

Abstract
Jiubiying, the dried barks of Ilex rotunda Thunb. (Aquifoliaceae), has been used as herbal tea and traditional Chinese medicine for heat-clearing, detoxifying, dehumidification, and odynolysis. Pedunculoside and syringin are two main bioactive components. For the new drug development, we tried to isolate and purify several chemical constituents from Jiubiying by high-speed counter-current chromatography (HSCCC). The two-phase solvent system used was composed of ethyl acetate-n-butanol-water (1:6:7, v/v/v). From 1.0 g of Jiubiying extracts syringaresinol 4′,4″-bis-O-β-D- glucopyranoside (I, 20.2 mg), syringin (II, 56.8 mg), sinapaldehyde glucoside (III, 26.2 mg), syringaresinol 4′-O-β-D-glucopyranoside (IV, 20.4 mg), and pedunculoside (V, 45.1 mg) were obtained by one run of TBE-1000A HSCCC machine with 1000 mL of column volume. Their structures were identified by IR, MS, and extensive NMR studies. Syringaresinol 4′,4″-bis-O-β-D-glucopyranoside (I) was isolated from this plant for the first time.
Keywords: Jiubiying, Counter-counter chromatography, Ilex rotunda, Syringaresinol 4′, 4″-bis-O-β-D-glucopyranoside
1. Introduction
Jiubiying, the dried barks of Ilex rotunda Thunb. (Aquifoliaceae),has been used as herbal tea and traditional Chinese medicine. It possesses variousmedicinal functions such as heat-clearing, detoxifying, dehumidification, and odynolysis, and used for the treatment of fever, throat-swell, eczema, diarrhea, and furuncle [1]. The n-butanol extracts of Jiubiying can depress both normal blood pressure and arterial hypertension in rats [2], and have protective effects on arrhythmia and the damage of myocardial ischemia in rats and mice [3]. The ethanol extracts of Jiubiying can reduce coronary blood flow, weaken myocardial contractility, slow down heart rate, prolong survival time, and prevent arrhythmias in mice [4]. A number of chemical components have been isolated from the parks of I. rotunda, including pedunculoside, syringin [5,6], 3β-hydroxy-oleanane, rotundic acid, rotundic acid isopropylidene, sinapaldehyde, sinapaldehyde glucoside, stearic acid, syringaldehyde [7], ilexrotunin, rotundanonic acid, rotundioic acid, rotungenic acid [8], 3β-[(α-L-arabinopyranosyl) oxy]-19α-hydroxyolean-12-en-28-oic acid 28-O-β-D-glucopyranosyl ester, caffeic acid 4-O-β-D-glucopyranoside, β-daucosterol, kudinoside H, pomolic acid, β-sitosterol, syringaresinol 4′-O-β-D-glucopyranoside, vanillic acid 4-O-β-D-glucopyranoside [9], abbeokutone, 3β,19α-dihydroxyurs-12-en-24,28-dioic acid, 19α,24-dihydroxyurs-12-en-3-one-28-oic acid, disyringin ether, friedelin, glucose, 28-hydroxy-friedelin, 3β-hydroxy-oleanane, inositol, nonadecylic acid, oleanolic acid, rotundaol, and sugereoside [10,11].
High-speed counter-current chromatography (HSCCC), a support-free liquid-liquid partition chromatographic technique, has been widely used for isolation and purification of active components from traditional Chinese medicine and natural products [12–14]. Most of the studies focus on several main compounds in some parts, such as flavones [15,16], phenols [17–20], saponins [21–23], alkaloids [24,25], and so on. However, only few studies have focused on the direct separation and purification of chemical constituents from crude extracts [26–28].
In this study, crude Jiubiying extracts with 50% ethanol were directly separated by HSCCC. using two separation columns with total capacities so 260 mL and 1000 mL.
2. Experimental
2.1. Apparatus
TBE-300A model HSCCC (Tauto Biotechnique Company, Shanghai, China) for semi-preparative HSCCC separation has three PTFE (polytetrafluoroethylene) preparative coils (I.D. of the tubing = 1.8 mm, column volume = 260 mL) and a 20 mL manual injection sample loop. The distance between the holder axis and central axis of the centrifuge (R) was 5 cm and the β value (β = r/R, where r was the distance from the coil to the holder shaft) of the multilayer coil varied from 0.6 (internal terminal) to 0.8 (external terminal). The revolution speed of the apparatus was regulated at 0–1000 rpm with an electronic speed controller. The solvent was pumped into the column with a Tauto TBP50A pump (Tauto Biotechnique Company, Shanghai, China) and the eluent was continuously monitored by a TBD-2000 UV detector (Tauto Biotechnique Company, Shanghai, China). The separation temperature was controlled by DTY-20A water-circulating constant temperature implement (Tauto Biotechnique Company, Shanghai, China). The chromatogram was recorded by a Jinda Biochemical Chromatography workstation V4.0 (Tauto Biotechnique Company, Shanghai, China).
TBE-1000A model HSCCC for preparative separation has three PTFE preparative coils (I.D. of the tubing = 3.0 mm, column volume = 1000 mL) and an 80mL manual injection sample loop. The distance between the holder axis and central axis of the centrifuge (R) was 5 cm and the β value of the multilayer coil varied from 0.6 (internal terminal) to 0.78 (external terminal). The revolution speed of the apparatus was regulated at 0–600 rpm with an electronic speed controller. The solvent was pumped into the column with a Tauto TBP50A pump (Tauto Biotechnique Company, Shanghai, China) and the eluent was continuously monitored by a TBD-2000 UV detector (Tauto Biotechnique Company, Shanghai, China). The separation temperature was controlled by a TC-1050 water-circulating constant temperature implement (Beijing Detianyou Science and Technology Development Company, Beijing, China). The chromatogram was recorded by a Jinda Biochemical Chromatography workstation V4.0 (Tauto Biotechnique Company, Shanghai, China).
Samples were analyzed by a Shimadzu LC-20AT high performance liquid chromatography (HPLC) (Shimadzu, Japan) instrument equipped with an SPD-M20A diode array detector (DAD), an SIL-20A auto sampler, a DGU-20As degasser, a CTO-10ASvp column oven, and a Shimadzu LC-solution workstation.
The 1H and 13C NMR spectra were measured by a Bruker AV400 spectrometer. The chemical shift values are reported as δ in ppm relative to tetramethylsilane (TMS) or sodium trimethylsilylpropionate (TSP), and the coupling constants (J) are in hertz (Hz). HR-ESI-MS mass spectra were recorded on a Waters Xevo G2 Qtof mass spectrometer. IR spectra were recorded on Shimadzu FTIR-8400s spectrometer with KBr pellets.
2.2. Reagents and materials
Acetonitrile, n-butanol, ethanol, ethyl acetate, n-heptane, and methanol, used for HSCCC separation were all of analytical grades, and purchased from Sinopharm Chemical Reagent Beijing Co., Ltd (Beijing, China). Acetonitrile used for HPLC was of chromatographic grade (Merck, Germany). Dried barks of I. rotunda were purchased from Guangzhou Caizhitang Pharmaceutical Co., Ltd (Guangdong Province, China) and identified by Prof Shilin Hu, Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences. A voucher specimen was deposited in Department of Chemistry, Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences with the specimen number of 20111016.
2.3. Preparation of Jiubiying extracts
The dried barks (1 kg) of I. rotunda were extracted 3 times with 10 L of 50% ethanol-water solution in a water bath at 80oC. The extract was concentrated to a volume of 5 L in a rotary evaporator (RE-201D, Henan Yuhua Instrument Co., Ltd, China) and centrifuged at 6000 rpm for 10 min using an LD5-10 centrifuge (Beijing Jinli Centrifuge Co., Ltd, China). The supernatant fluid was dried with a rotary evaporator to yield 175 g of Jiubiying extracts.
2.4. Measurement of partition coefficients (K)
The solvent mixtures were thoroughly equilibrated in a test tube and an upper phase and a lower phase were separated. The lower phase (2.0 mL) and 10 mg Jiubiying extracts were delivered into a 10 mL test tube, mixed thoroughly, and stood for several minutes. The solution (5 μL) was taken directly for HPLC determination and the peak area was recorded as Ainitial. Then, the upper phase (2.0 mL) was added to the solution, mixed thoroughly, and stood until two clear layers were formed. The lower phase solution (5 μL) was taken directly for HPLC determination and the peak area was recorded as Alower. The partition coefficients (K) of compounds were obtained by the following equation: K=(Ainitial−Alower)/Alower.
2.5. Preparation of two-phase solvent system and sample solution
The two-phase solvent system composed of ethyl acetate-n-butanol-water (1:6:7, v/v/v) were thorouthly mixed in a separatory funnel and allowed to stand until two clear layers were formed at 25oCThen, the upper phase and lower phase were separated. The sample solution (25 mg/mL) for HSCCC separation was made by dissolving 1.0 g of Jiubiying extracts in the mixture of 20 mL lower phase and 20 mL upper phase of the above solvent system.
2.6. Separation procedure
2.6.1. TBE-300A model HSCCC separation
A head-tail elution mode was selected for the initial separation. The upper phase was filled into the coil column as a stationary phase at a flow rate of 40 mL/min for 7 min. Then the apparatus was rotated at 850 rpm, meanwhile the lower mobile phase was pumped into the coil column at a flow rate of 1.0 mL/min. After hydrodynamic equilibrium was established, 12 mL of sample solution containing 300 mg of Jiubiying extracts was injected into the column. was After several peaks were elutedin 312 min, the elution mode was turned to the tail-head mode, and the upper phase was pumped into the coil column at a flow rate of 1.0 mL/min. During the separation process, the column temperature was controlled at 25oC. The effluent from the column was monitored at UV 254 nm. The fractions were collected manually according to the chromatogram displayed on the recorder.
2.6.2. TBE-1000A model HSCCC separation
A head-tail elution mode was applied for initial separation. The upper phase was filled into the coil column as a stationary phase at a flow rate of 50 mL/min for 20 min. Then the apparatus was rotated at 450 rpm, meanwhile the lower mobile phase was pumped into the coil column at a flow rate of 3.0 mL/min. After the hydrodynamic equilibrium was established, 40 mL of sample solution containing 1.0 g of Jiubiying extracts was injected into the column. After several peaks were elutedin 436 min, the elution mode was turned to the tail-head mode, and the upper phase was pumped into the coil column at a flow rate of 3.0 mL/min. During the separation process, the column temperature was controlled at 25oC. The effluent from the column was monitored at UV 254 nm. The fractions were collected manually according to the chromatogram displayed on the recorder.
2.7. HPLC analysis
Jiubiying extracts and HSCCC fractions were analyzed by HPLC method. The analysis was performed using a Shimadzu LC-20AT chromatographic system with a Shiseido UG120-C18 reversed-phase column (4.6 mm×150 mm, 5 μm, Shiseido Co. Ltd., Japan). The mobile phase was acetonitrile (A) - water (B) for gradient elution, and the elution system was: 0–10 min, 10% of B; 10–20 min, 10%–40% of B; 20–30 min, 40% of B; the flow rate of mobile phase was 1 mL/min. The column temperature was controlled at 30oC. The detection wavelength was 210 nm.
2.8. Identification of HSCCC fractions
The fractions from HSCCC were evaporated to dryness to measure infrared radiation (IR) spectra, high resolution electron spray ionization-mass (HR-ESI-MS) spectra, and extensive NMR (1H, 13C, 1H-1H COSY, DEPT, HMQC, HMBC, and NOESY). The chemical structures of chemical constituents were identified on the base of these spectra.
3. Results and discussion
3.1. Selection of two-phase solvent system
The most important factor for a successful HSCCC separation is the selection of a suitable two-phase solvent system [29], which provides an ideal range of the partition coefficient (K) for the target compounds. As shown on the HPLC chromatogram, syringin was a higher polar component and pedunculoside was a lower polar component in Jiubiying extracts. On the basis of these properties and HBAW method [30], medium polar or polar solvent systems were tested. A series of experiments were performed to determine the optimal solvent two-phase system for the HSCCC separation and the results are summarized in Table 1.
Table 1.
The K-values of main components in Jiubiying extracts in two-phase solvent systems.
| Solvent system (v/v) |
K
|
|||
|---|---|---|---|---|
| Syringin (II) | Syringaresinol 4′,4″-O-β-D-bis- glucopyranoside (I) | Syringaresinol 4′-O-β-D-glucopyranoside (IV) | Pedunculoside (V) | |
| n-Heptane-n-butanol-acetonitrile-water (2:3.8:1.2:4) | 0.15 | 0.01 | 0.75 | 15.32 |
| n-Heptane-n-butanol-acetonitrile-water (1:4.4:0.6:4) | 0.33 | 0.01 | 2.71 | 71.53 |
| n-Butanol-water (1:1) | 0.76 | 0.13 | 10.24 | 165.0 |
| Ethyl acetate-n-butanol-water (4:1:5) | 0.12 | 0.06 | 0.63 | 196.3 |
| Ethyl acetate-n-butanol-water (3:2:5) | 0.33 | 0.01 | 7.88 | 220.2 |
| Ethyl acetate-n-butanol-water (1:4:5) | 0.54 | 0.10 | 6.69 | 66.50 |
| Ethyl acetate-n-butanol-water (1:6:7) | 0.66 | 0.09 | 10.35 | 96.48 |
The ideal K-value is between 0.5 and 2.0. As shown in Table 1, the K-values of compounds I, II, IV, and V were very different. The K-value of compound I was very small and that of compound V was very large. Therefore, the head-tail elution mode was chosen to separate the polar compounds firstly and then the tail-head elution mode was chosen to obtain the lower polar compounds.
As shown in Table 1, the very small K-values of compounds I, II, and IV meant that they would be maintained in the upper phase of n-heptane-n-butanol-acetonitrile-water (2:3.8:1.2:4, v/v/v/v) used as the stationary phase of HSCCC. The K-values of these compounds increased in two-phase systems of n-heptane-n-butanol-acetonitrile-water (1:4.4:0.6:4, v/v/v/v) and n-butanol-water (1:1, v/v). The solvent system of n-butanol-water (1:1, v/v) system seemed to be suitable but the retention of the upper stationary phase would be unsatisfactory. Therefore, ethyl acetate was added into n-butanol-water system according to the proportion of Oka [31] and Ito [32] methods. As the decrease of the ethyl acetate proportion, the K-values were gradually become to more ideal. Finally, ethyl acetate-n-butanol-water (1:6:7, v/v/v) was chosen for HSCCC to separate the chemical constituents in Jiubiying extracts.
3.2. Optimization of operational parameters on semipreparative HSCCC
Besides compositions of the two-phase solvent system, the flow rate of mobile phase, the revolution speed of separation column, and the separation temperature were also investigated. The retention of stationary phase with 35% was obtained with revolution speed of 850 rpm, flow rate of 1.0 mL/min, and column temperature at 25 oC for TBE-300A model HSCCC separation with 260 mL of column volume. The result was shown in Fig. 2, compounds I, II, III, IV, and V were separated and obtained from 300 mg of Jiubiying extracts using the two-phase solvent system composed of ethyl acetate-n-butanol-water (1:6:7, v/v/v). However, the amount of these compounds was too small to be identified.
Fig. 2.
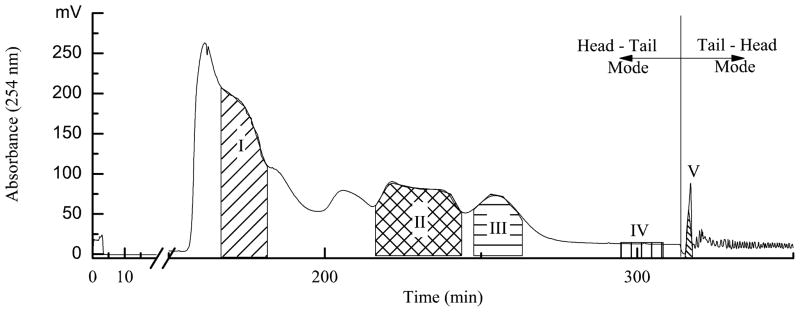
Chromatogram of Jiubiying extracts by semi-preparative HSCCC. Conditions: solvent system: ethyl acetate-n-butanol-water (1:6:7, v/v/v); stationary phase: upper organic phase (head-tail mode); mobile phase: lower aqueous phase (head-tail model); column volume: 260 mL; sample concentration: 25 mg/mL; sample volume: 12 mL; flow rate: 1.0 mL/min; revolution: 850 rpm; detection wavelength: 254 nm; temperature: 25oC; retention of stationary phase: 35%.
3.3. Optimization of operational parameters on preparative HSCCC
After being further investigated, a good retention of stationary phase with 50% was obtained using the solvent system of ethyl acetate-n-butanol-water (1:6:7, v/v/v) with revolution speed of 450 rpm, flow rate of 3.0 mL/min, and column temperature at 25 oC, for TBE-1000A model HSCCC instrumentwith 1000 mL of column volume. The HSCCC separation of the crude sample under this experimental condition yielded syringaresinol 4′,4″-bis-O-β-D-glucopyranoside (I, 20.2 mg) with 55.4% purity, syringin (II, 56.8 mg) with 98.1% purity, sinapaldehyde glucoside (III, 26.2 mg) with 68.6% purity, syringaresinol 4′-O-β-D-glucopyranoside (IV, 20.4 mg) with 95.3% purity, and pedunculoside (V, 45.1 mg) with 97.3% purity from 1.0 g of Jiubiying extracts in a single run. The fractions containing compounds I and III were further purified by preparative HPLC.
3.4. Structural identification
The chemical structures of each HSCCC fractions were identified on bases of IR, HR-ESI-MS (positive ion mode), and extensive NMR (1H, 13C, 1H-1H COSY, DEPT, HMQC, HMBC, and NOESY) studies.
Syringaresinol 4′,4″-bis-O-β-D-glucopyranoside (I) IR (KBr) νmax: 3420, 2966, 2941, 1635, 1458, 1419, 1384, 1126, 1076 cm−1. HR-ESI-MS [M+NH4]+ m/z: 760.3037 (C34H46O18NH4, Calcd. for 760.3028). 1H NMR (400 MHz, CD3OD) δ ppm: 6.71 (4H, s, H-2′, 2″, 6′, 6″), 4.94 (2H, d, J=6.4 Hz, Glc-H-1′, 1″), 4.80 (2H, d, J=3.2 Hz, H-2, 6), 4.23 (2H, m, H-4α, 8α), 3.90 (2H, m, H-4β, 8β), 3.80 (12H, s, OCH3×4), 3.73 (2H, dd, J=12.4, 2.4 Hz, Glc-H-6′, 6″), 3.64 (2H, dd, J=12.4, 5.2 Hz, Glc-H-6′, 6″), 3.49 (2H, m, Glc-H-2′, 2″), 3.48 (2H, m, Glc-H-3′, 3″), 3.42 (2H, m, Glc-H-4′, 4″), 3.26 (2H, m, Glc-H-5′, 5″), 3.18 (2H, m, H-1, 5). 13C NMR (100 MHz, CD3OD) δ ppm: 152.9 (C-3′, 3″, 5′, 5″), 137.3 (C-1′, 1″), 133.9 (C-4′, 4″), 104.0 (C-2′, 2″, 6′, 6″), 102.9 (Glc C-1′, 1″), 85.2 (C-2, 6), 77.4 (Glc C-5′, 5″), 76.4 (Glc C-3′, 3″), 73.8 (Glc C-2′, 2″), 71.5 (C-4, 8), 70.1 (Glc C-4′, 4″), 60.6 (Glc C-6′, 6″), 56.5 (3′, 3″, 5′, 5″, -OCH3×4), 53.5 (C-1, 5). The chemical structure was identified on the base of these spectra and references [33,34]
Syringin (II) IR (KBr) νmax: 3564, 3390, 1589, 1508, 1419, 1132, 1093, 1028, 966, 617 cm−1. HR-ESI-MS [M+NH4]+ m/z: 390.1774 (C17H24O9NH4, Calcd. for 390.1764). 1H NMR (400 MHz, CD3OD) δ ppm: 6.76 (2H, s, H-3, 5), 6.56 (1H, d, J = 16 Hz, H-7), 6.35 (1H, m, H-8), 4.89 (1H, d, J=7.2 Hz, Glc H-1′), 4.24 (2H, d, J=5.6 Hz, H-9), 3.87 (6H, s, 2, 6, OCH3×2), 3.80 (1H, dd, J=12.0, 2.0Hz, Glc H-6′), 3.69 (1H, dd, J=12, 5.2 Hz, Glc H-6′), 3.51 (1H, m, H-2′), 3.48 (1H, m, H-3′), 3.44 (1H, m, H-4′), 3.22 (1H, m, H-5′). 13C NMR (100 MHz, CD3OD) δ ppm: 152.9 (C-2, 6), 134.5 (C-1), 133.9 (C-4), 129.9 (C-8), 128.7 (C-7), 104.1 (C-3, 5), 103.9 (Glc C-1′), 76.9 (Glc C-3′), 76.4 (Glc C-5′), 74.3 (Glc C-2′), 69.9 (Glc C-4′), 62.2 (C-9), 61.1 (Glc C-6′), 55.7 (CH3O-2, 6). The chemical structure was identified on the base of these spectra and references [7,11].
sinapaldehyde glucoside (III) IR (KBr) νmax: 3420, 1683, 1506, 1456, 1419, 1338, 1245, 1132, 1070, 669 cm−1. HR-ESI-MS [M+H]+ m/z: 371.1282 (C17H22O9H, Calcd. for 371.1298). 1H NMR (400 MHz, D2O) δ ppm: 9.49 (1H, d, J=7.6 Hz, 9-H), 7.61 (1H, d, J=15.6, H-7), 7.01 (2H, s, H-3, 5), 6.72 (1H, dd, J=15.6, 7.6 Hz, H-8), 3.84 (6H, s, 2, 6, OCH3×2), 5.06 (1H, d, J=7.2 Hz, Glc H-1′), 3.76 (1H, dd, J=12.4, 2 Hz, H-6′), 3.65 (1H, dd, J=12.4, 5.2 Hz, H-6′), 3.51 (1H, m, H-2′), 3.50 (1H, m, H-3′), 3.43 (1H, m, H-4′), 3.31 (1H, m, H-5′). 13C NMR (100 MHz, D2O) δ ppm: 195.1 (C-9), 155.8 (C-7), 153.4 (C-3, 5), 138.8 (C-4), 131.6 (C-1), 127.9 (C-8), 106.9 (C-2, 6), 103.1 (Glc C-1′), 76.5 (Glc C-3′), 75.8 (Glc C-5′), 73.8 (Glc C-2′), 69.4 (Glc C-4′), 60.2 (Glc C-6′). The chemical structure was identified on the base of these spectra and reference [11].
Syringaresinol 4′-O-β-D-glucopyranoside (IV) IR: 3390, 1635, 1456, 1418, 1385, 1120, 1078 cm−1. HR-ESI-MS [M+NH4]+ m/z: 598.2493 (C28H36O13NH4, Calcd. for 598.2500). 1H NMR (400 MHz, CD3OD) δ ppm: 6.74 (2H, s, H-2″, 6″), 6.67 (2H, s, H-2′, 6′), 4.86 (1H, m, Glc H-1′), 4.78 (1H, d, J=4.0 Hz, H-2), 4.74 (1H, d, J=4.0 Hz, H-6), 4.29 (1H, m, H-4β), 3.93 (2H, dd, J=2.8, 9.2 Hz, H-4α, 8α), 3.88 (6H, s, 3″, 5″, OCH3×2), 3.86 (6H, s, 3′, 5′, OCH3×2), 3.79 (1H, dd, J=11.6, 2 Hz, Glc H-6′), 3.66 (1H, dd, J=11.6, 4.4 Hz, Glc H-6′), 3.50 (1H, m, Glc H-2′), 3.48 (1H, m, Glc H-3′), 3.44 (1H, m, Glc H-4′), 3.42 (1H, d, J=2.4 Hz, H-8β), 3.24 (1H, m, Glc H-5′), 3.15 (2H, m, H-1, 5). 13C NMR (100 MHz, CD3OD) δ ppm: 152.6 (C-3″, 5″), 147.6 (C-3′, 5′), 137.3 (C-1″), 134.3 (C-4′), 133.8 (C-4″), 128.9 (C-1′), 104.2 (C-2″, 6″), 103.1 (C-2′, 6′), 102.8 (Glc C-1′), 86.8 (C-6), 81.5 (C-2), 77.0 (Glc C-5′), 76.4 (Glc C-3′), 74.2 (Glc-2′), 70.4 (Glc C-4′), 70.0 (C-4), 69.1 (C-8), 61.0 (Glc C-6′), 55.7 (3″, 5″, -OCH3×2), 55.4 (3′, 5′, -OCH3×2), 53.9 (C-5), 49.4 (C-1). The chemical structure was identified on the base of these spectra and references [9,35,36].
Pedunculoside (V) IR (KBr) νmax: 3420, 2932, 1734, 1652, 1471, 1386, 1074, 1047, 650 cm−1. HR-ESI-MS [M+Na]+ m/z: 673.3923 (C36H58O10Na, Calcd. for 673.3928 ). 1H NMR (400 MHz, C5D5N) δ ppm: 6.34 (1H, d, J=7.6 Hz, Glc H-1′), 5.59 (1H, s, H-12), 4.50 (1H, m, H-6′), 4.45 (1H, m, H-6′), 4.40 (1H, t, J=8.6 Hz Glc H-4′), 4.34 (1H, t, J=8.6 Hz, Glc H-3′), 4.26 (1H, t, J=8.6 Hz, Glc H-2′), 4.22 (1H, d, J=6 Hz, H-3), 4.19 (1H, d, J=10 Hz, H-23), 4.08 (1H, t, J=8.6 Hz, Glc H-5′), 3.73 (1H, d, J=10 Hz, H-23), 3.10 (1H, td, J=13.2, 3.2 Hz, H-16), 2.96 (1H, s, H-18), 2.50 (1H, td, J=13.2, 3.2 Hz, H-15), 2.09 (2H, m, H-11), 2.07 (1H, m, H-1), 2.02 (3H, m, H-16, 20α, 20β), 1.94 (1H, m, H-9), 1.92 (2H, m, H-2), 1.87 (1H, m, H-1), 1.72 (1H, td, J=11.2, 2.8Hz, 7-H), 1.67 (1H, s, H-27), 1.65 (1H, m, H-6), 1.59 (1H, m, H-22), 1.55 (1H, m, H-5), 1.46 (1H, m, H-7), 1.42 (3H, s, H-29; 1H, m, H-6), 1.38 (1H, m, H-20), 1.26 (3H, s, H-26), 1.24 (1H, m, H-15), 1.09 (3H, s, H-24; 2H, m, H-22), 1.08 (3H, d, J=5.6 Hz, H-30), 1.06 (3H, s, H-25). 13C NMR (100 MHz, C5D5N) δ ppm: 177.9 (C-28), 140.2 (C-13), 129.4 (C-12), 96.8 (C-1′), 80.2 (C-5′), 79.9 (C-3′), 75.0 (C-2′), 74.5 (C-3), 73.6 (C-19), 72.2 (C-4′), 69.0 (C-23), 63.3 (C-6′), 55.4 (C-18), 49.6 (C-5), 49.6 (C-17), 48.8 (C-9), 43.8 (C-4), 43.0 (C-14), 43.0 (C-20), 41.5 (C-8), 39.8 (C-22), 38.6 (C-1), 38.1 (C-10), 34.2 (C-7), 30.2 (C-15), 28.6 (C-2), 27.9 (C-29), 27.6 (C-21), 27.0 (C-16), 25.5 (C-27), 25.0 (C-11), 19.7 (C-6), 18.4 (C-26), 17.6 (C-30), 17.0 (C-25), 14.0 (C-24). The chemical structure was identified on the base of these spectra and references [7, 37].
4. Conclusion
Five compounds in 50% ethanol extracts of dried barks of I. rotunda were successfully separated by semi-preparative (260 mL column volume) and preparative HSCCC (1000 mL column volume) with the two-phase solvent system of ethyl acetate-n-butanol-water (1:6:7, v/v/v). From 1.0 g of Jiubiying extracts syringaresinol 4′,4″-bis-O-β-D-glucopyranoside (I, 20.20 mg) with 55.41% purity, syringin (II, 56.81 mg) with 98.1% purity, sinapaldehyde glucoside (III, 26.21 mg) with 68.6% purity, syringaresinol 4′-O-β-D-glucopyranoside (IV, 20.37 mg) with 95.3% purity, and pedunculoside (V, 45.09 mg) with 97.3% purity were obtained in 550 min by a single separation procedure of preparative HSCCC with a 1000 mL column volume. Their chemical structures were identified by means of IR, MS, and extensive NMR studies.
This was the first report that chemical constituents were isolated from the plant of I. rotunda by high-speed counter-current chromatography method, and syringaresinol 4′,4″-bis-O-β-D-glucopyranoside (I) was found from this plant for the first time.
Fig. 1.

Chemical structures of syringaresinol 4′,4″-O-β-D-bis-glucopyranoside (I), syringin (II), sinapaldehyde glucoside (III), syringaresinol 4′-O-β-D-glucopyranoside (IV), and pedunculoside (V).
Fig. 3.
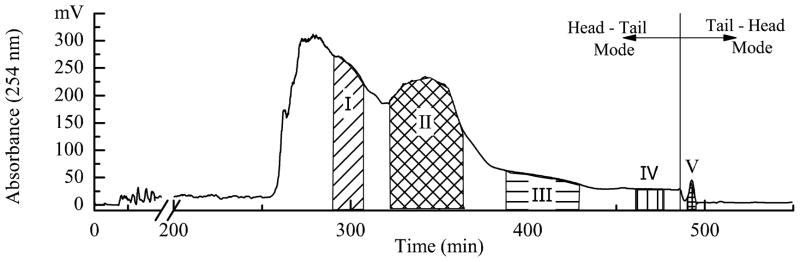
Chromatogram of Jiubiying extracts by preparative HSCCC. Conditions: solvent system: ethyl acetate-n-butanol-water (1:6:7, v/v/v); stationary phase: upper organic phase (head-tail mode); mobile phase: lower aqueous phase (head-tail mode); column volume: 1000 mL; sample concentration: 25 mg/mL; sample volume: 40 mL; flow-rate: 3.0 mL/min; revolution: 450 rpm; detection wavelength: 254 nm; temperature: 25oC; retention of stationary phase: 50%.
Fig. 4.

HPLC chromatogram of Jiubiying extracts and fractions I, II, III, IV, and V. Conditions: column: Shiseido UG120-C18 reversed-phase column (4.6 mm×150 mm, 5 μm); mobile phase: acetonitrile (A) -water (B) for gradient elution, and the elution system was: 0–10 min, 10% of B; 10–20 min, 10%–40% of B; 20–30 min, 40% of B; flow rate: 1.0 mL/min; detection wavelength: 210 nm; column temperature: 30oC.
Acknowledgments
The authors gratefully acknowledge the financial support by the public welfare research special project in General Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China (No.201210209).
References
- 1.China Pharmacopoeia Committee, Pharmacopoeia of the People’s Republic of China, First Division. 2010. People’s Medical Publishing House; Beijing: 2010. p. 293. [Google Scholar]
- 2.Dong YF, Liang YL, Luo JP. Zhong Yao Cai. 1997;20:406. [PubMed] [Google Scholar]
- 3.Chen XX, He B, Xu YF, Li JH, Luo JP. Zhong Yao Yao Li Yu Lin Chuang. 1998;14:22. [Google Scholar]
- 4.He B, Chen XX, Li JH, Jiang T, Luo JP. Zhong Yao Cai. 1997;20:303. [PubMed] [Google Scholar]
- 5.Chu JH, Hung SH, Wang YH. Acta Chim Sin. 1956;14:128. [Google Scholar]
- 6.Xie PS, Yang ZX. Yao Xue Xue Bao. 1980;15:303. [PubMed] [Google Scholar]
- 7.Wen DX, Chen ZL. Zhong Cao Yao. 1991;22:246. [Google Scholar]
- 8.Wen DX, Chen ZL. Phytochemistry. 1996;41:657. [Google Scholar]
- 9.Sun H, Zhang XQ, Cai Y, Han WL, Wang Y, Ye WC. Lin Chan Hua Xue Yu Gong Ye. 2009;29:111. [Google Scholar]
- 10.Xu R. Doctoral dissertation. Guangzhou University of Chinese Medicine; 2009. p. 26. [Google Scholar]
- 11.Xu R, Gao YH, Wei ZX, Zhu SH. Zhong Cao Yao. 2011;42:2389. [Google Scholar]
- 12.Wei Y, Xie Q, Fisher D, Sutherland IA. J Chromatogr A. 2011;1218:6206. doi: 10.1016/j.chroma.2011.01.058. [DOI] [PubMed] [Google Scholar]
- 13.Han LF, Ji LN, Boakye-Yiadom M, Wei L, Song XB, Gao XM. Molecules. 2012;17:8276. doi: 10.3390/molecules17078276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye HY, Chen LJ, Li YF, Peng AH, Fu AF, Song H, Tang MH, Luo HD, Luo YF, Xu YB, Shi JY, Wei YQ. J Chromatogr A. 2008;1178:101. doi: 10.1016/j.chroma.2007.11.060. [DOI] [PubMed] [Google Scholar]
- 15.Peng JY, Fan GR, Hong ZY, Chai YF, Wu YT. J Chromatogr A. 2005;1074:111. doi: 10.1016/j.chroma.2005.03.067. [DOI] [PubMed] [Google Scholar]
- 16.Wu SJ, Sun AL, Liu RM. J Chromatogr A. 2005;1066:243. doi: 10.1016/j.chroma.2005.01.054. [DOI] [PubMed] [Google Scholar]
- 17.Chen L, Han YS, Yang FQ, Zhang TY. J Chromatogr A. 2001;907:343. doi: 10.1016/s0021-9673(00)00960-2. [DOI] [PubMed] [Google Scholar]
- 18.Li HB, Chen F. J Chromatogr A. 2004;1052:229. doi: 10.1016/j.chroma.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Lu JJ, Wei Y, Yuan QP. J Chromatogr B. 2007;857:175. doi: 10.1016/j.jchromb.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Geng YL, Li FW, Gao QS, Shi XG. J Chromatogr A. 2006;1103:166. doi: 10.1016/j.chroma.2005.11.092. [DOI] [PubMed] [Google Scholar]
- 21.Qi XC, Ignatova S, Luo GA, Liang QL, Jun FW, Wang YM, Sutherland I. J Chromatogr A. 2010;1217:1995. doi: 10.1016/j.chroma.2010.01.057. [DOI] [PubMed] [Google Scholar]
- 22.Cao XL, Tian Y, Zhang TY, Liu QH, Jia LJ, Ito Y. J Liq Chromatogr & Rel Technol. 2003;26:1579. [Google Scholar]
- 23.Du QZ, Yuan J. J Liq Chromatogr & Rel Technol. 2005;28:1717. [Google Scholar]
- 24.Wu SH, Sun CR, Cao XJ, Zhou H, Zhang H, Pan YJ. J Chromatogr A. 2004;1041:153. doi: 10.1016/j.chroma.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Tang QF, Yang CH, Ye WC, Liu JH, Zhao SX. J Chromatogr A. 2007;1144:203. doi: 10.1016/j.chroma.2007.01.058. [DOI] [PubMed] [Google Scholar]
- 26.Sun QH, Sun AL, Liu RM. J Chromatogr A. 2006;1104:69. doi: 10.1016/j.chroma.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 27.Liu RM, Chu X, Sun AL, Kong LY. J Chromatogr A. 2005;1074:139. doi: 10.1016/j.chroma.2005.03.099. [DOI] [PubMed] [Google Scholar]
- 28.Yao S, Li Y, Kong LY. J Chromatogr A. 2006;1115:64. doi: 10.1016/j.chroma.2006.02.071. [DOI] [PubMed] [Google Scholar]
- 29.Ito Y. J Chromatogr A. 2005;1065:145. doi: 10.1016/j.chroma.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 30.Foucault AP. Solvent System in Cenreifugal Partition Chromatography. In: Foucault AP, editor. Chromatography Science Series. Vol. 68. Marcel Dekker Inc; New York: 1995. p. 71. [Google Scholar]
- 31.Oka F, Oka H, Ito Y. J Chromatogr A. 1991;538:99. doi: 10.1016/s0021-9673(01)91626-7. [DOI] [PubMed] [Google Scholar]
- 32.Ito Y. Journal of Chromatography Library Series. 51A. Elsevier; Amsterdam: 1992. Countercurrent Chromatography; p. 69. [Google Scholar]
- 33.Lami N, Kadota S, Kikuchi T, Momose Y. Chem Pharm Bull. 1991;39:1551. doi: 10.1248/cpb.39.1551. [DOI] [PubMed] [Google Scholar]
- 34.Jiang YP, Feng F, Xie N, Chen L, Zhu MX. Pharm & Clin Res. 2008;16:163. [Google Scholar]
- 35.Li XC, Barnes DL, Khan IA. Planta Med. 2001;67:776. doi: 10.1055/s-2001-18352. [DOI] [PubMed] [Google Scholar]
- 36.Wu JJ, Wang H, Ye WC, Zuo XF, Zhao SX. Zhongguo Yao Ke Da Xue Xue Bao. 2006;37:487. [Google Scholar]
- 37.Nakatani M, Hatanaka S, Komura H, Kubota T, Hase T. Bull Chem Soc. 1989;62:469. [Google Scholar]