group, piperidine ring formation is indeed successful. Thus, generation of the sodium enolate of 29 (with sodium hydride) in the presence of allyl bromide yielded a mixture of the C-allylated (32) and O-allylated (not shown) adducts. The mixture is easily converged to 32 upon heating (μW, 160 °C) through a Claisen rearrangement. At this stage, reduction of the carbonyl and cyano groups of 32 (with LiAlH4) followed by Boc-protection of the resulting primary amine provides 33 as a single diastereomer (the stereochemistry of the C20 center was not determined). Exposure of Boc-protected amino alcohol 33 to thionyl chloride effects clean cyclization to forge the piperidine ring to provide 34 in 72% yield. In contrast, treatment of the free amino alcohol (generated by LiAlH4 reduction of 32) with thionyl chloride led only to decomposition. The structure of 34 was unambiguously confirmed by X-ray analysis of the p-nitrobenzoyl derivative 35.
Scheme 6. Installation of the Allyl Group and Piperidine Ring formation.

At this stage, we next planned the conversion of the angular allyl group to a vinyl group, which was accomplished in six steps as illustrated in Scheme 7. The sequence commenced with an UpJohn dihydroxylation27 of 34 followed by periodate cleavage of the resulting diol to afford aldehyde 36. Reduction of the aldehyde group using sodium borohydride gives alcohol 37. A modified Mitsunobu displacement of the hydroxyl group of 37 with a phenyl sulfide group is readily accomplished with tributylphosphine and diphenyl sulfide. Selective oxidation of the phenyl sulfide group to the corresponding sulfoxide (38) is effected with H2O2 and Sc(OTf)3.28 Sulfoxide extrusion was then accomplished by heating (μW, 180 °C) to afford 39 in 80% yield.
Scheme 7. Conversion of the Allyl Substituent to the Vinyl Group.

With access to methoxyarene 39, the stage was set to attempt the installation of the [2.2.2] bicycle using a sequence analogous to that employed by Gin and Peese in their synthesis of nominine.8 Thus, subjection of arene 39 to dissolving metal reduction conditions (Na, i-PrOH, liquid NH3) followed by hydrolysis of the resulting methyl enol ether under acidic conditions (with citric or oxalic acid) gives a mixture of enones (40 and 41) that proved to be unstable and could not be easily advanced. This is in marked contrast to the observations of Gin and Peese on the Birch reduction of 42 to give 43 in high yield as illustrated in Scheme 8B.8
Scheme 8. Reductive Dearomatization Studies.

The lengthy sequence required for the installation of the vinyl group in 39 from the corresponding allyl compound (34; see Scheme 7) as well as the instability of the Birch reduction products (40 and 41) caused us to consider an alternative strategy for the preparation of the hetidine core, especially with regard to installing the [2.2.2] bicycle.
Revised Approach to the Hetidine Framework
In a revised approach to the hetidine framework, we sought to address both challenges inherent in the preparation of the hetidine core. That is, the installation of the [2.2.2] bicycle (which had been the focal point of our first approach) as well as the position selective introduction of hydroxylation to address targets including navirine (5), guan-fu base A (13), and spiramine H (10). In our new approach, an alternative synthetic equivalent to the hetidine framework (14, Scheme 9) was identified. We envisioned 14 arising from enone aldehyde 44, where in the forward sense the [2.2.2] bicycle would be forged using a Michael/aldol sequence. In turn, 44 would be formed from allyl methoxyarene 34 by employing a key oxidative dearomatization reaction. This strategy would address several shortcomings of our initial approach. First, the laborious conversion of the allyl group to a vinyl group (Scheme 7) would be avoided. Second, the programmed installation of hydroxylation at C11 and C14 (see 14) would be achieved from the allyl methoxyarene precursor (34) using the oxidative dearomatization and the aldol synthetic sequences.
Scheme 9. Revised Retrosynthesis to the Hetidine Framework.

The synthesis of 14 (Scheme 10) began with the demethylation of methoxyarene 34 using sodium ethanethiolate (μW, 180 °C) to provide phenol 45 in 85% yield. Subjecting phenol 45 to oxidative dearomatization conditions29 with phenyliodone diacetate (PIDA) in a mixture of water and acetonitrile (1:1) yielded cyclic carbamate 47 as the sole product in 15% yield. The oxazolidinone moiety in 47 is likely formed by nucleophilic attack of the Boc carbonyl on the activated phenol (see 46). Although low yielding, the defined trajectory of approach for the Boc group as a nucleophile controls the newly introduced stereocenter at C14 in 47. In efforts to optimize this transformation, the more activated hypervalent iodine reagent, phenyliodine bis(trifluoroacetate) (PIFA; 1 equiv) was employed in the oxidative dearomatization along with a buffer (NaHCO3). In addition, trifluoroethanol was used in place of water as the cosolvent. These modifications increased the yield of 47 to 54%. Subsequently, we have also found that iodosobenzene (2 equiv) may be used in place of PIFA to obtain 47 in similar yields.
Scheme 10. Oxidative Dearomatization of Methoxyarene 34 To Obtain 47.

With cyclohexanedienone 47 in hand, a dihydroxylation of the allyl group followed by periodate cleavage leads to aldehyde 48 (Scheme 11). Simply stirring this aldehyde with silica gel in methylene chloride promotes a Michael addition to yield 49. A selective reduction of the enone double bond at this stage affords ketoaldehyde 50, which upon exposure to mildly basic conditions (K2CO3 in methanol) affords synthetic hetidine framework equivalent 14. The structure of 14 was unambiguously confirmed by an X-ray crystallographic study (see ORTEP, Scheme 11).
Scheme 11. Synthesis of Hetidine Core Structure 14.

Synthesis of Dihydronavirine
In an initial demonstration of the utility of 14 in the synthesis of diterpenoid alkaloids, we chose to focus on the synthesis of navirine (5) and the related derivative dihydronavirine (8). Navirine was isolated in 2004 from Aconitum naviculare, a perennial herb found in the Himalayas of Nepal and Tibet.30 Navirine is unusual among diterpenoid alkaloids in that it is a conjugate of the diterpenoid alkaloid core and hordenine (56, Scheme 13), a phenolic natural product, which is also coisolated with 5. In 2008, congeners of navirine that vary in their oxidation level (navirine B, 6, and navirine C, 7)31 were isolated from the same plant source and would seem to suggest that like many diterpenoid alkaloid families, additional navirines of other oxidation levels (e.g., dihydronavirine, 8) may exist and are yet to be isolated. For our immediate plans, the hydroxy group at C11 of 14, although useful in the context of an eventual synthesis of kobusine (11) and guan-fu base A (13), would have to be removed in order to achieve a synthesis of dihydronavirine (8) and navirine (5).
Scheme 13. Synthesis of Dihydronavirine (8).

Our planned synthesis of 5 and 8 commenced with the deoxygenation of 14 at C11, which was accomplished using the Barton–McCombie protocol32 to give 51 (Scheme 12) in 65% yield over the two steps. Deprotonation of ketone 51 with LiHMDS followed by the addition of Comins’ reagent (52)33 to the resulting enolate gives vinyl triflate 53. At this point, the C17 methylene group of the diterpenoid alkaloid core architecture was installed using a Stille cross-coupling reaction with tributylstannyl methanol (54) to yield 55.
Scheme 12. Preparation of Advanced Hetidine Framework 55.

To arrive at the complete framework of dihydronavirine (8) and navirine (5), the hordenine moiety had to be appended. This was accomplished by first activating the C17 allylic alcohol group of 55 (Scheme 13) toward nucleophilic displacement by converting it to the corresponding mesylate (not shown). Treatment of this compound with the sodium salt of hordenine (56) yields the desired adduct (57). Notably, an attempted Mitsunobu reaction to link 55 to 56 directly did not result in any conversion and only returned the starting material. With 57 in hand, hydrolytic cleavage of the cyclic carbamate group (using barium hydroxide; μW, 160 °C)34 provided dihydronavirine (8).
A conversion of dihydronavirine (8) to navirine (5) would require a dehydrogenation of the secondary amine to the corresponding imine. This type of oxidation has been studied previously in many contexts, and various oxidants including MnO2 and CrO3 have been employed for this purpose.35 In a closely related precedent in the diterpenoid alkaloid series, Benn and co-workers showed during their partial synthesis studies in the delphinine series that the piperidine moiety of des-N-methyllycaconitine (58, Scheme 14A) is converted to the corresponding imine (anhweidelphinine, 59) by the action of MnO2.36 However, in our hands, attempts using various oxidants to convert dihydronavirine (8) to navirine (5) resulted in nonspecific decomposition in many cases, whereas for a few of the oxidants (e.g., N-iodosuccinimide and MnO2) trace amounts of the C14–C20 bond cleavage product (60) were isolated. Given that 60 represents the core of the atisane diterpenoid alkaloids, we investigated the optimization of the conversion of dihydronavirine to 60. Following an extensive survey of reagents and reaction conditions, it was found that exposure of 8 to iodosobenzene (1.5 equiv) in the presence of NaHCO3 in methylene chloride for 1 h provides 60 in 75% yield. Various thermal and photochemical isomerization conditions were attempted in order to convert 60 to navirine (e.g., via an aza-Prins-type process) but led only to nonspecific decomposition in all cases. Thus, to date, we have not been able to convert dihydronavirine or related derivatives to navirine.
Scheme 14. Attempted Dehydrogenation of Dihydronavirine (8) to Navirine (5).

Synthesis of the Atisine Skeleton
Even though our attempted conversion of dihydronavirine (8) to navirine (5) was ultimately not successful, the formation of 60 demonstrates that the hetidine framework can be converted to the atisine skeleton as had been designed at the outset of our studies (Figure 2).37 While the conversion 8 → 60 is in essence a complexity-minimizing transformation, it achieves an important goal of installing oxygenation at C11 of the atisine core, which is required for the synthesis of natural products such as spiramine H (10). As such, we sought to investigate this C–C bond cleaving transformation on our synthetic equivalent to the hetidine framework (14). In this regard (Scheme 15), treatment of cyclic carbamate 14 with barium hydroxide cleaves the oxazolidinone to yield amino diol 61. Subjecting 61 to the optimized conditions identified for the oxidative Grob-like fragmentation of 8 (PhIO, 1.5 equiv and NaHCO3) leads to 63 in 75% yield. The structure of 63 was unambiguously confirmed by an X-ray crystallographic study (see ORTEP, Scheme 15). Two possible intermediates (62A and 62B) may be envisioned en route to 63. Given the inherently higher nucleophilicity of secondary amines compared to hydroxyl groups, intermediate 62A would seem more likely, although 62B cannot be ruled out.
Scheme 15. Conversion of the Hetidine Framework to the Atisine Skeleton.

Conclusion
Our studies on the C20-diterpenoid alkaloids with a focus on a general approach to the hetidine and atisine subclasses have identified a polycylic scaffold (14) that provides synthetic access to the hetidine framework and demonstrates its conversion to the atisine framework using an oxidative Grob-like fragmentation. Furthermore, 14 has been advanced to a synthesis of dihydronavirine (8), which is formally the hydrogenation product of navirine (5). These synthetic studies were enabled by a Ga(III)-catalyzed cycloisomerization reaction of alkynylindenes to build a 6–7–6 carbocyclic tricycle, a Michael/aldol sequence to forge the [2.2.2] bicycle, and a Stille/etherification synthetic sequence to append a hordenine fragment. It is our anticipation that these synthetic studies will set the stage for further studies on the interconversion of the carbon frameworks of diterpenoid alkaloid congeners and provide access to unique derivatives of these architecturally fascinating natural products to aid in future synthetic and biological studies.
Experimental Section
Experimental procedures and characterization data for the preparation of 22, 23, 19, 18, 27–29, 32–35, 45, 47–50, and 14 have been reported previously.16
Methyl 2-(4-Cyano-4-methylhex-5-yn-1-yl)-6-methoxy-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (22)
Alkyl iodide 21 (3.3 g, 13 mmol, 1 equiv) and K2CO3 (5.6 g, 40 mmol, 3 equiv) were added to a round-bottomed flask containing β-ketoester 20 (3.0 g, 13 mmol, 1 equiv) in acetone (67 mL, 0.2 M). The reaction vessel was fitted with a reflux condenser, and the reaction mixture was heated to 65 °C and stirred. After 18 h, the mixture was cooled to room temperature, diluted with water (75 mL), and extracted with EtOAc (3 × 75 mL). The combined organics were washed with brine (1 × 50 mL), dried over MgSO4, and concentrated. The resulting solid was purified by flash chromatography (9:1 hexanes/EtOAc to 4:1 hexanes/EtOAc) to obtain 3.5 g (10 mmol, 77% yield) of β-ketoester 22 as a pale orange solid (1:1 mixture of diastereomers): mp 99–102 °C; Rf 0.48 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.38 (d, J = 8.3 Hz, 1H), 7.22 (dd, J = 8.4, 2.6 Hz, 1H), 7.18 (d, J = 2.6 Hz, 1H), 3.83 (s, 3H), 3.68 (s, 3H), 3.63 (dd, J = 17.0, 3.9 Hz, 1H), 3.03 (d, J = 17.0 Hz, 1H), 2.35 (d, J = 20.2 Hz, 1H), 2.23–2.12 (m, 1H), 1.94–1.65 (m, 4H), 1.60 (d, J = 3.0 Hz, 3H) 1.58–1.48 (m, 1H); 13C NMR (151 MHz, CDCl3) δ 202.2, 202.2, 171.4, 171.4, 159.9, 145.9, 145.9, 136.4, 136.3, 127.3, 127.2, 125.2, 125.2, 120.2, 106.0, 80.8, 80.8, 72.4, 72.4, 61.3, 61.3, 55.8, 52.9, 52.9, 41.2, 41.1, 36.1, 34.3, 34.2, 31.3, 27.3, 27.2, 21.2, 21.1; IR (NaCl, thin film) νmax 3267, 2953, 2360, 1740, 1708, 1616 cm–1; HRMS (ESI) calcd for [C20H22NO4] ([M + H]+) m/z 340.1543, found 340.1543.
2-Ethynyl-5-(6-methoxy-1-oxo-2,3-dihydro-1H-inden-2-yl)-2-methylpentanenitrile (23)
Lithium hydroxide monohydrate (1.1 g, 26 mmol, 2.5 equiv) was added to a stirred solution of β-ketoester 22 (3.5 g, 10 mmol, 1 equiv) in a 4:1 mixture of THF/H2O (52 mL, 0.2 M). The heterogeneous mixture was heated to 65 °C in the reaction vessel fitted with a reflux condenser. After 12 h, the reaction mixture was cooled to room temperature, acidified with 1 N HCl (50 mL), and extracted with EtOAc (3 × 50 mL). The combined organics were washed with brine (1 × 25 mL), dried over MgSO4, and concentrated to yield 2.9 g (10 mmol, 100% yield) of indanone 23 as a yellow oil that was used without further purification (1:1 mixture of diastereomers based on 1H NMR): Rf 0.61 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.37–7.33 (m, 1H), 7.21–7.17 (m, 2H), 3.83 (s, 3H), 3.30 (dd, J = 16.6, 7.6 Hz, 1H), 2.76 (dtd, J = 16.9, 3.1, 1.6 Hz, 1H), 2.73–2.67 (m, 1H), 2.40 (d, J = 9.5 Hz, 1H), 2.06–1.96 (m, 1H), 1.89–1.73 (m, 4H), 1.64 (s, 3H), 1.58–1.46 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 208.5, 159.6, 146.5, 137.9, 137.8, 127.4, 124.4, 124.4, 120.4, 105.2, 81.0, 72.4, 72.3, 55.7, 48.2, 48.0, 41.0, 41.0, 32.3, 32.2, 31.4, 31.4, 31.1, 30.9, 27.3, 27.3, 24.0, 23.7; IR (NaCl, thin film) νmax 3285, 2838, 1705, 1614 cm–1; HRMS (ESI) calcd for [C18H20NO2] ([M + H]+) m/z 282.1489, found 282.1489.
2-Ethynyl-5-(5-methoxy-1H-inden-2-yl)-2-methylpentanenitrile (19)
Sodium borohydride (0.39 g, 10 mmol, 1 equiv) was added to a stirred solution of indanone 23 (2.9 g, 10 mmol, 1 equiv) in EtOH (95%, 50 mL, 0.2 M) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h. The reaction mixture was quenched by the slow addition of satd aqueous NH4Cl (25 mL). Ethanol was removed using a rotary evaporator, and the resulting concentrate was extracted with EtOAc (3 × 50 mL). The combined organics were washed with brine (1 × 25 mL), dried over MgSO4, and concentrated to give 2.9 g (10 mmol, 100% yield) of the desired indanol as a yellow oil that was used without further purification. 1H NMR analysis showed a 3:2 dr (major diastereomer not determined); Rf 0.36 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); major diastereomer 1H NMR (600 MHz, CDCl3) δ 7.10 (d, J = 8.2 Hz, 1H), 6.92 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 8.2, 2.5 Hz, 1H), 4.79 (d, J = 7.2 Hz, 1H), 3.80 (s, 3H), 3.07 (dd, J = 15.2, 7.9 Hz, 1H), 2.42 (d, J = 8.5 Hz, 1H), 2.40 (s, 1H), 2.24–2.15 (m, 2H), 1.92–1.71 (m, 4H), 1.65 (m, 3H) 1.61–1.52 (m, 1H); minor diastereomer 1H NMR (600 MHz, CDCl3) δ 7.14 (d, J = 8.3, 1H), 6.96 (d, J = 2.4 Hz, 1H), 6.83 (dd, J = 8.3, 2.5 Hz, 1H), 4.99 (d, J = 5.7 Hz, 1H), 3.80 (s, 3H), 2.90 (dd, J = 15.3, 7.5 Hz, 1H), 2.68 (dd, J = 15.3, 8.7 Hz, 1H), 2.45 (d, J = 8.7 Hz, 1H), 2.24–2.15 (m, 2H), 1.92–1.71 (m, 4H), 1.65 (m, 3H), 1.61–1.52 (m, 1H); 13C NMR (151 MHz, CDCl3) δ 159.3, 159.1, 146.3, 146.2, 135.3, 133.4, 125.8, 125.8, 125.6, 120.6, 120.5, 115.3, 114.9, 114.9, 109.8, 109.8, 108.7, 81.8, 81.8, 81.2, 81.1, 72.3, 72.2, 72.2, 55.6, 51.4, 51.3, 45.6, 45.6, 41.4, 41.3, 41.2, 41.2, 35.4, 35.4, 35.3, 35.3, 33.0, 33.0, 31.5, 31.5, 31.5, 31.5, 28.5, 28.5, 27.4, 27.3, 24.5, 24.4, 24.4, 24.3; IR (NaCl, thin film) νmax 3281, 2992, 2938, 2242, 1613 cm–1; HRMS (ESI) calcd for [C18H21NO2Na] ([M + Na]+) m/z 306.1465, found 306.1462.
Pyridinium p-toluenesulfonate (PPTS) (3.1 g, 12 mmol, 1.2 equiv) was added to a stirred solution of the crude indanol (2.9 g, 10 mmol, 1 equiv) in benzene (70 mL, 0.15 M), and the mixture was heated to 80 °C in a flask fitted with a reflux condenser for 24 h. The reaction mixture was cooled to room temperature, filtered through a pad of Celite, washed with EtOAc (200 mL), and concentrated. The crude yellow oil was purified by flash chromatography (9:1 hexanes/EtOAc) to yield 1.9 g (7.1 mmol, 71% yield) of indene 19 as a white solid. mp 59–60 °C; Rf 0.62 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.26 (d, J = 8.1 Hz, 1H), 6.85 (d, J = 2.4 Hz, 1H), 6.68 (dd, J = 8.1, 2.4 Hz, 1H), 6.51 (t, J = 1.7 Hz, 1H), 3.82 (s, 3H), 3.29–3.24 (m, 2H), 2.57 (t, J = 7.3 Hz, 2H), 2.40 (s, 1H), 2.03–1.88 (m, 2H), 1.87–1.74 (m, 2H), 1.65 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 159.1, 150.6, 146.8, 135.2, 127.2, 123.9, 120.4, 109.8, 106.1, 81.0, 72.3, 55.6, 40.6, 40.3, 31.4, 30.7, 27.3, 24.9; IR (NaCl, thin film) νmax 3283, 2929, 2241, 1615, 1578 cm–1; HRMS (ESI) calcd for [C18H20NO] ([M + H]+) m/z 266.1539, found 266.1539.
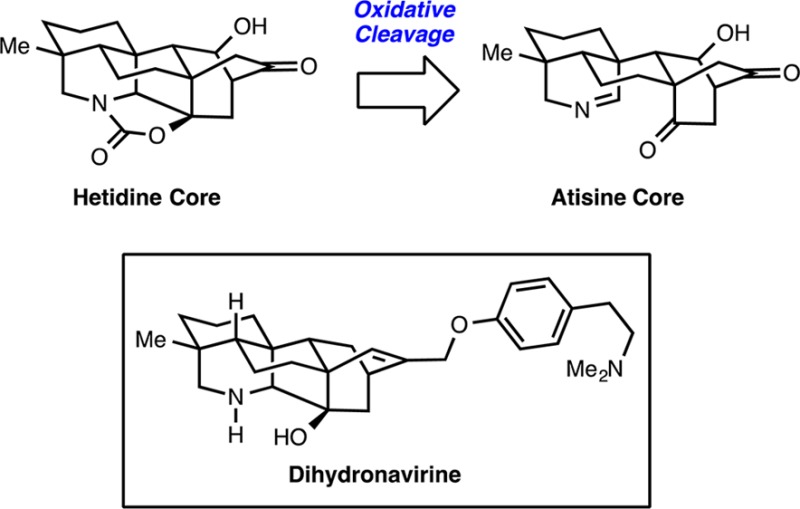
8-Methoxy-1-methyl-2,3,4,5-tetrahydro-1H-dibenzo[a,d][7]annulene-1-carbonitrile (18)
Indene 19 was dried by azeotrope with benzene (3 × 5 mL) and dried under vacuum for 12 h before use. Gallium(III) iodide (0.80 g, 1.8 mmol, 0.25 equiv) and 4 Å activated powdered molecular sieves (1.6 g, 2.0 g per gram of GaI3) were added to a Schlenk flask in a N2 drybox. The Schlenk flask was removed from the drybox, and a solution of indene 19 (1.9 g, 7.1 mmol, 1 equiv) in freshly distilled toluene (36 mL, 0.2 M) was added to the flask via cannula. The Schlenk flask was sealed, and the pale yellow mixture was heated to 100 °C and stirred vigorously for 48 h. The reaction mixture was cooled room to temperature, filtered through a pad of Celite, washed with EtOAc (200 mL), and concentrated. If the reaction mixture turns red in within the first few minutes of the reaction, it should be stopped immediately in order to minimize the decomposition of the material. The crude product was immediately purified by flash chromatography (9:1 hexanes/EtOAc) to yield 1.7 g (6.3 mmol, 89% yield) of cycloheptadiene 18 as a clear oil: Rf 0.62 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 7.05 (d, J = 6.8 Hz, 1H), 7.03 (d, J = 3.6 Hz, 1H), 6.89 (dd, J = 8.3, 2.6 Hz, 1H), 6.83 (d, J = 2.5 Hz, 1H), 6.63 (d, J = 11.7 Hz, 1H), 3.80 (s, 3H), 2.98–2.88 (m, 2H), 2.39 (qt, J = 18.8, 6.1 Hz, 2H), 2.08 (ddd, J = 12.3, 9.1, 2.6 Hz, 1H), 1.77 (dtd, J = 14.7, 5.8, 2.4 Hz, 1H), 1.72 (ddd, J = 13.2, 8.8, 2.4 Hz, 1H), 1.69–1.61 (m, 1H), 1.41 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 157.9, 137.3, 136.5, 133.1, 129.6, 128.7, 128.1, 126.9, 124.8, 115.2, 112.1, 55.5, 40.2, 35.5, 35.5, 31.3, 26.7, 19.0; IR (NaCl, thin film) νmax 3441, 2940, 2229, 1606 cm–1; HRMS (ESI) calcd for [C18H19NONa] ([M + Na]+) m/z 288.1359, found 288.1357.
8-Methoxy-1-methyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[a,d][7]annulene-1-carbonitrile (27)
p-Toluenesulfonyl hydrazide (11 g, 61 mmol, 8 equiv) and Et3N (17 mL, 120 mmol, 16 equiv) were added to a solution of cycloheptadiene 18 (2.0 g, 7.7 mmol, 1 equiv) in 1,2-dichloroethane (31 mL, 0.25 M). The reaction mixture was heated to 65 °C and stirred. After 24 h, the reaction was observed to have stalled. The mixture was filtered through a pad of Celite, washed with EtOAc (50 mL), and concentrated. The unreacted cycloheptadiene (18) and the product (27) were isolated together by column chromatography (100% hexanes to 4:1 hexanes/EtOAc) and resubmitted to the reaction conditions for another 24 h. The final product was isolated by flash chromatography (100% hexanes to 9:1 hexanes/EtOAc) to give 1.7 g (6.3 mmol, 82% yield) of cycloheptene 27 as a white solid: mp 63–68 °C; Rf 0.62 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 6.97 (d, J = 8.2 Hz, 1H), 6.71 (d, J = 2.7 Hz, 1H), 6.65 (dd, J = 8.2, 2.7 Hz, 1H), 3.79 (s, 3H), 3.59 (d, J = 16.5 Hz, 1H), 3.16 (ddd, J = 14.3, 10.9, 3.3 Hz, 1H), 3.08 (d, J = 16.6 Hz, 1H), 2.84 (ddd, J = 14.5, 7.5, 3.5 Hz, 1H), 2.61 (ddt, J = 17.1, 7.4, 2.5 Hz, 1H), 2.48–2.39 (m, 1H), 2.15 (tt, J = 5.8, 2.6 Hz, 2H), 2.06 (ddt, J = 11.7, 6.8, 3.0 Hz, 1H), 1.82–1.70 (m, 2H), 1.59 (dd, J = 23.9, 3.1 Hz, 1H), 1.39 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 158.5, 142.3, 134.6, 131.1, 129.3, 128.2, 124.9, 114.2, 110.7, 55.3, 39.6, 37.7, 36.1, 33.0, 32.2, 29.6, 25.3, 19.6; IR (NaCl, thin film) νmax 2936, 2227, 1609, 1503 cm–1; HRMS (EI) calcd for [C18H21NO] ([M]+) m/z 267.1623, found 267.1629.
8-Methoxy-1-methyl-5-oxo-2,3,4,5,10,11-hexahydro-1H-dibenzo[a,d][7]annulene-1-carbonitrile (28)
Ceric ammonium nitrate (CAN) (3.6 g, 3.4 mmol, 5 equiv), silica (5.3 g, 2.0 g per gram of CAN), and water (2.6 mL, 0.5 M) were added to a 50 mL beaker and stirred with a spatula until a uniform yellow powder formed. The beaker was cooled to 0 °C, a solution of cycloheptene 27 (350 mg, 1.3 mmol, 1 equiv) in CH2Cl2 (10 mL, 0.125 M) was added slowly, and the resulting orange slurry was vigorously stirred for 5 min with a spatula. The mixture was filtered through a pad of Celite, washed with CH2Cl2 (100 mL), and concentrated. The crude orange oil was purified by flash chromatography (9:1 hexanes/EtOAc) to give 190 mg (0.68 mmol, 52% yield) of enone 28 as a yellow solid: mp 105–106 °C; Rf 0.36 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 7.82 (d, J = 8.7 Hz, 1H), 6.76 (dd, J = 8.7, 2.6 Hz, 1H), 6.62 (d, J = 2.6 Hz, 1H), 3.79 (s, 3H), 3.13 (dd, J = 15.7, 11.2 Hz, 1H), 2.92 (dd, J = 15.8, 8.3 Hz, 1H), 2.75–2.66 (m, 1H), 2.61 (ddd, J = 17.0, 11.0, 2.5 Hz, 1H), 2.47 (dd, J = 18.3, 6.0 Hz, 1H), 2.38–2.28 (m, 1H), 2.16–2.07 (m, 1H), 1.79–1.64 (m, 3H), 1.50 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 194.0, 162.6, 143.9, 143.9, 138.6, 133.0, 130.6, 123.5, 113.5, 112.2, 55.3, 38.3, 35.6, 34.5, 30.7, 26.5, 24.8, 18.6; IR (NaCl, thin film) νmax 2942, 2230, 1626, 1596 cm–1; HRMS (ESI) calcd for [C18H20NO2] ([M + H]+) m/z 282.1489, found 282.1496.
(1R,4aS,11aS)-8-Methoxy-1-methyl-5-oxo-2,3,4,4a,5,10,11,11a-octahydro-1H-dibenzo[a,d][7]annulene-1-carbonitrile (29)
Palladium on carbon (10 wt %, 200 mg) was added to a stirred solution of enone 28 (720 mg, 2.6 mmol, 1 equiv) in EtOAc (26 mL, 0.1 M). The flask was evacuated under vacuum, backfilled with H2 (3×), and stirred under a balloon of H2 for 48 h. The reaction mixture was filtered through a pad of Celite, washed with EtOAc (50 mL), and concentrated to give 720 mg of ketone 29 (2.6 mmol, quantitative yield) as a white solid that was used without further purification. A single diastereomer (determined by 1H NMR) was isolated. Slow diffusion crystallization from benzene and hexanes provided X-ray quality crystals: mp 154–156 °C; Rf 0.25 (2:1 hexanes/EtOAc, UV); 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.7 Hz, 1H), 6.78 (dd, J = 8.9, 2.7 Hz, 1H), 6.70 (d, J = 2.7 Hz, 1H), 3.82 (s, 3H), 3.20–3.12 (m, 1H), 3.02 (ddd, J = 41.6, 16.2, 9.1 Hz, 2H), 2.51 (dt, J = 14.6, 7.4 Hz, 1H), 2.40 (ddd, J = 16.8, 10.0, 3.4 Hz, 1H), 2.31–2.17 (m, 1H), 2.05 (dt, J = 13.4, 3.9 Hz, 1H), 1.95 (dt, J = 11.7, 7.1 Hz, 1H), 1.77–1.62 (m, 2H), 1.47–1.36 (m, 2H), 1.35 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 199.8, 162.3, 146.4, 132.1, 131.1, 122.5, 115.2, 111.9, 55.4, 46.2, 44.3, 38.1, 37.8, 32.9, 29.7, 27.0, 26.5, 20.6; IR (NaCl, thin film) νmax 2941, 2232, 1670, 1600 cm–1; HRMS (ESI) calcd for [C18H21NO2Na] ([M + Na]+) m/z 306.1465, found 306.1470.
(1R,4aR,11aR)-8-Methoxy-1-methyl-5-oxo-4a-vinyl-2,3,4,4a,5,10,11,11a-octahydro-1H-dibenzo[a,d][7]annulene-1-carbonitrile (31)
Sodium hydride (60 wt % dispersion in mineral oil, 120 mg, 3.1 mmol, 5 equiv) was added to a Schlenk flask followed by a solution of ketone 29 (170 mg, 0.61 mmol, 1 equiv) and phenyl vinyl sulfoxide (80 μL, 0.61 mmol, 1 equiv) in dry DMF (6.0 mL, 0.1 M). The Schlenk flask was sealed and heated to 50 °C for 20 min. The reaction mixture was cooled to room temperature and quenched by dropwise addition of water until gas evolution ceased (∼0.5 mL). The reaction mixture was diluted with EtOAc (50 mL), washed with water (3 × 25 mL) and brine (1 × 25 mL), dried over MgSO4, and concentrated. The crude yellow oil was purified by flash chromatography (4:1 hexanes/EtOAc to 100% EtOAc) to give 230 mg (0.53 mmol, 87% yield) of sulfoxide 30 as a clear oil that was immediately carried on to the sulfoxide extrusion: Rf 0.07 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain).
Sulfoxide 30 (230 mg, 0.53 mmol, 1 equiv) was dried by azeotrope with benzene (1 × 10 mL) and transferred to a microwave vial with freshly distilled toluene (11 mL, 0.05 M). The solution was sparged with argon for 1 h, and then the reaction mixture was heated in the microwave to 160 °C for 2 h. The reaction mixture was diluted with EtOAc (75 mL), washed with water (3 × 50 mL) and brine (1 × 25 mL), dried over MgSO4, and concentrated. The crude yellow oil was purified by flash chromatography (4:1 hexanes/EtOAc) to give 115 mg (0.37 mmol, 70% yield) of ketone 31 as a white solid. Slow diffusion crystallization from benzene and hexanes provided X-ray quality crystals: mp 127–128 °C; Rf 0.47 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.7 Hz, 1H), 6.80 (dd, J = 8.8, 2.6 Hz, 1H), 6.63 (d, J = 2.6 Hz, 1H), 5.88 (dd, J = 17.6, 10.7 Hz, 1H), 4.93 (d, J = 10.8 Hz, 1H), 4.77 (d, J = 17.6 Hz, 1H), 3.82 (s, 3H), 3.31–3.20 (m, 1H), 3.07 (ddd, J = 16.7, 6.8, 2.3 Hz, 1H), 2.50 (tdd, J = 9.4, 8.2, 5.1 Hz, 1H), 2.21–2.09 (m, 2H), 2.02–1.94 (m, 3H), 1.67 (dddt, J = 13.9, 9.7, 6.0, 2.5 Hz, 1H), 1.56 (t, J = 13.4 Hz, 1H), 1.52–1.45 (m, 1H), 1.42 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 201.8, 162.0, 143.6, 143.1, 133.5, 131.5, 123.6, 114.9, 114.8, 112.2, 58.0, 55.4, 48.6, 38.8, 37.0, 33.6, 29.2, 26.5, 19.8; IR (NaCl, thin film) νmax 2938, 1662, 1599 cm–1; HRMS (ESI) calcd for [C20H23NO2Na] ([M + Na]+) m/z 332.1621, found 332.1623.
(1R,4aR,11aR)-4a-Allyl-8-methoxy-1-methyl-5-oxo-2,3,4,4a,5,10,11,11a-octahydro-1H-dibenzo[a,d][7]annulene-1-carbonitrile (32)
Sodium hydride (60 wt % dispersion in mineral oil, 0.51 g, 13 mmol, 5 equiv) was added to a Schlenk flask followed by a solution of ketone 29 (720 mg, 2.6 mmol, 1 equiv) and allyl bromide (0.22 mL, 2.6 mmol, 1 equiv) in dry DMF (25 mL, 0.1 M). The flask was sealed and heated to 50 °C for 45 min. The reaction mixture was cooled to room temperature and quenched by dropwise addition of water until bubbling ceased (∼0.5 mL). The reaction mixture was diluted with EtOAc (100 mL), washed with water (3 × 50 mL) and brine (1 × 25 mL), dried over MgSO4, and concentrated to give a 96:4 mixture of O-allylated product to C-allylated product 32 (based on 1H NMR) as a clear oil that was carried on crude: Rf 0.47 (2:1 hexanes/EtOAc, UV); 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 8.6 Hz, 1H), 6.79 (dd, J = 8.6, 2.7 Hz, 1H), 6.68 (d, J = 2.7 Hz, 1H), 5.93 (ddt, J = 17.3, 10.4, 5.8 Hz, 1H), 5.23–5.17 (m, 1H), 5.17–5.13 (m, 1H), 4.00 (dt, J = 5.8, 1.3 Hz, 2H), 3.82 (s, 3H), 2.92 (dt, J = 13.3, 6.4 Hz, 1H), 2.72–2.63 (m, 1H), 2.63–2.55 (m, 1H), 2.36 (dddd, J = 17.4, 14.0, 9.0, 5.4 Hz, 2H), 2.12 (dd, J = 12.5, 7.0 Hz, 1H), 2.00–1.77 (m, 4H), 1.54–1.46 (m, 1H), 1.32 (s, 3H).
The crude mixture of products was dried by azeotrope with benzene (1 × 10 mL) and transferred to a microwave vial with dry DMF (25 mL, 0.1 M). The reaction mixture was heated in the microwave to 160 °C for 45 min. The reaction mixture was diluted with EtOAc (100 mL), washed with water (3 × 50 mL) and brine (1 × 25 mL), dried over MgSO4, and concentrated. The crude yellow oil was purified by flash chromatography (4:1 hexanes/EtOAc) to give 570 mg (1.8 mmol, 69% over two steps) of C-allylated ketone 32 as a clear oil: Rf 0.33 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 7.55 (d, J = 8.7 Hz, 1H), 6.76 (dd, J = 8.7, 2.5 Hz, 1H), 6.64 (d, J = 2.5 Hz, 1H), 5.51 (dddd, J = 16.4, 9.4, 8.1, 6.7 Hz, 1H), 5.01 (dd, J = 10.0, 1.8 Hz, 1H), 4.85 (dd, J = 17.0, 2.0 Hz, 1H), 3.82 (s, 3H), 3.76 (ddd, J = 16.4, 13.8, 2.6 Hz, 1H), 2.78 (ddd, J = 16.4, 6.2, 2.5 Hz, 1H), 2.47 (ddt, J = 16.5, 13.6, 3.1 Hz, 1H), 2.43–2.29 (m, 3H), 2.29–2.21 (m, 1H), 2.09 (dd, J = 13.9, 8.0 Hz, 1H), 2.04 (dq, J = 12.9, 2.7 Hz, 1H), 1.72 (dt, J = 14.8, 4.1 Hz, 1H), 1.68–1.66 (m, 1H), 1.46 (s, 3H), 1.46–1.41 (m, 1H), 1.12 (td, J = 14.0, 13.5, 4.3 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 206.1, 161.8, 139.8, 133.7, 133.6, 132.1, 124.3, 119.2, 115.5, 111.8, 55.4, 54.7, 50.0, 45.1, 40.7, 36.0, 33.7, 32.4, 26.7, 24.1, 20.5; IR (NaCl, thin film) νmax 2946, 2227, 1666, 1601 cm–1; HRMS (ESI) calcd for [C21H26NO2] ([M + H]+) m/z 324.1958, found 324.1967.
(1R,4aR,11aR)-4a-Allyl-8-methoxy-1-methyl-5-oxo-2,3,4,4a,5,10,11,11a-octahydro-1H-dibenzo[a,d][7]annulene-1-carbonitrile (33)
LiAlH4 (330 mg, 8.8 mmol, 5 equiv, from Aldrich) was added to a flame-dried Schlenk flask followed by a solution of ketone 32 (570 mg, 1.8 mmol, 1 equiv) in THF (35 mL, 0.05 M) and heated to 65 °C for 4 h. The reaction mixture was cooled to room temperature, slowly quenched with powdered Na2SO4·10H2O (1.8 g, 0.2 g per mmol LiAlH4), and stirred until the gray color disappeared and the white precipitate clumped to the bottom of flask (∼12 h). The clear solution was filtered through a pad of Celite, washed with Et2O (75 mL), and concentrated to give an amino alcohol that was immediately subjected to the Boc-protection conditions: Rf 0.02 (100% EtOAc, UV and p-anisaldehyde stain).
Di-tert-butyl dicarbonate (Boc2O) (580 mg, 2.6 mmol, 1.5 equiv) and Et3N (0.32 mL, 2.3 mmol, 1.3 equiv) were added to a solution of the crude amino alcohol (580 mg, 1.8 mmol, 1 equiv) in CH2Cl2 (44 mL, 0.04 M). After 45 min, the solution was concentrated and the crude clear oil was purified by flash chromatography (4:1 hexanes/EtOAc) to give 700 mg (1.6 mmol, 93% yield over two steps) of Boc-protected amino alcohol 33 as a clear oil: Rf 0.47 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 7.01 (d, J = 8.0 Hz, 1H), 6.66–6.53 (m, 2H), 5.74 (dt, J = 17.1, 8.0 Hz, 1H), 5.00 (dd, J = 9.9, 2.6 Hz, 1H), 4.87 (d, J = 16.9 Hz, 1H), 4.72 (t, J = 6.4 Hz, 1H), 4.38 (s, 1H), 3.76 (s, 4H), 3.63 (d, J = 13.3 Hz, 1H), 3.35–3.18 (m, 2H), 2.68–2.55 (m, 1H), 2.36–1.96 (m, 4H), 1.93–1.69 (m, 5H), 1.43 (s, 9H), 1.10–0.91 (m, 5H).; 13C NMR (151 MHz, CDCl3) δ 159.0, 156.4, 142.9, 135.3, 132.9, 131.9, 117.8, 115.5, 110.3, 78.8, 55.2, 55.2, 50.5, 44.4, 44.0, 38.8, 34.3, 34.1, 31.7, 28.9, 28.6, 28.5, 22.8, 22.7, 18.8, 14.2; IR (NaCl, thin film) νmax 3459, 2930, 2248, 1701, 1608 cm–1; HRMS (ESI) calcd for [C26H39NO4Na] ([M + Na]+) m/z 452.2771, found 452.2780.
(1R,4aR,11aR)-tert-Butyl 4a-Allyl-8-methoxy-1-methyl-2,3,4,4a,5,10,11, 11a-octahydro-1H-5,1-(epiminomethano)dibenzo[a,d][7]annulene-12-carboxylate (34)
Boc-protected amino alcohol 33 was dried by azeotrope with benzene (1 × 10 mL) before use. A solution of Boc-protected amino alcohol 33 (430 mg, 1.0 mmol, 1 equiv) in CH2Cl2 (13 mL, 0.08 M) was added dropwise over 20 min to a stirred solution of SOCl2 (0.22 mL, 3.0 mmol, 3 equiv) in CH2Cl2 (13 mL, 0.08 M) and stirred at room temperature. After 1 h, water (10 mL) was added, and stirring was continued for an additional 15 min. The mixture was poured into water (40 mL) and extracted with CH2Cl2 (3 × 40 mL). The combined organics were washed with brine (1 × 20 mL), dried over MgSO4, and concentrated. The crude oil was purified by flash chromatography (9:1 hexanes/EtOAc) to give 300 mg (0.72 mmol, 72% yield) of 34 as a clear oil: Rf 0.62 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3, mixture of 2 rotamers) δ 7.31–7.27 (m), 7.08 (d, J = 8.1 Hz), 6.66–6.54 (m), 5.73 (dtd, J = 17.2, 7.2, 3.6 Hz), 5.02–4.94 (m), 4.89–4.81 (m), 4.86 (s) 4.60 (s), 3.76 (s), 3.75 (s), 3.72 (s), 3.65 (d, J = 13.9 Hz), 3.39–3.28 (m), 2.55 (tt, J = 16.6, 4.0 Hz), 2.08–2.00 (m), 1.91–1.80 (m), 1.80–1.68 (m), 1.64 (dd, J = 12.9, 5.5 Hz), 1.62–1.47 (m), 1.43 (s), 1.41 (s), 1.04 (s), 1.02 (s), (1.74:1 ratio of major to minor rotamer); major rotamer 1H NMR (600 MHz, CDCl3) δ 7.08 (d, J = 8.1 Hz, 1H), 6.66–6.54 (m, 2H), 5.73 (dtd, J = 17.2, 7.2, 3.6 Hz, 1H), 5.02–4.94 (m, 1H), 4.89–4.81 (m, 1H), 4.60 (s, 1H), 3.76 (s, 3H), 3.75–3.72 (m, 1H), 3.39–3.28 (m, 2H), 2.55 (tt, J = 16.6, 4.0 Hz, 1H), 2.08–2.00 (m, 2H), 1.91–1.80 (m, 3H), 1.80–1.68 (m, 2H), 1.64 (dd, J = 12.9, 5.5 Hz, 1H), 1.62–1.47 (m, 3H), 1.43 (s, 9H), 1.04 (s, 3H); minor rotamer 1H NMR (600 MHz, CDCl3) δ 7.31–7.27 (m, 1H), 6.66–6.54 (m, 2H), 5.73 (dtd, J = 17.2, 7.2, 3.6 Hz, 1H), 5.02–4.94 (m, 1H), 4.89–4.81 (m, 1H), 4.86 (s, 1H), 3.75 (s, 3H), 3.65 (d, J = 13.9 Hz, 1H), 3.39–3.28 (m, 2H), 2.55 (tt, J = 16.6, 4.0 Hz, 1H), 2.08–2.00 (m, 2H), 1.91–1.80 (m, 3H), 1.80–1.68 (m, 2H), 1.64 (dd, J = 12.9, 5.5 Hz, 1H), 1.62–1.47 (m, 3H), 1.41 (s, 9H), 1.02 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 158.5, 155.1, 141.5, 141.1, 134.8, 134.2, 134.0, 134.0, 133.2, 133.1, 117.9, 117.9, 117.0, 116.7, 109.9, 109.8, 79.8, 79.4, 65.5, 64.0, 55.12 55.14 53.0, 52.6, 45.8, 45.5, 45.1, 44.8, 43.9, 43.9, 39.7, 39.3, 38.5, 38.4, 34.6, 34.5, 33.7, 33.5, 28.8, 26.6, 26.5, 23.5, 21.3, 21.2; IR (NaCl, thin film) νmax 2931, 1684 cm–1; HRMS (ESI) calcd for [C26H38NO3] ([M + H]+) m/z 412.2846, found 412.2862.
(1R,4aR,11aR)-4-Nitrophenyl 4a-Allyl-8-methoxy-1-methyl-2,3,4,4a,5,10,11,11a-octahydro-1H-5,1-(epiminomethano)dibenzo[a,d][7]annulene-12-carboxylate (35)
Trifluoroacetic acid (1.3 mL, 0.2 M) was added to a stirred solution of Boc-protected amine 34 (110 mg, 0.26 mmol, 1 equiv) in CH2Cl2 (1.3 mL, 0.2 M) at room temperature. After 30 min, 1 N NaOH was slowly added (10 mL), and the mixture was extracted with CH2Cl2 (3 × 15 mL). The combined organics were washed with brine (1 × 10 mL), dried over MgSO4, and concentrated. The crude amine was purified by flash chromatography (9:1 CH2Cl2/MeOH, 1% NH4OH) to provide 64 mg (0.21 mmol, 81% yield) of the free amine as a clear oil: Rf 0.50 (9:1 CH2Cl2/MeOH 1% NH4OH, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 6.96 (d, J = 8.2 Hz, 1H), 6.68–6.64 (m, 1H), 6.61 (dd, J = 8.2, 2.8 Hz, 1H), 5.74 (ddt, J = 17.2, 10.0, 7.3 Hz, 1H), 4.99–4.95 (m, 1H), 4.83 (ddt, J = 16.9, 2.4, 1.3 Hz, 1H), 3.79 (s, 1H), 3.77 (s, 3H), 3.48 (td, J = 15.9, 14.2, 4.0 Hz, 1H), 3.32 (dd, J = 12.4, 2.9 Hz, 1H), 2.72 (d, J = 12.5 Hz, 1H), 2.62 (dt, J = 16.6, 4.1 Hz, 1H), 2.54 (dq, J = 19.5, 7.2 Hz, 1H), 2.24 (s, 1H), 2.12 (tt, J = 14.9, 3.7 Hz, 1H), 2.07–1.99 (m, 1H), 1.85 (d, J = 7.3 Hz, 2H), 1.72 (dd, J = 13.7, 5.9 Hz, 1H), 1.67–1.53 (m, 2H), 1.47–1.44 (m, 1H), 1.43–1.39 (m, 1H), 0.95 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 158.5, 141.4, 137.0, 134.4, 132.9, 117.6, 117.6, 110.7, 67.9, 55.3, 53.2, 46.0, 44.6, 44.5, 39.5, 38.5, 35.6, 34.1, 27.0, 23.5, 22.9; IR (NaCl, thin film) νmax 3353, 2919, 1606 cm–1; HRMS (ESI) calcd for [C21H30NO] ([M + H]+) m/z 312.2322, found 312.2332.
Triethylamine (5.0 μL, 0.035 mmol, 1.5 equiv) and 4-nitrobenzoyl chloride (4.7 mg, 0.025 mmol, 1.1 equiv) were added to a stirred solution of the free amine (7.2 mg, 0.023 mmol, 1 equiv) in CH2Cl2 (0.46 mL, 0.05 M) at room temperature. After 1 h, the solution was diluted with CH2Cl2 (5 mL), washed with 1 N HCl (1 × 1 mL), saturated NaHCO3 (1 × 1 mL), and brine (1 × 1 mL), dried over MgSO4, and concentrated. The resulting white solid was purified by flash chromatography (4:1 hexanes/EtOAc) and slow evaporation of the column eluent provided X-ray quality crystals of 35: mp 88–89 °C; Rf 0.47 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3, mixture of 2 rotamers) δ 8.30 (dd, J = 7.3, 1.6 Hz), 8.23 (dd, J = 7.3, 1.5 Hz), 7.44 (d, J = 8.1 Hz), 7.41 (dd, J = 6.9, 1.8 Hz), 7.37 (d, J = 7.8 Hz), 6.70–6.67 (m), 6.67 (d, J = 2.7 Hz), 6.61 (d, J = 2.7 Hz), 6.28 (dd, J = 8.4, 2.7 Hz), 5.76 (ddt, J = 17.3, 10.1, 7.3 Hz), 5.61–5.57 (m), 5.55 (s), 5.53 (d, J = 8.4 Hz), 5.04 (dd, J = 10.2, 2.2 Hz), 4.97 (dd, J = 10.2, 2.1 Hz), 4.90 (dq, J = 16.7, 1.5 Hz), 4.81 (dq, J = 16.9, 1.6 Hz), 4.46 (s), 4.17 (d, J = 14.9 Hz), 3.79 (s), 3.73 (s), 3.67 (dd, J = 13.5, 2.4 Hz), 1.55–1.51 (m), 3.53 (dd, J = 14.9, 2.6 Hz), 3.46–3.33 (m), 3.14 (d, J = 13.5 Hz), 2.72 (dt, J = 16.7, 3.9 Hz), 2.59 (dt, J = 16.4, 4.0 Hz), 2.18 (tt, J = 15.1, 3.8 Hz), 2.13–1.99 (m), 1.94 (d, J = 7.4 Hz), 1.92–1.88 (m), 1.87–1.82 (m), 1.77 (p, J = 7.6 Hz), 1.75–1.68 (m), 1.67–1.58 (m), 1.57–1.55 (m), 1.51–1.43 (m), 1.43–1.36 (m), 1.31–1.27 (m), 1.26–1.23 (m), 1.16 (s), 0.95 (s), 0.88 (t, J = 7.3 Hz). (1.6:1 ratio of major to minor rotamer); 13C NMR (151 MHz, CDCl3) δ 169.8, 168.0, 159.1, 158.8, 148.1, 148.1, 144.1, 144.0, 141.8, 140.9, 135.3, 133.5, 133.0, 133.0, 131.9, 131.5, 127.4, 124.0, 123.8, 118.8, 118.5, 117.8, 117.6, 110.5, 109.9, 68.9, 62.7, 56.9, 55.3, 52.2, 45.9, 45.7, 44.6, 44.1, 43.8, 43.6, 39.2, 39.1, 39.1, 38.4, 36.8, 35.0, 34.7, 34.4, 34.3, 33.6, 26.5, 26.2, 23.4, 23.2, 22.5, 21.6, 21.5, 14.2; IR (NaCl, thin film) νmax 2919, 1606, 1500 cm–1; HRMS (ESI) calcd for [C28H33N2O4] ([M + H]+) m/z 461.2435, found 461.2441.
(1R,4aR,11aR)-tert-Butyl 8-Methoxy-1-methyl-4a-(2-oxoethyl)-2,3,4,4a,5,10,11,11a-octahydro-1H-5,1-(epiminomethano)dibenzo[a,d][7]annulene-12-carboxylate (36)
Osmium(IV) tetraoxide (2.5 wt % in t-butanol, 0.10 mL, 0.008 mmol, 0.01 equiv) was added to a stirred solution of 34 (340 mg, 0.84 mmol, 1 equiv) in 2:1 acetone/H2O (21 mL, 0.04 M). After 5 min, 4-methylmorpholine N-oxide (NMO, 200 mg, 1.7 mmol, equiv) was added. The reaction mixture was stirred at room temperature for 18 h, diluted with EtOAc (100 mL), washed with water (1 × 50 mL) and brine (1 × 50 mL), dried over MgSO4, and concentrated. Silica plug (10% MeOH in CH2Cl2) provided 350 mg (0.79 mmol, 94% yield) of the diol that was immediately carried onto the oxidative cleavage: Rf 0.13 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain).
NaIO4 on silica38 (1.6 g, 2 g/mmol diol) was added to a stirred solution of the diol (350 mg, 0.79 mmol, 1 equiv) in CH2Cl2 (8 mL, 0.1 M). After 1 h, the mixture was filtered through a pad of Celite, washed with CH2Cl2 (30 mL), and concentrated to give 320 mg (0.77 mmol, 97% yield) of aldehyde 36 as a clear oil that was used without further purification: Rf 0.43 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 9.72 (dd, J = 3.4, 1.9 Hz), 9.68 (dd, J = 2.8, 1.9 Hz), 7.27–7.24 (m), 7.04 (d, J = 8.3 Hz), 6.61–6.55 (m), 5.05 (s), 4.86 (s), 3.73 (s), 3.73–3.71 (m), 3.65 (d, J = 14.0 Hz), 3.38–3.29 (m), 2.54 (tt, J = 16.1, 4.0 Hz), 2.24–2.17 (m), 2.14–2.04 (m), 2.03–1.83 (m), 1.66–1.57 (m), 1.57–1.50 (m), 1.40 (s), 1.39 (s), 1.03 (s), 1.02 (s), (1.75:1 ratio of major to minor rotamer); ajor rotamer 1H NMR (600 MHz, CDCl3) δ 9.68 (dd, J = 2.8, 1.9 Hz, 1H), 7.04 (d, J = 8.3 Hz, 1H), 6.61–6.55 (m, 2H), 4.86 (s, 1H), 3.75–3.71 (m, 1H), 3.73 (s, 3H), 3.38–3.29 (m, 2H), 2.54 (tt, J = 16.1, 4.0 Hz, 1H), 2.24–2.17 (m, 1H), 2.14–2.04 (m, 2H), 2.03–1.83 (m, 2H), 1.66–1.57 (m, 3H), 1.57–1.50 (m, 3H), 1.40 (s, 9H), 1.03 (s, 3H); minor rotamer 1H NMR (600 MHz, CDCl3) δ 9.72 (dd, J = 3.4, 1.9 Hz, 1H), 7.27–7.24 (m, 1H), 6.61–6.55 (m, 2H), 5.05 (s, 1H), 3.72 (s, 3H), 3.65 (d, J = 14.0 Hz, 1H), 3.38–3.29 (m, 2H), 2.54 (tt, J = 16.1, 4.0 Hz, 1H), 2.24–2.17 (m, 1H), 2.14–2.04 (m, 2H), 2.03–1.83 (m, 2H), 1.66–1.57 (m, 3H), 1.57–1.50 (m, 3H), 1.39 (s, 9H), 1.02 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 202.5, 202.1, 158.7, 158.6, 155.0, 154.5, 141.1, 140.8, 134.5, 134.0, 132.7, 132.5, 117.3, 117.0, 110.2, 110.0, 80.2, 79.7, 64.3, 63.3, 55.1, 55.1, 52.9, 52.9, 52.7, 52.5, 46.0, 45.9, 44.7, 44.5, 40.2, 40.1, 39.3, 39.3, 34.3, 34.1, 33.5, 33.3, 28.7, 26.6, 26.5, 23.8, 23.8, 21.0, 21.0; IR (NaCl, thin film) νmax 2931, 1719, 1683, 1607 cm–1; HRMS (ESI) calcd for [C25H36NO4] ([M + H]+) m/z 414.2639, found 414.2648.
(1R,4aR,11aR)-tert-Butyl 8-Methoxy-1-methyl-4a-(2-(phenylthio)ethyl)-2,3,4,4a,5,10,11,11a-octahydro-1H-5,1-(epiminomethano)dibenzo[a,d][7]annulene-12-carboxylate (S1)
Sodium borohydride (29 mg, 0.77 mmol, 1 equiv) was added to a stirred solution of aldehyde 36 (320 mg, 0.77 mmol, 1 equiv) in MeOH (30 mL, 0.025 M) at 0 °C. The reaction mixture was allowed to slowly warm to room temperature and stirred for 1 h. The reaction was quenched by slow addition of saturated aq NH4Cl (10 mL) and then concentrated to a volume of 20 mL. The heterogeneous mixture was diluted with water (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organics were washed with brine (1 × 20 mL), dried over MgSO4, and concentrated to 300 mg (0.72 mmol, 94% yield) of alcohol 37 as a clear oil that was immediately used without further purification: Rf 0.29 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain).
Alcohol 37 (300 mg, 0.72 mmol, 1 equiv) was dried by azeotrope with benzene (3 × 5 mL). Diphenyl disulfide (0.79 g, 3.6 mmol, 5 equiv) was added to the flask, and the headspace was evacuated and backfilled with N2 (3×). Acetonitrile (15 mL, 0.05 M) was then added followed by a dropwise addition of tributylphosphine (0.9 mL, 3.6 mmol, 5 equiv). The reaction mixture was allowed to stir at room temperature for 18 h, 1 N NaOH (10 mL) was added, and the mixture was stirred for 30 min. The mixture was then poured into 1 N NaOH (100 mL) and extracted with EtOAc (3 × 75 mL). The combined organics were washed with 1 N NaOH (1 × 75 mL) and brine (1 × 75 mL), dried over MgSO4, and concentrated. The crude sulfide was purified by flash chromatography (9:1 hexanes/EtOAc) to give 370 mg (0.72 mmol, 100% yield) of sulfide S1 as a clear oil: Rf 0.57 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (600 MHz, CDCl3) δ 7.35–7.31 (m), 7.17–7.06 (m), 7.01–6.97 (m), 6.96–6.92 (m), 6.64 (dd, J = 8.3, 2.8 Hz), 6.61–6.55 (m), 4.94 (s), 4.65 (s), 3.80 (s), 3.79 (s), 3.74 (d, J = 14.1 Hz), 3.65 (d, J = 13.9 Hz), 3.35–3.24 (m), 2.86–2.77 (m), 2.55–2.44 (m), 2.01–1.93 (m), 1.93–1.81 (m), 1.76–1.66 (m), 1.64–1.48 (m), 1.44 (s), 1.41 (s), 1.02 (s), 1.01 (s), (1.75:1 ratio of major to minor rotamer); major rotamer 1H NMR (600 MHz, CDCl3) δ 7.35–7.31 (m, 1H), 7.17–7.06 (m, 3H), 7.01–6.97 (m, 2H), 6.64 (dd, J = 8.3, 2.8 Hz, 1H), 6.61–6.55 (m, 1H), 4.65 (s, 1H), 3.80 (s, 3H), 3.74 (d, J = 14.1 Hz, 1H), 3.35–3.24 (m, 2H), 2.86–2.77 (m, 2H), 2.55–2.44 (m, 1H), 2.01–1.93 (m, 2H), 1.93–1.81 (m, 3H), 1.76–1.66 (m, 1H), 1.64–1.48 (m, 5H), 1.44 (s, 9H), 1.02 (s, 3H); minor rotamer 1H NMR (600 MHz, CDCl3) δ 7.17–7.06 (m, 4H), 6.96–6.92 (m, 2H), 6.61–6.55 (m, 2H), 4.94 (s, 1H), 3.79 (s, 3H), 3.65 (d, J = 13.9 Hz, 1H), 3.35–3.24 (m, 2H), 2.86–2.77 (m, 2H), 2.01–1.93 (m, 2H), 1.93–1.81 (m, 3H), 1.76–1.66 (m, 1H), 1.64–1.48 (m, 5H), 2.55–2.44 (m, 1H), 1.41 (s, 9H), 1.01 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 158.6, 158.5, 155.0, 154.7, 141.5, 136.4, 134.5, 134.0, 132.7, 132.6, 129.0, 128.9, 128.8, 128.7, 125.8, 125.6, 117.3, 117.0, 110.2, 110.1, 80.0, 79.6, 64.8, 63.1, 55.2, 55.2, 52.9, 52.5, 46.7, 46.1, 44.9, 44.7, 39.6, 39.5, 39.3, 38.9, 38.7, 38.6, 34.6, 34.4, 33.6, 33.4, 28.8, 28.8, 27.5, 26.6, 26.5, 23.3, 23.2, 21.2, 21.2; IR (NaCl, thin film) νmax 2930, 2871, 1738, 1684, 1606 cm–1; HRMS (ESI) calcd for [C31H42NO3S] ([M + H]+) m/z 508.2880, found 508.2899.
(1R,4aR,11aR)-tert-Butyl 8-Methoxy-1-methyl-4a-vinyl-2,3,4,4a,5,10,11,11a-octahydro-1H-5,1-(epiminomethano)dibenzo[a,d][7]annulene-12-carboxylate (39)
Scandium(III) triflate (71 mg, 0.14 mmol, 0.2 equiv) was added to a stirred solution of hydrogen peroxide (30 wt %, 0.82 mL, 7.2 mmol, 10 equiv) in 9:1 CH2Cl2/MeOH (35 mL, 0.02 M). After the mixture was stirred at room temperature for 5 min, sulfide S1 (370 mg, 0.72 mmol, 1 equiv) in 9:1 CH2Cl2/MeOH (35 mL, 0.02 M) was added. After 18 h, the reaction mixture was poured into water (100 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organics were washed with brine (1 × 100 mL), dried over MgSO4, and concentrated to give sulfoxide 38 that was immediately used without further purification: Rf 0.07 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain).
Crude sulfoxide 38 was dried by azeotrope with benzene (1 × 20 mL) and transferred to two separate microwave vials with freshly distilled toluene (29 mL, 0.025 M). The solutions were sparged with argon for 2 h and then heated in the microwave to 180 °C for 2 h. The reaction mixtures were recombined and concentrated. Flash chromatography (9:1 hexanes/EtOAc) provided 190 mg (0.47 mmol, 65% yield over two steps) of vinyl compound 39 as a clear oil: Rf 0.62 (2:1 hexanes/EtOAc, UV and p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 8.3 Hz), 7.10 (d, J = 8.1 Hz), 6.62 (dd, J = 8.3, 2.7 Hz), 6.61–6.55 (m), 5.67–5.56 (m), 5.03 (s), 4.94–4.83 (m), 4.74 (s), 3.78 (s), 3.76 (s), 3.75 (s), 3.74 (s), 3.66 (d, J = 13.8 Hz), 3.40–3.27 (m), 2.58–2.46 (m), 2.17 (tt, J = 14.8, 3.5 Hz), 2.07–1.97 (m), 1.96–1.80 (m), 1.68–1.52 (m), 1.43 (s), 1.42 (s), 1.07 (s), 1.05 (s), (1.88:1 ratio of major to minor rotamer); major rotamer 1H NMR (500 MHz, CDCl3) δ 7.10 (d, J = 8.1 Hz, 1H), 6.61–6.55 (m, 2H), (dd, J = 17.8, 11.3 Hz, 1H), 4.94–4.85 (m, 2H), 4.74 (s, 1H), 3.76 (d, J = 19.7 Hz, 1H), 3.76 (s, 3H), 3.40–3.27 (m, 2H), 2.58–2.46 (m, 1H), 2.17 (tt, J = 14.8, 3.5 Hz, 1H), 2.07–1.97 (m, 1H), 1.96–1.80 (m, 2H), 1.68–1.52 (m, 5H), 1.43 (s, 9H), 1.07 (s, 3H); minor rotamer 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 8.3 Hz, 1H), 6.62 (dd, J = 8.3, 2.7 Hz, 1H), 6.61–6.55 (m, 1H), 5.63 (dd, J = 17.9, 11.3 Hz, 1H), 5.03 (s, 1H), 4.91–4.85 (m, 2H), 3.75 (s, 3H), 3.66 (d, J = 13.8 Hz, 1H), 3.40–3.27 (m, 2H), 2.58–2.46 (m, 1H), 2.17 (tt, J = 14.8, 3.5 Hz, 1H), 2.07–1.97 (m, 1H), 1.96–1.80 (m, 2H), 1.68–1.52 (m, 5H), 1.42 (s, 9H), 1.05 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 158.4, 158.2, 155.0, 154.5, 145.5, 145.4, 141.4, 141.1, 134.9, 134.3, 133.7, 133.6, 116.8, 116.6, 112.9, 112.7, 110.0, 109.9, 79.9, 79.5, 64.8, 63.4, 55.1, 55.1, 52.7, 52.3, 46.5, 46.4, 44.9, 44.7, 42.1, 42.1, 42.0, 42.0, 34.5, 34.4, 33.7, 33.5, 28.7, 26.6, 26.4, 24.2, 24.1, 21.4, 21.4; IR (NaCl, thin film) νmax 2931, 1684, 1607 cm–1; HRMS (ESI) calcd for [C25H36NO3] ([M + H]+) m/z 398.2690, found 398.2705.
(4R,4aR,10aR,13bR)-13b-Allyl-4-methyl-2,3,4,4a,5,6-hexahydro-1H-4,13-methanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazole-8,12(13aH,13bH)-dione (45)
Sodium hydride (60 wt % dispersion in mineral oil, 52 mg, 1.3 mmol, 10 equiv) was added to a microwave vial, washed with hexanes (1 × 1 mL), and dried under vacuum. Dry DMF (2.5 mL, 0.05 M) was added, and the vial was cooled to 0 °C. Ethanethiol (0.10 mL, 1.3 mmol, 10 equiv) was added dropwise, and the solution was warmed to room temperature and stirred until the solution turned clear (∼5 min), at which time 34 (54 mg, 0.13 mmol, 1 equiv) in dry DMF (2.5 mL, 0.05 mmol) was added. The reaction mixture was heated in the microwave to 180 °C for 15 min. The mixture was diluted with EtOAc (50 mL), washed with water (3 × 15 mL) and brine (1 × 10 mL), dried over MgSO4, and concentrated. The crude oil was purified by flash chromatography (4:1 hexanes/EtOAc) to give 42 mg (0.11 mmol, 85% yield) of 45 as a clear oil: Rf 0.47 (2:1 hexanes/EtOAc, p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3, mixture of 2 rotamers) δ 7.00 (dd, J = 10.3, 7.9 Hz), 6.92 (s), 6.70 (s), 6.59 (d, J = 2.4 Hz), 6.57 (s), 6.15 (dd, J = 8.2, 2.6 Hz), 5.71 (dp, J = 16.5, 8.9, 8.2 Hz), 5.00 (dd, J = 10.3, 2.4 Hz,), 4.95 (dd, J = 9.9, 2.5 Hz), 4.87 (d, J = 16.9 Hz), 4.83–4.77 (m), 4.58 (s), 3.70 (d, J = 13.9 Hz), 3.66 (d, J = 13.9 Hz), 3.35 (ddd, J = 19.3, 13.8, 2.6 Hz), 3.30–3.19 (m), 2.51 (dt, J = 16.1, 4.0 Hz), 2.41 (dt, J = 16.3, 3.9 Hz), 2.00 (dt, J = 11.4, 3.8 Hz), 1.82 (dtd, J = 30.2, 13.9, 7.4 Hz), 1.72–1.60 (m), 1.59–1.47 (m), 1.43 (d, J = 2.7 Hz), 1.37–1.28 (m), 1.26 (q, J = 5.3, 3.6 Hz), 1.02 (s), 1.00 (s); major rotamer 1H NMR (500 MHz, CDCl3) δ 7.00 (d, J = 10.3 Hz, 1H), 6.70 (s, 1H), 6.57 (s, 2H) 5.71 (dp, J = 16.5, 8.9, 8.2 Hz, 1H), 5.00 (dd, J = 10.3, 2.4 Hz, 1H), 4.87 (d, J = 16.9 Hz, 1H), 4.58 (s, 1H), 3.70 (d, J = 13.9 Hz, 1H), 3.35 (ddd, J = 19.3, 13.8, 2.6 Hz, 1H), 3.30–3.19 (m, 1H), 2.41 (dt, J = 16.3, 3.9 Hz, 1H), 2.00 (dt, J = 11.4, 3.8 Hz, 2H), 1.82 (dtd, J = 30.2, 13.9, 7.4 Hz, 4H), 1.72–1.60 (m, 1H), 1.59–1.47 (m, 3H), 1.43 (s, 9H), 1.37–1.28 (m, 1H), 1.00 (s, 3H); minor rotamer 1H NMR (500 MHz, CDCl3) δ 7.00 (d, J = 7.9 Hz, 1H), 6.92 (s, 1H), 6.59 (d, J = 2.4 Hz, 1H), 6.15 (dd, J = 8.2, 2.6 Hz, 1H), 5.71 (dp, J = 16.5, 8.9, 8.2 Hz, 1H), 4.95 (dd, J = 9.9, 2.5 Hz, 1H), 4.83–4.77 (m, 2H), 3.66 (d, J = 13.9 Hz, 1H), 3.35 (ddd, J = 19.3, 13.8, 2.6 Hz, 1H), 3.30–3.19 (m, 1H), 2.51 (dt, J = 16.1, 4.0 Hz, 1H), 2.00 (dt, J = 11.4, 3.8 Hz, 2H), 1.82 (dtd, J = 30.2, 13.9, 7.4 Hz, 3H), 1.72–1.60 (m, 2H), 1.59–1.47 (m, 3H), 1.43 (s, 9H), 1.26 (q, J = 5.3, 3.6 Hz, 1H), 1.02 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 155.7, 155.2, 155.1, 154.8, 141.4, 141.2, 135.9, 134.3, 134.0, 132.5, 131.9, 118.3, 118.0, 117.9, 112.4, 112.0, 80.5, 78.0, 65.6, 64.3, 53.0, 52.8, 45.6, 45.3, 45.0, 44.8, 43.8, 43.8, 39.7, 39.2, 38.5, 38.4, 34.4, 34.2, 33.7, 33.5, 28.8, 28.8, 26.6, 26.4, 23., 23.4, 21.3, 21.2; IR (NaCl, thin film) νmax 3339, 2928, 1651, 1583 cm–1; HRMS (ESI) calcd for [C25H35NO3Na] ([M + Na]+) m/z 420.2509, found 420.2519.
(4R,4aR,10aR,13bR)-13b-Allyl-4-methyl-2,3,4,4a,5,6-hexahydro-1H-4,13-methanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazole-8,12(13aH,13bH)-dione (47)
A solution of phenol 45 (20 mg, 0.050 mmol, 1 equiv) in a 1:1 mixture of acetonitrile/2,2,2-trifluoroethanol (2.5 mL, 0.02 M) was added dropwise over 20 min to a stirred solution of bis(trifluoroacetoxy)iodobenzene (PIFA) (32 mg, 0.075 mmol, 1.5 equiv) and NaHCO3 (21 mg, 0.25 mmol, 5 equiv) in acetonitrile/2,2,2-trifluoroethanol (2.5 mL, 0.02 M) at 0 °C and stirred for an additional 1 h. The reaction mixture was poured into brine (10 mL), extracted with EtOAc (3 × 20 mL), dried over MgSO4, and concentrated. The crude product was purified by flash chromatography (4:1 hexanes/EtOAc) to give 9.1 mg (0.027 mmol, 54% yield) of quinol 47 as a clear oil: Rf 0.20 (2:1 hexanes/EtOAc, UV and KMnO4 stain); 1H NMR (500 MHz, CDCl3) δ 6.85 (d, J = 10.0 Hz, 1H), 6.36 (dd, J = 10.0, 1.9 Hz, 1H), 6.11 (s, 1H), 5.62 (ddt, J = 17.1, 10.1, 7.2 Hz, 1H), 5.08 (dd, J = 10.1, 1.9 Hz, 1H), 4.97 (dd, J = 16.9, 2.1 Hz, 1H), 3.92 (s, 1H), 3.27–3.14 (m, 2H), 2.50 (dt, J = 14.5, 4.5 Hz, 1H), 2.42 (dd, J = 13.8, 6.9 Hz, 1H), 2.28–2.15 (m, 2H), 1.84 (dd, J = 13.9, 7.6 Hz, 1H), 1.76 (tt, J = 14.4, 3.9 Hz, 1H), 1.68–1.55 (m, 1H), 1.46 (dq, J = 9.6, 2.9, 2.5 Hz, 3H), 1.40–1.32 (m, 1H), 1.27–1.15 (m, 2H), 1.07 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 185.2, 158.7, 157.6, 144.8, 132.1, 131.9, 129.1, 119.7, 80.1, 66.9, 51.4, 44.5, 44.0, 40.6, 38.6, 38.4, 33.5, 32.5, 28.2, 25.8, 19.4; IR (NaCl, thin film) νmax 2926, 1756, 1671 cm–1; HRMS (ESI) calcd for [C21H26NO3] ([M + H]+) m/z 340.1907, found 340.1914. Alternative procedure: A solution of phenol 45 (160 mg, 0.40 mmol, 1 equiv) in a 1:1 mixture of acetonitrile/2,2,2-trifluoroethanol (8 mL, 0.05 M) was added dropwise over 5 min to a stirred solution of iodosobenzene (200 mg, 0.88 mmol, 2.2 equiv) in acetonitrile/2,2,2-trifluoroethanol (8 mL, 0.05 M) at room temperature and stirred for an additional 30 min. The cloudy mixture was filtered through a pad of Celite, washed with EtOAc (20 mL), and concentrated. Flash chromatography (4:1 hexanes/EtOAc) gave 73 mg (0.22 mmol, 55% yield) of quinol 47 as a clear oil.
2-((4R,4aR,10aR,13bR)-4-Methyl-8,12-dioxo-2,3,4,4a,5,6,8,12,13a,13b-decahydro-1H-4,13-methanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazol-13b-yl)acetaldehyde (48)
4-Methylmorpholine N-oxide (28 mg, 0.24 mmol, 2 equiv) and OsO4 (2.5 wt % in tert-butyl alcohol, 15 μL, 0.0012 mmol, 0.01 equiv) was added to a stirred solution of 47 (41 mg, 0.12 mmol, 1 equiv) in a 2:1 mixture of THF/H2O (4 mL, 0.03 M). The mixture was stirred at room temperature for 24 h, at which time the reaction mixture was diluted with EtOAc (20 mL), washed with water (1 × 10 mL) and brine (1 × 5 mL), dried over MgSO4, and concentrated. The resulting diol was filtered through a pad of silica with 9:1 CH2Cl2/MeOH (10 mL), concentrated, and used without any further purification: Rf 0.05 (100% EtOAc, UV and KMnO4 stain).
NaIO4 on silica (240 mg, 2.0 g/mmol diol) was added to a stirred solution of the diol (45 mg, 0.12 mmol, 1 equiv) in CH2Cl2 (1.2 mL, 0.1 M). After 30 min, the mixture was filtered through a pad of Celite, washed with CH2Cl2 (10 mL), and concentrated. The crude aldehyde was purified by flash chromatography (1:1 hexanes/EtOAc) to give 25 mg (0.070 mmol, 58% yield over two steps) of aldehyde 48 as a clear oil: Rf 0.14 (1:1 hexanes/EtOAc, UV and KMnO4 stain); 1H NMR (500 MHz, CDCl3) δ 9.63 (t, J = 1.5 Hz, 1H), 6.81 (d, J = 10.0 Hz, 1H), 6.33 (dd, J = 10.1, 1.8 Hz, 1H), 6.14 (d, J = 2.0 Hz, 1H), 4.20 (s, 1H), 3.29–3.18 (m, 2H), 2.67–2.55 (m, 2H), 2.55–2.48 (m, 1H), 2.34–2.19 (m, 2H), 1.99 (dq, J = 13.1, 2.7 Hz, 1H), 1.78 (ddt, J = 14.4, 12.6, 3.7 Hz, 1H), 1.72–1.57 (m, 4H), 1.56–1.42 (m, 2H), 1.35–1.22 (m, 2H), 1.11 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 199.7, 184.8, 158.3, 156.9, 145.4, 131.8, 129.7, 79.8, 65.3, 51.2, 48.9, 46.3, 43.9, 40.7, 38.6, 33.5, 32.3, 28.4, 26.1, 19.4; IR (NaCl, thin film) νmax 2929, 2873, 1755, 1719, 1671, 1636 cm–1; HRMS (ESI) calcd for [C20H24NO4] ([M + H]+) m/z 342.1700, found 342.1708.
(4R,4aR,6aS,10aR,13bS,14S)-4-Methyl-8,12-dioxo-2,3,4,4a,5,6,7,8,12,13a-decahydro-1H-4,13:6a,13b-dimethanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazole-14-carbaldehyde (49)
Silica (86 mg, 10 mg per 1 mg 3.52) was added to a stirred solution of aldehyde 48 (8.6 mg, 0.025 mmol, 1 equiv) in CH2Cl2 (0.5 mL, 0.05 M). After 36 h, the mixture was filtered through a pad of Celite, washed with EtOAc (20 mL), and concentrated to give 6.9 mg (0.020 mmol, 80% yield) of 49 as 2.5:1 mixture of epimers α to the aldehyde (based in 1H NMR) as a clear oil that was used without further purification: Rf 0.16 (major), 0.15 (minor) (1:1 hexanes/EtOAc, UV and KMnO4 stain); major epimer 1H NMR (600 MHz, CDCl3) δ 9.88 (d, J = 1.7 Hz, 1H), 6.59 (d, J = 10.1 Hz, 1H), 6.19 (d, J = 10.0 Hz, 1H), 3.66 (d, J = 14.2 Hz, 1H), 3.63 (s, 1H), 3.00 (d, J = 14.1, 1H), 2.60 (d, J = 16.8, 1H), 2.58 (d, J = 16.8. 1H), 2.27 (t, J = 2.1 Hz, 1H), 2.10–2.03 (m, 1H), 1.89–1.81 (m, 3H), 1.81–1.77 (m, 1H), 1.74 (dddd, J = 14.7, 12.3, 6.3, 3.2 Hz, 2H), 1.68 (dq, J = 13.8, 4.6 Hz, 1H), 1.48–1.39 (m, 3H), 1.03 (s, 3H); minor epimer 1H NMR (600 MHz, CDCl3) δ 9.62 (d, J = 5.3 Hz, 1H), 6.73 (d, J = 10.1 Hz, 1H), 6.21 (d, J = 10.1 Hz, 1H), 4.10 (s, 1H), 3.72 (d, J = 14.3 Hz, 1H), 3.01 (d, J = 14.1 Hz, 1H), 2.43 (d, J = 18.0, 1H), 2.28 (d, J = 5.4 Hz, 1H), 2.20 (dd, J = 14.3, 9.3 Hz, 1H), 2.10–2.03 (m, 1H), 1.89–1.81 (m, 3H), 1.81–1.77 (m, 1H), 1.74 (dddd, J = 14.7, 12.3, 6.3, 3.2 Hz, 1H), 1.68 (dq, J = 13.8, 4.6 Hz, 2H), 1.48–1.39 (m, 3H),1.05 (s, 3H); major epimer 13C NMR (151 MHz, CDCl3) δ 200.6, 196.2, 158.7, 144.9, 130.6, 81.8, 67.1, 60.4, 49.1, 47.8, 45.6, 43.4, 41.3, 40.5, 36.4, 35.6, 28.6, 27.6, 20.1, 19.9; minor epimer 13C NMR (151 MHz, CDCl3) δ 200.8, 195.1, 158.8, 144.4, 132.5, 82.4, 70.5, 67.9, 48.8, 46.1, 44.8, 42.7, 41.1, 36.1, 35.9, 33.2, 29.9, 27.8, 20.5, 19.8; IR (NaCl, thin film) νmax 2924, 1755, 1714 cm–1; HRMS (ESI) calcd for [C20H24NO4] ([M + H]+) m/z 342.1700, found 342.1710.
(4R,4aR,6aS,10aR,13bS,14S)-4-Methyl-8,12-dioxododecahydro-1H-4,13:6a,13b-dimethanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazole-14-carbaldehyde (50)
Rhodium on alumina (5 wt %, 9.5 mg) was added to a solution of enone 49 (19 mg, 0.055 mmol, 1 equiv) in benzene (1.8 mL, 0.03 M). The flask was evacuated under vacuum, backfilled with hydrogen (3×), and stirred vigorously under a balloon of hydrogen for 24 h. The reaction mixture was filtered through a pad of Celite, washed with EtOAc (25 mL), and concentrated. The crude ketone was purified by flash chromatography (1:1 hexanes/EtOAc) to obtain 10 mg (0.030 mmol, 55% yield) of ketone 50 as a clear oil: Rf 0.38 (100% EtOAc, KMnO4 stain); 1H NMR (500 MHz, CDCl3) δ 9.92 (d, J = 2.1 Hz, 1H), 3.65 (d, J = 14.2 Hz, 1H), 3.43 (s, 1H), 2.96 (d, J = 14.2 Hz, 1H), 2.77–2.68 (m, 1H), 2.57 (d, J = 15.6 Hz, 1H), 2.38–2.29 (m, 3H), 2.27 (t, J = 2.2 Hz, 1H), 2.17–2.07 (m, 2H), 1.87–1.65 (m, 5H), 1.44 (ddd, J = 13.2, 4.1, 2.3 Hz, 2H), 1.28 (dddd, J = 26.8, 13.7, 11.7, 6.6 Hz, 3H), 1.02 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 208.3, 201.5, 159.0, 84.4, 66.8, 62.8, 51.2, 48.8, 47.9, 46.1, 41.3, 40.3, 36.7, 35.7, 35.6, 34.4, 29.8, 27.7, 20.2, 20.0; IR (NaCl, thin film) νmax 2924, 1747 cm–1; HRMS (ESI) calcd for [C20H26NO4] ([M + H]+) m/z 344.1856, found 344.1861.
(4R,4aR,6aS,10aR,13bS,14S)-15-Hydroxy-4-methyloctahydro-1H-6a,13b,9-(epiethane[1,1,2]triyl)-4,13-methanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazole-8,12(7H,13aH)-dione (14)
Potassium carbonate (30 mg, 0.23 mmol, 10 equiv) was added to a stirred solution of keto-aldehyde 50 (8.0 mg, 0.023 mmol, 1 equiv) in 9:1 MeOH/CH2Cl2 (2.3 mL, 0.01 M). After 4 h at room temperature, the mixture was poured into water (10 mL), extracted with CH2Cl2 (3 × 10 mL), dried over MgSO4, and concentrated. The crude product was purified by flash chromatography (100% EtOAc) to give 6.3 mg (0.018 mmol, 78% yield) of hetidine core 14 as a white solid. Slow diffusion crystallization from benzene and hexanes provided X-ray quality crystals: mp 160 °C dec; Rf 0.42 (100% EtOAc, KMnO4 stain); 1H NMR (600 MHz, CDCl3) δ 4.43 (d, J = 4.1 Hz, 1H), 3.56 (d, J = 14.1 Hz, 1H), 3.40 (s, 1H), 2.88 (d, J = 14.1 Hz, 1H), 2.61–2.54 (m, 2H), 2.46 (s, 1H), 2.23 (d, J = 14.4, 1H), 2.15 (d, J = 19.8 Hz, 1H), 2.09 (dd, J = 14.5, 5.1 Hz, 1H), 2.03–1.98 (m, 1H), 1.95 (dd, J = 14.7, 9.3 Hz, 1H), 1.81–1.71 (m, 2H), 1.66–1.60 (m, 2H), 1.55 (t, J = 2.2 Hz, 1H), 1.52 (s, 1H), 1.48–1.40 (m, 2H), 1.32 (td, J = 13.4, 4.5 Hz, 1H), 1.16–1.07 (m, 1H), 1.04 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 210.7, 158.6, 85.5, 70.5, 69.7, 58.0, 51.3, 48.6, 45.5, 44.9, 42.6, 41.8, 41.4, 36.5, 34.95, 31.9, 30.0, 28.1, 20.5, 20.3; IR (NaCl, thin film) νmax 3399, 2923, 1735 cm–1; HRMS (ESI) calcd for [C20H26NO4] ([M + H]+) m/z 344.1856, found 344.1867.
(4R,4aR,6aS,10aR,13bS,14S)-4-Methyloctahydro-1H-6a,13b,9-(epiethane[1,1,2]triyl)-4,13-methanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazole-8,12(7H,13aH)-dione (51)
Alcohol 14 (66 mg, 0.19 mmol, 1 equiv) was added to a flame-dried round-bottom flask with THF (19 mL, 0.01 M). The flask was cooled to 0 °C, and NaH (60 wt %, 76 mg, 1.9 mmol, 10 equiv) was added in one portion. The suspension was stirred at 0 °C for 1 h, and then CS2 (0.75 mL, 13 mmol, 66 equiv) was added dropwise. The mixture was stirred at 0 °C for 1 h then MeI (0.38 mL, 6.1 mmol, 32 equiv) was added dropwise. The mixture was stirred at 0 °C for 1 h, quenched dropwise with water (1.0 mL), and then warmed to room temperature. The mixture was diluted with brine (30 mL), extracted with ethyl acetate (3 × 30 mL), dried over MgSO4, and concentrated. The resulting xanthate (S2) was purified by flash chromatography (2:1 hexanes/EtOAc) to give 59 mg (0.14 mmol, 74% yield) of the xanthate as a white solid: mp 123–124 °C; Rf 0.30 (1:1 hexanes/EtOAc, KMnO4 stain); 1H NMR (600 MHz, CDCl3) δ 6.28 (dd, J = 4.1, 1.4 Hz, 1H), 3.56 (d, J = 14.1 Hz, 1H), 3.49 (s, 1H), 2.93–2.90 (m, 1H), 2.89 (d, J = 14.2 Hz, 1H), 2.64 (d, J = 20.0 Hz, 1H), 2.50 (s, 3H), 2.27 (dd, J = 14.6, 1.2 Hz, 1H), 2.22 (d, J = 19.9 Hz, 1H), 2.19 (dd, J = 14.7, 5.2 Hz, 1H), 2.05–2.02 (m, 1H), 1.99 (dd, J = 14.8, 9.5 Hz, 1H), 1.81–1.72 (m, 3H), 1.63–1.56 (m, 2H), 1.45 (ddt, J = 13.6, 4.2, 2.1 Hz, 1H), 1.38 (td, J = 13.5, 4.1 Hz, 1H), 1.28 (td, J = 13.5, 4.5 Hz, 1H), 1.25–1.23 (m, 1H), 1.14 (ddt, J = 15.5, 11.5, 9.7 Hz, 1H), 1.03 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 214.4, 207.5, 158.3, 85.2, 79.7, 69.5, 55.6, 48.5, 46.9 45.9, 44.9, 43.0, 41.8, 41.3, 36.5, 34.7, 32.6, 29.8, 28.0, 20.3, 20.3, 19.1; FTIR (NaCl, thin film) νmax 2925, 1747 cm–1; HRMS (EI) calcd for [C22H27NO4S2] ([M]+) m/z 433.1382, found 433.1380.
The xanthate (S2, 59 mg, 0.14 mmol, 1 equiv) was added to a round-bottom flask and dried via azeotrope with benzene (1 × 1 mL). AIBN (5.7 mg, 0.035 mmol, 0.25 equiv) was added followed by toluene (5.6 mL, 0.025 M). The flask was evacuated and backfilled with nitrogen (3×), and nBu3SnH (1.0 M in cyclohexane, 0.25 mL, 0.25 mmol, 1.8 equiv) was added. The mixture was heated to 95 °C under nitrogen. After 1 h, the reaction mixture was cooled to room temperature and concentrated. The crude product was purified by flash chromatography (100% hexanes then 2:1 hexanes/EtOAc then 100% EtOAc) to give 40 mg (0.12 mmol, 86% yield) of 51 as a clear oil: Rf 0.59 (100% EtOAc, KMnO4 stain); 1H NMR (500 MHz, CDCl3) δ 3.56 (d, J = 14.4 Hz, 1H) 3.55 (s, 1H), 2.87 (d, J = 14.0 Hz, 1H), 2.56 (d, J = 19.7 Hz, 1H), 2.37 (td, J = 4.4, 2.5 Hz, 1H), 2.34–2.27 (m, 1H), 2.21 (dd, J = 14.2, 5.2 Hz, 1H), 2.08 (dt, J = 15.4, 3.3 1H), 2.05 (d, J = 19.8 Hz, 1H), 1.99–1.84 (m, 3H), 1.74 (dtd, J = 14.8, 6.3, 5.2, 3.8 Hz, 2H), 1.63–1.56 (m, 1H), 1.51–1.40 (m, 3H), 1.25 (dtd, J = 20.5, 13.5, 4.4 Hz, 3H), 1.11 (ddt, J = 15.4, 11.5, 9.7 Hz, 1H), 1.02 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 212.9, 158.7, 85.9, 69.5, 48.5, 47.6, 46.4, 44.7, 43.1, 42.6, 42.2, 41.4, 38.3, 36.5, 32.3, 30.1, 28.0, 25.6, 20.6, 20.3; FTIR (NaCl, thin film) νmax 2924, 1744 cm–1; HRMS (ESI) calcd for [C20H26O3N] ([M + H]+) m/z 328.1907, found 328.1911.
(4R,4aR,6aR,10aR,13bS,14S)-4-Methyl-12-oxo-2,3,4,4a,5,6,9,10,12,13a-decahydro-1H-6a,13b,9-(epiethane[1,1,2]triyl)-4,13-methanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazol-8-yl Trifluoromethanesulfonate (53)
Ketone 51 (40 mg, 0.12 mmol, 1 equiv) was added to a round-bottom flask and dried via azeotrope with benzene (1 × 1 mL). THF (3.0 mL, 0.04 M) was added, the flask was cooled to −78 °C, and LiHMDS (1.0 M in THF, 0.25 mL, 0.25 mmol, 2 equiv) was added. After 1 h, N-(5-chloro-2-pyridyl)bis(trifluoromethanesulfonimide) (52, 94 mg, 0.12 mmol, 2 equiv) in THF (3.0 mL, 0.04 M) was added, and the reaction mixture was stirred at −78 °C for an additional 3 h. The reaction mixture was quenched with saturated aq NaHCO3 (3 mL) at −78 °C and warmed to room temperature. The mixture was diluted with water (20 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organics were washed with 10% aq NaOH (1 × 20 mL) and brine (1 × 20 mL), dried over MgSO4, and concentrated. The crude vinyl triflate was purified by flash chromatography (4:1 hexanes/EtOAc) to give 46 mg (0.10 mmol, 83% yield) of vinyl triflate 53 as a white solid: mp 192–193 °C; Rf 0.50 (1:1 hexanes/EtOAc, KMnO4 stain); 1H NMR (500 MHz, CDCl3) δ 5.45 (d, J = 2.8 Hz, 1H), 3.56 (d, J = 14.1 Hz, 1H), 3.51 (d, J = 1.2 Hz, 1H), 2.86 (d, J = 14.1 Hz, 1H), 2.67 (tt, J = 2.9, 1.6 Hz, 1H), 2.29 (dd, J = 14.5, 9.5 Hz, 1H), 2.13–2.09 (m, 1H), 2.08–2.03 (m, 1H), 1.92–1.84 (m, 1H), 1.84–1.76 (m, 4H), 1.74–1.69 (m, 1H), 1.60 (dd, J = 9.1, 4.8 Hz, 2H), 1.42 (dt, J = 4.2, 2.5 Hz, 1H), 1.33–1.23 (m, 3H), 1.15 (ddt, J = 15.4, 11.2, 9.5 Hz, 1H), 1.03 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 158.7, 154.3, 119.8, 118.7 (q, JC–F = 321 Hz, CF3) 87.0, 69.4, 50.2, 50.0, 48.5, 46.5, 44.8, 41.5, 41.4, 36.5, 34.4, 34.0, 29.0, 28.3, 28.1, 20.7, 20.2; FTIR (NaCl, thin film) νmax 2924, 1748, 1673 cm–1; HRMS (ESI) calcd for [C21H25O5NF3S] ([M + H]+) m/z 460.1400, found 460.1409.
(4R,4aR,6aS,10aR,13bS,14S)-8-(Hydroxymethyl)-4-methyl-2,3,4,4a,5,6,9,10-octahydro-1H-6a,13b,9-(epiethane[1,1,2]triyl)-4,13-methanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazol-12(13aH)-one (55)
Triflate 53 (13 mg, 0.028 mmol, 1 equiv) was added to a 4 mL vial and dried via azeotrope with benzene (1 × 0.5 mL). Flame-dried lithium chloride (3.6 mg, 0.084 mmol, 3 equiv) and Pd(PPh3)4 (3.3 mg, 0.0028 mmol, 0.1 equiv) were added to the vial followed by a solution of (tributylstannyl)methanol39 (54, 27 mg, 0.086 mmol, 3 equiv) in THF (0.6 mL, 0.05 M). The vial was sealed with a Teflon cap and heated to 70 °C for 1 h. The reaction mixture was concentrated, and the crude allylic alcohol was purified by flash chromatography (2:1 hexanes/EtOAc) to give 9.2 mg (0.027 mmol, 96% yield) of allylic alcohol 55 as a white solid: mp 204–207 °C; Rf 0.13 (1:1 hexanes/EtOAc, KMnO4 stain); 1H NMR (500 MHz, CDCl3) δ 5.47 (q, J = 1.7 Hz, 1H), 4.25–4.20 (m, 2H), 3.56 (d, J = 14.0 Hz, 1H), 3.49 (s, 1H), 2.85 (d, J = 14.0 Hz, 1H), 2.49 (dd, J = 4.4, 2.4 Hz, 1H), 2.24 (dd, J = 14.0, 9.4 Hz, 1H), 1.96 (dd, J = 12.4, 4.4 Hz, 1H), 1.88–1.83 (m, 1H), 1.82–1.76 (m, 1H), 1.76–1.73 (m, 1H), 1.71 (q, J = 2.4 Hz, 1H), 1.69 (dd, J = 5.0, 2.5 Hz, 1H), 1.62 (ddd, J = 17.9, 8.8, 4.1 Hz, 1H), 1.45–1.35 (m, 4H), 1.35–1.31 (m, 1H), 1.29–1.21 (m, 3H), 1.19–1.09 (m, 1H), 1.03 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 159.2, 149.3, 126.9, 87.9, 69.9, 63.2, 51.0, 48.6, 47.7, 46.1, 45.2, 41.7, 41.5, 36.6, 33.9, 31.8, 28.9, 28.2, 27.4, 20.8, 20.4; FTIR (NaCl, thin film) νmax 3427, 2924, 1731 cm–1; HRMS (ESI) calcd for [C21H28O3N] ([M + H]+) m/z 342.2064, found 342.2069.
(4R,4aR,6aS,10aR,13bS,14S)-8-((4-(2-(Dimethylamino)ethyl)phenoxy)methyl)-4-methyl-2,3,4,4a,5,6,9,10-octahydro-1H-6a,13b,9-(epiethane[1,1,2]triyl)-4,13-methanodibenzo[2,3:6,7]cyclohepta[1,2-d]oxazol-12(13aH)-one (57)
Triethylamine (28 μL, 0.20 mmol, 2 equiv) was added to a stirred solution of allylic alcohol 55 (35 mg, 0.10 mmol, 1 equiv) in CH2Cl2 (3.0 mL, 0.03 M) followed by methanesulfonyl chloride (12 μL, 0.15 mmol, 1.5 equiv). The reaction mixture was stirred at 0 °C for 15 min and then filtered through a pad of Celite and washed with 10 mL (1:1 hexanes/EtOAc) while still cold. The resulting mesylate was concentrated and immediately carried on to the next reaction. If the reaction was allowed to warm above 0 °C before filtration through silica the allylic chloride was obtained as the major product: allylic mesylate Rf 0.15 (9:1 CH2Cl2/MeOH, 1% aq NH4OH, p-anisaldehyde stain); allylic chloride Rf 0.50 (9:1 CH2Cl2/MeOH, 1% aq NH4OH, p-anisaldehyde stain);
Sodium hydride (60 wt % dispersion in mineral oil, 4.0 mg, 0.10 mmol, 1 equiv) was added to a stirred solution of the mesylate (41 mg, 0.10 mmol, 1 equiv) and hordenine (56, 16 mg, 0.10 mmol, 1 equiv) in THF (4.0 mL, 0.025 M). After 1 h at room temperature, wet CH2Cl2 (1 mL) was added, and the reaction mixture was concentrated. Flash chromatography (19:1 CH2Cl2/MeOH, 1% NH4OH) provided 24 mg (0.050 mmol, 50% yield) of 57 as a white solid. The product 57 can also be obtained from the allylic chloride in diminished yields using the same conditions reported above except the reaction must be heated to 70 °C for 1 h: mp 132–137 °C; Rf 0.13 (9:1 CH2Cl2/MeOH, 1% aq NH4OH, p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.10 (dd, J = 6.7, 1.9 Hz, 2H), 6.82 (dd, J = 6.5, 2.0 Hz, 2H), 5.59 (d, J = 2.0 Hz, 1H), 4.56 (t, J = 1.9 Hz, 2H), 3.56 (d, J = 14.0 Hz, 1H), 3.49 (s, 1H), 2.85 (d, J = 13.9 Hz, 1H), 2.76–2.70 (m, 2H), 2.61–2.57 (m, 1H), 2.57–2.50 (m, 2H), 2.32 (s, 6H), 2.24 (dd, J = 14.1, 9.4 Hz, 1H), 1.94 (dd, J = 12.5, 4.3 Hz, 1H), 1.88–1.81 (m, 2H), 1.80–1.66 (m, 3H), 1.60 (tdd, J = 13.8, 9.4, 4.0 Hz, 1H), 1.45–1.39 (m, 3H), 1.36 (td, J = 10.6, 2.4 Hz, 1H), 1.25 (tdd, J = 13.4, 9.2, 4.4 Hz, 3H), 1.19–1.08 (m, 1H), 1.02 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 159.2, 157.3, 145.5, 132.5, 129.7, 129.5, 114.8, 87.9, 69.9, 68.1, 61.7, 50.8, 48.6, 47.8, 45.8, 45.4, 45.1, 41.6, 41.5, 36.5, 33.8, 33.3, 31.7, 28.8, 28.2, 27.3, 20.8, 20.4; FTIR (NaCl, thin film) νmax, 2925, 1748 cm–1; HRMS (ESI) calcd for [C31H41O3N2] ([M + H]+) m/z 489.3116, found 489.3112.
(1R,4aS,5aR,9aS,11aR,12S)-8-((4-(2-(Dimethylamino)ethyl)phenoxy)methyl)-1-methyl-2,3,4,5,5a,6,7,10,11,11a-decahydro-1H-4a,9a,7-(epiethane[1,1,2]triyl)-5,1-(epiminomethano)dibenzo[a,d][7]annulen-5a-ol (8)
Barium hydroxide octahydrate (28 mg, 0.09 mmol, 10 equiv) and cyclic carbamate 57 (4.4 mg, 0.009 mmol, 1 equiv) were added to a microwave vial. The vial was sealed, and EtOH (95%, 0.6 mL, 0.015 M) was added followed by water (0.3 mL, 0.03 M). The headspace was evacuated and backfilled with N2 (3×), and then the reaction mixture was heated in the microwave to 160 °C for 90 min. The reaction mixture was concentrated, and the crude amino alcohol was purified by flash chromatography (19:1 CH2Cl2/MeOH, 1% NH4OH) to provide 3.7 mg (0.008 mmol, 89% yield) of amino alcohol 8 as a clear oil: Rf 0.03 (9:1 CH2Cl2:MeOH, 1% aq NH4OH, p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.09 (dd, J = 6.3, 2.1 Hz, 2H), 6.84 (dd, J = 6.6, 2.0 Hz, 2H), 5.66 (d, J = 2.0 Hz, 1H), 4.57–4.49 (m, 2H), 2.99 (d, J = 12.1 Hz, 1H), 2.82 (s, 1H), 2.79 (d, J = 12.1 Hz, 1H), 2.73 (dd, J = 10.0, 6.3 Hz, 2H), 2.53 (dd, J = 10.2, 6.1 Hz, 2H), 2.45 (s, 1H), 2.32 (s, 6H), 2.13 (dd, J = 12.9, 7.4 Hz, 1H), 1.88 (d, J = 13.4 Hz, 1H), 1.66 (ddt, J = 28.8, 17.3, 4.6 Hz, 4H), 1.49–1.38 (m, 3H), 1.34–1.17 (m, 9H), 0.87 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 157.6, 145.1, 132.4, 131.6, 129.6, 115.0, 78.2, 68.8, 67.4, 61.9, 51.0, 50.5, 50.4, 46.6, 45.6, 45.1, 42.5, 40.0, 35.5, 33.9, 33.6, 32.2, 30.3, 28.1, 27.7, 21.8, 21.2; FTIR (NaCl, thin film) νmax 3329, 2925, 2853, 1511 cm–1; HRMS (ESI) calcd for [C30H43N2O2] ([M + H]+) m/z 463.3319, found 463.3313.
(4bS,8R,8aR)-2-((4-(2-(Dimethylamino)ethyl)phenoxy)methyl)-8-methyl-3,4,4a,5,6,7,8,8a,9,10-decahydro-3,10a-ethano-8,4b-(methanoazenometheno)phenanthren-11-one (60)
Iodosobenzene (3.3 mg, 0.015 mmol, 1.5 equiv) was added to a stirred suspension of amino alcohol 8 (4.6 mg, 0.010 mmol, 1 equiv) and NaHCO3 (2.5 mg, 0.03 mmol, 3 equiv) in CH2Cl2 (1.0 mL, 0.01 M) at room temperature. After 1 h, the reaction mixture as filtered through a pad of Celite, washed with CH2Cl2 (5 mL), and concentrated. Flash chromatography (19:1 CH2Cl2/MeOH, 1% aq NH4OH) provided 3.4 mg (0.0075 mmol, 75% yield) of 60 as a clear oil: Rf 0.07 (9:1 CH2Cl2/MeOH, 1% aq NH4OH, p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.56 (d, J = 2.8 Hz, 1H), 7.11 (d, J = 8.1 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 5.81 (d, J = 2.0 Hz, 1H), 4.55 (d, J = 1.6 Hz, 2H), 3.46–3.35 (m, 2H), 3.04 (q, J = 2.8 Hz, 1H), 2.81–2.75 (m, 2H), 2.65–2.55 (m, 2H), 2.42 (dd, J = 9.5, 3.0 Hz, 2H), 2.37 (s, 6H), 2.18 (d, J = 2.7 Hz, 2H), 1.94 (dd, J = 8.1, 2.7 Hz, 2H), 1.73–1.59 (m, 3H), 1.52–1.41 (m, 2H), 1.34–1.17 (m, 3H), 1.06 (ddd, J = 19.8, 11.9, 6.1 Hz, 2H), 0.83 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 211.2, 162.5, 157.1, 145.6, 131.5, 129.7, 114.9, 68.4, 61.7, 60.9, 52.7, 48.2, 47.4, 45.5, 42.3, 42.1, 35.3, 33.7, 33.4, 33.3, 29.9, 28.7, 27.0, 26.0, 20.7, 20.3; FTIR (NaCl, thin film) νmax 3399, 2927, 1715, 1652, 1512 cm–1; HRMS (ESI) calcd for [C30H41N2O2] ([M + H]+) m/z 461.3163, found 461.3166.
(1R,4aS,5aR,9aS,11aR,12S)-5a,13-Dihydroxy-1-methyldecahydro-1H-4a,9a,7-(epiethane[1,1,2]triyl)-5,1-(epiminomethano)dibenzo[a,d][7]annulen-8(2H)-one (61)
Barium hydroxide octahydrate (85 mg, 0.27 mmol, 5 equiv) and cyclic carbamate 14 (18 mg, 0.053 mmol, 1 equiv) were added to a microwave vial. The vial was sealed, and EtOH (95%, 6.6 mL, 0.008 M) was added followed by water (3.3 mL, 0.016 M). The headspace was evacuated and backfilled with N2 (3×), and then the reaction mixture was heated in the microwave to 160 °C for 30 min. The reaction mixture was concentrated, and the crude amino alcohol was purified by flash chromatography (9:1 CH2Cl2/MeOH, 1% aq NH4OH) to provide 9.7 mg (0.031 mmol, 58% yield) of amino diol 61 as a white solid: mp 226–229 °C; Rf 0.03 (9:1 CH2Cl2/MeOH, 1% aq NH4OH, p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 4.38 (d, J = 4.2 Hz, 1H), 2.99 (d, J = 12.1 Hz, 1H), 2.78 (d, J = 12.1 Hz, 1H), 2.67 (s, 1H), 2.52 (d, J = 19.8 Hz, 1H), 2.46 (t, J = 4.6 Hz, 1H), 2.07 (d, J = 19.8 Hz, 1H), 2.07–2.02 (m, 1H), 1.89 (d, J = 14.3 Hz, 1H), 1.80–1.74 (m, 1H), 1.74–1.61 (m, 3H), 1.48–1.37 (m, 3H), 1.36 (s, 1H), 1.33–1.23 (m, 4H), 0.86 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 213.7, 75.6, 70.7, 67.2, 57.8, 52.0, 45.0, 44.6, 43.6, 42.4, 42.2, 40.8, 40.4, 33.7, 33.4, 31.3, 28.0, 21.4, 21.0; FTIR (NaCl, thin film) νmax 3350, 2913, 1712 cm–1; HRMS (ESI) calcd for [C19H28NO3] ([M + H]+) m/z 318.2064, found 318.2061.
(4bS,8R,8aR)-4-Hydroxy-8-methyl-2,3,4,4a,6,7,8,8a,9,10-decahydro-3,10a-ethano-8,4b-(methanoazenometheno)phenanthrene-1,12(5H)-dione (63)
Iodosobenzene (4.0 mg, 0.018 mmol, 1.5 equiv) was added to a stirred suspension of amino alcohol 61 (3.7 mg, 0.012 mmol, 1 equiv) and NaHCO3 (3.0 mg, 0.036 mmol, 3 equiv) in CH2Cl2 (1.2 mL, 0.01 M) at room temperature. After 30 min, the reaction mixture as filtered through a pad of Celite, washed with CH2Cl2 (5 mL), and concentrated. Flash chromatography (19:1 CH2Cl2/MeOH, 1% aq NH4OH) provided 3.0 mg (0.009 mmol, 75% yield) of 63 as a white solid. Slow diffusion of isopropyl ether into 3:1 ethanol/acetonitrile provided X-ray quality crystals: mp 245–246 °C; Rf 0.33 (9:1 CH2Cl2/MeOH, 1% NH4OH, p-anisaldehyde stain); 1H NMR (500 MHz, CDCl3) δ 7.59 (s, 1H), 4.68 (t, J = 3.8 Hz, 1H), 3.47–3.34 (m, 2H), 2.92 (q, J = 3.1 Hz, 1H), 2.63 (dd, J = 19.9, 2.8 Hz, 1H), 2.48 (dd, J = 20.0, 3.2 Hz, 1H), 2.38–2.27 (m, 2H), 2.18 (dt, J = 13.4, 3.0 Hz, 1H), 1.99–1.90 (m, 1H), 1.78 (d, J = 3.9 Hz, 1H), 1.67–1.61 (m, 1H), 1.61–1.56 (m, 1H), 1.48 (t, J = 12.7 Hz, 1H), 1.36–1.23 (m, 3H), 1.20 (dd, J = 12.8, 3.5 Hz, 1H), 1.16 (s, 1H), 1.05 (td, J = 13.3, 4.1 Hz, 1H), 0.86 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 209.8, 209.2, 164.0, 68.4, 60.0, 55.7, 54.0, 49.2, 48.6, 46.7, 43.7, 42.1, 37.1, 34.3, 33.1, 29.8, 25.8, 20.4 20.0.; FTIR (NaCl, thin film) νmax 3400, 2926, 1726 cm–1; HRMS (ESI) calcd for [C19H26NO3] ([M + H]+) m/z 316.1907, found 316.1894.
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS RO1 084906). D.L. thanks the FQNRT Canada for a postdoctoral fellowship. We thank A. DiPasquale for solving the crystal structures of 29, 31, 35, 14, and 63 (supported by NIH Shared Instrumentation Grant No. S10-RR027172).
Supporting Information Available
Details on general experimental methods; copies of 1H and 13C NMR spectra for compounds 31, 36, S1, 39, S2, 51, 53, 55, 57, 8, 60, 61, and 63. X-ray crystal structures of 31 and 63 (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For a review on the diterpenoid alkaloids, see:; a Wang F.-P.; Liang X.-T. In The Alkaloids: Chemistry and Biology; Cordell G. A., Ed.; Elsevier Science: New York, 2002; 59, 1. [DOI] [PubMed] [Google Scholar]; b Wang F.-P.; Chen Q.-H.; Liang X.-T. Nat. Prod. Rep. 2010, 27, 529. [DOI] [PubMed] [Google Scholar]
- a Gao X.; Zhu J.; Yang Y. M.; et al. Zhonghua Xinxueguanbing Zazhi 2007, 35, 151.17445412 [Google Scholar]; b Huang X.; Yang Y.; Zhu J.; Xu D.; Peng J.; Liu J. J. Cardiovasc. Pharmacol. 2012, 59, 77. [DOI] [PubMed] [Google Scholar]; c Huang X.; Yang Y.; Zhu J.; Dai Y.; Pu J. Basic Clin. Pharmacol. Toxicol. 2009, 104, 145. [DOI] [PubMed] [Google Scholar]
- For lappaconitine, see:; a Mashkovskii M. D. Lek. Sredstva, Torsing, Khar′kov 1998, 1, 380. [Google Scholar]; (in Russian).; b Dzhakhangirov F. N.; Sultankhodzhaev M. N.; Tashkodzhaev B.; Salimov B. T. Khim. Prir. Soedin. 1997, 33, 254. [Google Scholar]; Chem. Nat. Compd. 1997, 33, 190. (Engl. Transl.).
- For guan-fu base A, see:; a Yang X. J.; Wang G. J.; Ling S. S.; Qiu N. Y.; Wang G. G.; Zhu J.; Liu J. H. J. Chromatogr. B. Biomed. Sci. Appl. 2000, 740, 273. [DOI] [PubMed] [Google Scholar]; b Chin. J. Nat. Med. 2006, 4, 1. [Google Scholar]
- For other classifications, see:; a Pelletier S. W.; Keith L. H. In The Alkaloids; Manske R. H. F., Ed.; Academic Press: New York, 1970; Vol. 12, Chapter 2. [Google Scholar]; b Pelletier S. W. J. Nat. Prod. 1980, 43, 41. [Google Scholar]; c Pelletier S. W.; Page S. W. In The Alkaloids; Manske R. H. F., Rodrigo R. G. A., Eds; Academic Press: New York, 1981; Vol. 18, Chapter 2. [Google Scholar]
- For a discussion on pattern recognition, see:Wilson R. M.; Danishefsky S. J. J. Org. Chem. 2007, 72, 4293. [DOI] [PubMed] [Google Scholar]
- a Wiesner K.; Tsai T. Y. R.; Huber K.; Bolton S. E.; Vlahov R. J. Am. Chem. Soc. 1974, 96, 4990. [Google Scholar]; b Guthrie R. W.; Valenta Z.; Wiesner K. Tetrahedron Lett. 1966, 38, 4645. [DOI] [PubMed] [Google Scholar]
- a Peese K. M.; Gin D. Y. J. Am. Chem. Soc. 2006, 128, 8734. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Peese K. M.; Gin D. Y. Chem.—Eur. J. 2008, 14, 1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Muratake H.; Natsume M. Angew. Chem., Int. Ed. 2004, 43, 4646. [DOI] [PubMed] [Google Scholar]; b Muratake H.; Natsume M. Tetrahedron 2006, 62, 7056. [Google Scholar]; c Muratake H.; Natsume M. Tetrahedron 2006, 62, 7071. [Google Scholar]; d Muratake H.; Natsume M. Tetrahedron 2006, 62, 7093. [Google Scholar]
- Shi Y.; Wilmot J. T.; Nordstrøm L. U.; Tan D. S.; Gin D. Y. J. Am. Chem. Soc. 2013, 135, 14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyana Y.; Han-ya Y.; Yokoshima S.; Fukuyama T. J. Am. Chem. Soc. 2014, 136, 6598. [DOI] [PubMed] [Google Scholar]
- For a recent review on approaches to the hetidine and hetisine alkaloids, see:Hamlin A. M.; Kisunzu J. K.; Sarpong R. Org. Biomol. Chem. 2014, 12, 1846. [DOI] [PubMed] [Google Scholar]
- For reviews of the HLF reaction, see:; a Wolff M. E. Chem. Rev. 1963, 63, 55. [Google Scholar]; b Majetich G.; Wheless K. Tetrahedron 1995, 51, 7095. [Google Scholar]; A related HLF-type reaction has been achieved by Okamoto:; c Yatsunami T.; Isono T.; Hayakawa I.; Okamoto T. Chem. Pharm. Bull. 1975, 23, 3030. [Google Scholar]; d Yatsunami T.; Furuya S.; Okamoto T. Chem. Pharm. Bull. 1978, 26, 3199. [Google Scholar]
- Prantz K.; Mulzer J. Chem. Rev. 2010, 110, 3741. [DOI] [PubMed] [Google Scholar]
- Conversion of the hetidine core to a mixture of the hetisine and atisine cores was achieved in low 13% and 38% yield, respectively. See ref (13c,13d).
- Hamlin A. M.; Cortez F.; de J.; Lapointe D.; Sarpong R. Angew. Chem., Int. Ed. 2013, 52, 4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chatani N.; Inoue H.; Kotsuma T.; Murai S. J. Am. Chem. Soc. 2002, 124, 10294. [DOI] [PubMed] [Google Scholar]; b Simmons E. M.; Sarpong R. Org. Lett. 2006, 8, 2883. [DOI] [PubMed] [Google Scholar]
- Hale K. J.; Grabski M.; Flasz J. T. Org. Lett. 2013, 15, 370. [DOI] [PubMed] [Google Scholar]
- a Cortez F. de J.; Sarpong R. Org. Lett. 2010, 12, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cortez F. de J.; Lapointe D.; Hamlin A. M.; Simmons E. M.; Sarpong R. Tetrahedron 2013, 69, 5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sawamura M.; Hamashima H.; Ito Y. Tetrahedron: Asymmetry 1991, 2, 593. [Google Scholar]; b Sawamura M.; Hamashima H.; Sugawara M.; Kuwano R.; Ito Y. Organometallics 1995, 14, 4549. [Google Scholar]; c Sawamura M.; Hamashima H.; Ito Y. J. Am. Chem. Soc. 1992, 114, 8295. [Google Scholar]; d Sawamura M.; Hamashima H.; Ito Y. Tetrahedron 1994, 50, 4439. [Google Scholar]
- Seminal publications:; a Corey E. J.; Mock W. L.; Pasto D. J. Tetrahedron Lett. 1961, 2, 347. [Google Scholar]; b Hünig S.; Muller H.-R.; Their W. Tetrahedron Lett. 1961, 2, 353. [Google Scholar]; c Corey E. J.; Pasto D. J.; Mock W. L. J. Am. Chem. Soc. 1961, 83, 2957. [Google Scholar]
- Mascitti V.; Corey E. J. J. Am. Chem. Soc. 2004, 126, 15564. [DOI] [PubMed] [Google Scholar]
- Eliel E. L.; Wilen S. H.; Mander L. N. In Stereochemistry of Organic Compounds; John Wiley & Sons: New York, 2009. [Google Scholar]
- Conformational searches (B3LYP 6-31G) did not reveal an apparent conformational preference for 28.
- See p 6 in:Woodward R. B.; Bader F. E.; Bickel H.; Frey A. J.; Kierstead R. W. Tetrahedron 1958, 2, 1–57. [Google Scholar]
- a This argument would assume the reacting palladium hydride is also considered an electron-deficient reagent.; b For a recent example in epoxidation where this argument is invoked, see:Peng F.; Danishefsky S. J. J. Am. Chem. Soc. 2012, 134, 18860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRheenen V.; Kelly R. C.; Cha D. Y. Tetrahedron Lett. 1976, 17, 1973. [Google Scholar]
- Matteucci M.; Bhalay G.; Bradley M. Org. Lett. 2003, 5, 235. [DOI] [PubMed] [Google Scholar]
- For pertinent reviews on oxidative dearomatization processes, see:; a Roche S. P.; Porco J. A. Jr. Angew. Chem., Int. Ed. 2011, 50, 4068. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Magdziak D.; Meek S. J.; Pettus T. R. R. Chem. Rev. 2004, 104, 1383. [DOI] [PubMed] [Google Scholar]
- Gao L.; Wei X.; Yang L. J. Chem. Res. 2004, 307. [Google Scholar]
- Dall’Acqua S.; Shrestha B. B.; Gewali M. B.; Jha P. K.; Carrara M.; Innocenti G. Nat. Prod. Commun. 2008, 3, 1985. [PubMed] [Google Scholar]
- Barton D. H. R.; McCombie S. W. J. Chem. Soc., Perkin Trans. 1 1975, 1574. [Google Scholar]
- Comins D. L.; Dehghani A. Tetrahedron Lett. 1992, 33, 6299. [Google Scholar]
- For a recent example of a hydrolysis of a hindered cyclic carbamate using Ba(OH)2, see:Huters A. D.; Quasdorf K. W.; Styduhar E. D.; Garg N. K. J. Am. Chem. Soc. 2011, 133, 15797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples of piperidine dehydrogenation, see:; a Ogawa K.; Nomura Y.; Takeuchi Y.; Tomoda S. J. Chem. Soc., Perkin Trans. 1 1982, 3031. [Google Scholar]; b Davis B. G.; Maughan M. A. T.; Chapman T. M.; Villard R.; Courtney S. Org. Lett. 2002, 4, 103. [DOI] [PubMed] [Google Scholar]; c Movassaghi M.; Chen B. Angew. Chem., Int. Ed. 2007, 46, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Murahashi S.; Mitsui H.; Shiota T.; Tsuda T.; Watanabe S. J. Org. Chem. 1990, 55, 1736. [Google Scholar]
- Sun F.; Bai Y.; Benn M. Heterocylces 1991, 32, 1137–1141. [Google Scholar]
- Okamoto has shown previously that the C14–C20 bond can be cleaved in the context of a degradation study on kobusine; see ref (13c,13d).
- Zhong Y.-L.; Shing T. K. M. J. Org. Chem. 1997, 62, 2622–2624. [DOI] [PubMed] [Google Scholar]
- Danheiser R. L.; Romines K. R.; Koyana H.; Gee S. K.; Johnson C. R.; Medich J. R. Org. Synth. 1993, 71, 133. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.