

Abstract
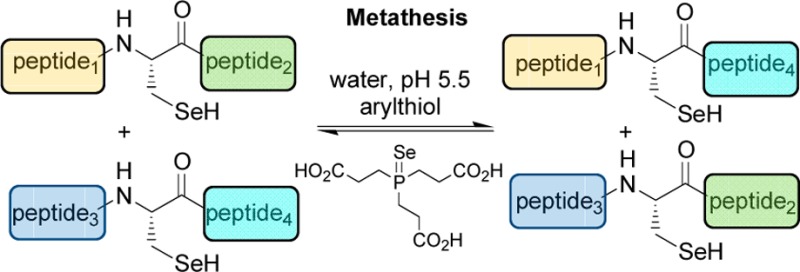
Selenopeptides can be transamidated by cysteinyl peptides in water using mild conditions (pH 5.5, 37 °C) in the presence of an arylthiol catalyst. Similar conditions also catalyze the metathesis of selenopeptides. The usefulness of the selenophosphine derived from TCEP (TCEP=Se) for inhibiting the TCEP-induced deselenization of selenocysteine residue is also reported.
Selenocysteine (Sec/U), i.e., the selenium analogue of cysteine (Cys/C), is a naturally occurring amino acid found in selenoproteins. The human genome encodes 25 selenoproteins1 into which one or several Sec residues are inserted cotranslationally. Most of the selenoproteins are redox enzymes that feature a Sec residue in their catalytic site. These proteins are collectively essential for living organisms as they participate in a large array of biological processes such as the control of the cellular redox balance.2 The loss of activity observed upon mutation of catalytic Sec residue into Cys in some selenoenzymes3,4 originates, at least in part, from the large difference in pKa and in reducing potential between Sec and Cys.5 Indeed, although selenium and sulfur are neighboring members of the chalcogen family, the pKa of the selenol group in Sec (pKa ∼5.5) is about 3 units less than the pKa of the thiol group (pKa 8.3) in Cys. Also, the redox properties of disulfides and diselenides differ significantly, the redox potential of diselenides being significantly lower than those of disulfides. Accordingly, the replacement of Cys by Sec is increasingly used in protein engineering approaches as a tool for modulating the stability,6 the physicochemical properties7 or the folding pathways of polypeptides.8
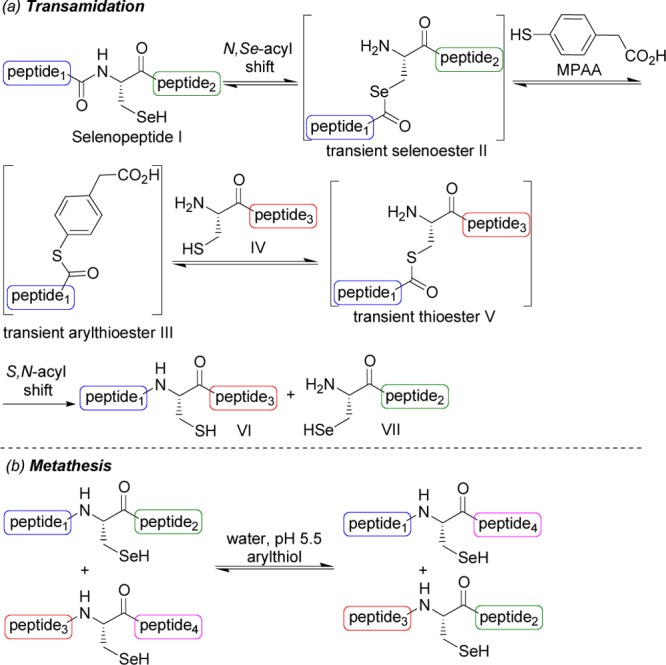
Recent studies have shown that the properties of selenopeptides have not been fully explored.9 Indeed, we report here a novel property for this important class of biomolecules by showing that selenopeptides featuring an internal or C-terminal Sec residue can participate in a transamidation reaction with cysteinyl peptides or in a metathesis reaction (Scheme 1). Both processes rely on an amine-carboxamide exchange reaction. Transamidation of amides usually requires harsch conditions (>250 °C) or metal catalysis10 in an organic solvent to occur due to the high stability of the carboxamide bond. In contrast, the transamidation and metathesis reactions of selenopeptides reported here take place in water at 37 °C at mild acidic pH (pH 5.5) in the presence of 4-mercaptophenylacetic acid (MPAA).11 Previous studies have demonstrated the metathesis of peptidomimetics featuring a reversible thioester bond12−14 or very recently an α-aminoacyl N-alkylcysteine15 reversible latent thioester.
Scheme 1. Transamidation or Metathesis of Selenopeptides.
Reversible covalent bonds have a great potential in dynamic covalent chemistry (DCC).16−18 We show for the first time that the field of DCC can be potentially extended to native peptide structures by exploiting the reversibility of the peptide bond to selenocysteine in aqueous solution.
The first steps of the transamidation reaction depicted in Scheme 1 proceed presumably through a reverse native chemical ligation (NCL).19 In particular, an N,Se-acyl migration reaction produces the transient selenoester20 II which enters into a series of exchanges with MPAA and the cysteinyl peptide to ultimately produce the transient thioester V. A related N,S-acyl shift of peptides featuring a C-terminal Cys residue, and the subsequent displacement of the transient thioester by an excess of an alkylthiol21 or hydrazine22 nucleophile has been shown to occur under forcing conditions. Interestingly, the displacement of a C-terminal Sec residue by an excess of an alkylthiol could be performed using milder conditions (40 °C).23 We reasoned that incubating the selenopeptides in water in the presence of MPAA would enable the formation of the transient arylthioesters of type III (Scheme 1), which are known to be much better acyl donor components in the NCL reaction than alkylthioesters.24 Once produced, the transient thioester V was expected to rearrange spontaneously into peptide VI by an S,N-acyl shift mechanism. The last step is poorly reversible in water at 37 °C at mildly acidic pH and therefore drives the reaction toward the formation of peptide VI.15 Overall, the transamidation reaction shown in Scheme 1 proceeds by using the mild experimental conditions designed for the NCL reaction.
The selenopeptides 3 needed for this study were produced by reacting chemoselectively the cyclic disulfide form of bis(2-sulfanylethyl)amido (SEA) peptides 1(25) with selenocystine 2 in the presence of MPAA (200 mM) (Scheme 2). Activation of the latent SEA thioester and of the Sec component required to perform the reaction in the presence of a disulfide and diselenide reducing agent. In a first approach, the reaction of peptide 1a (X = Phe) with selenocystine 2 in the presence of tris(2-carboxyethyl)phosphine (TCEP, 200 mM) furnished the selenopeptide 3a together with the corresponding deselenized product (Sec → Ala, ∼10% by HPLC). The TCEP-induced deselenization of selenopeptides is reminiscent of the desulfurization of thiols by phosphites.26,27 It has been mentioned by several authors as a serious side-reaction during the study of selenopeptides or proteins in the presence of TCEP.28,29 Alternately, it has been combined with the NCL reaction of C-terminal peptide thioesters with N-terminal Sec peptides30,31 to produce a native peptide bond to alanine.32,33 This problem was solved by using dithiothreitol (DTT, 200 mM) which instead furnished successfully diselenides 3a–h or 5d,e in good yields, showing that SEA native peptide ligation is complementary to the NCL reaction for accessing to selenopeptides.
Scheme 2. Synthesis of Model Selenopeptides 3 or 5.

The first step of the amine–amide exchange processes depicted in Scheme 1 involves the N,Se-acyl shift of the Sec residue, which proceeds through the nucleophilic attack of the free selenol group on the carbonyl group of the preceding amino acid residue. The reduction of the diselenides and the maintenance of Sec residue in the selenol form required the presence of a strong reducing agent in the mixture throughout the process. In our hands, the reduction of diselenides 3 or 5 by an excess of DTT was found to be sluggish and often incomplete after several hours, in accord with previous reports.8 Moreover, the recent use of DTT with reversible tertiary amide thioester surrogates resulted in acyl-DTT thioester and/or O-ester side-product formation.15 Therefore, the use of TCEP, a powerful irreversible reducing agent for disulfides and diselenides, was envisioned as an alternative. However, we had to find a solution for avoiding the TCEP-induced deselenization upon extended reaction times. MPAA is known to inhibit this process but failed to protect Sec residue against deselenization when used alone. Extensive experimentation led us to the conclusion that the water-soluble selenophosphine derived from TCEP, i.e., TCEP=Se, is very efficient in inhibiting the deselenization of Sec residue by TCEP in the conditions used for the transamidation process, provided it is used at concentration greater than 150 mM (see the Supporting Information). It is produced in one step and good yield by reacting metallic selenium and TCEP (see the Supporting Information). It inhibits also the desulfurization of Cys peptides by TCEP. Its inhibitory properties are specific of the phosphine selenide bond since the phosphine sulfide analogue, i.e., TCEP=S, had no inhibitory activity. The mode of action of TCEP=Se is still under investigation. It acts synergistically with sodium ascorbate, another well-known inhibitor of the TCEP-induced dechalcogenation process.34,35 The combined use of TCEP=Se and sodium ascorbate was found to be particularly useful for preventing Sec deselenization during the metathesis reactions which required several days for the equilibration to occur.
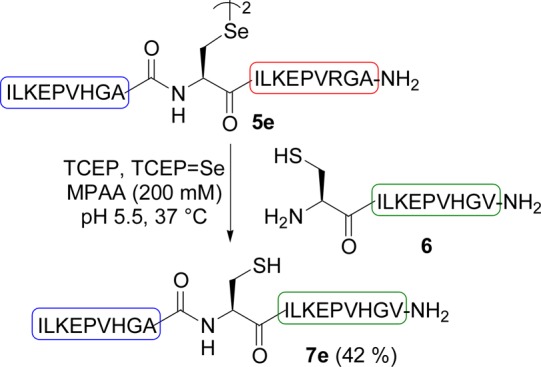
The transamidation process was first examined using selenopeptides 3 featuring a C-terminal Sec residue and differing by the penultimate amino acid X (Scheme 3). The exchange reaction with Cys peptide 6 proceeded faster for Ala, Arg, Glu, and Phe as the penultimate amino acid than for Gly, Cys, Ser, and Thr (Table 1, see also Figure S17 in the Supporting Information). A partial racemization of X residue was observed, especially for Phe (3a → 7a, Table 1). Importantly, the exchange reaction proceeded also with an internal Sec residue as for peptides 5e (X = Ala, Scheme 4), albeit with an initial kinetic rate of only 3% that of C-terminal Sec analog 3e (Table 1). An even larger difference in the initial transamidation kinetic rate was observed between selenopeptides 5d and 3d (X = Arg, Table 1).
Scheme 3. Transamidation of C-Terminal Sec Peptides 3.
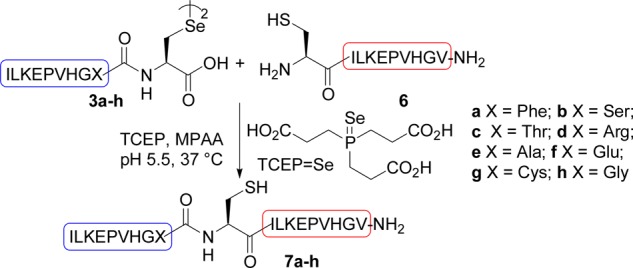
Table 1. Study of the Transamidation Reactiona.
| Sec peptide 3 | X | 3 D-AA content for X (%) | initial reaction rateb | yieldc (%) of 7 | 7 D-Aa content for X (%) |
|---|---|---|---|---|---|
| 3a | Phe | 0.34 | 0.30 | 7a: 41 | 4.5 |
| 3b | Ser | d | 0.17 | ||
| 3c | Thr | 0.04 | |||
| 3d | Arg | 0.19 | 0.97 | 7d: 34 | 1.1 |
| 3e | Ala | 0.36 | 1 | 7e: 43 | 2.2 |
| 3f | Glu | 0.95 | 0.66 | 7f: 36 | 1.4 |
| 3g | Cys | 0.26 | |||
| 3h | Gly | 0.27 | |||
| 5d | Arg | 0.005 | |||
| 5e | Ala | 0.36 | 0.03 | 7e: 42 | 0.86 |
Diselenide 3.5 mM, peptide 6 7 mM, 200 mM MPAA, 70 mM TCEP, 210 mM TCEP=Se, pH 5.5, 37 °C under nitrogen atmosphere.
Relative to peptide 3e (X = Ala), initial rate of peptide 7e formation is 1.17 mM/h (see the Supporting Information).
Isolated by HPLC. Identical by LC–MS to authentic samples obtained by SPPS.
Not determined.
Scheme 4. Transamidation of Internal Sec Peptides 5e.
We next examined the possibility to perform a metathesis reaction using selenopeptides 5e (X = Ala) and 3d (X = Arg) and MPAA as catalyst (Scheme 5). Hopefully, the reaction showed the formation of two novel selenopeptide products, peptides 5d and 3e (Figure 1a), which coeluted with authentic samples by HPLC. The identity of these compounds was further confirmed by extensive mass spectrometry fragmentation analysis. We observed also two other products, i.e., peptides ILKEPVHGA-OH and ILKEPVHGR-OH, arising from the X-Sec peptide bond hydrolysis (peptides 8d,e, Figure 1a). The level of hydrolysis products 8d and 8e remained below 10% after 100 h (see Figures S24 and S27 in the Supporting Information).
Scheme 5. Metathesis of selenopeptides.
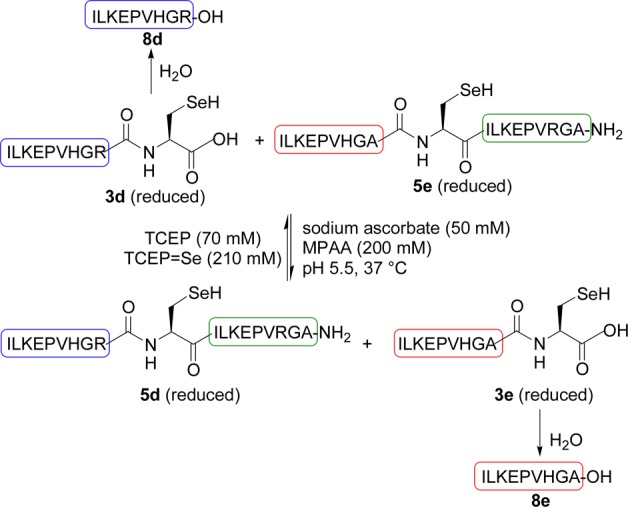
Figure 1.

Kinetic rates of the selenopeptide metathesis reactions and HPLC profiles of the crude metathesis reactions.
Importantly, the reversibility of the metathesis reaction was demonstrated by performing another metathesis experiment starting from selenopeptides 5d and 3e, which showed the formation of peptides 5e and 3d (Figure 1b). After ∼95–100 h, both forward and reverse metathesis reactions showed similar proportions for selenopeptides 3d and 3e but not for peptides 5d and 5e. The equilibrium is probably perturbed by the concomitant hydrolysis of the selenopeptides. The hydrolysis probably proceeded through C-terminal Sec peptides 3. Indeed, peptide 8d was the major hydrolysis product in the first metathesis reaction starting from 5e and 3d (Figure 1a), whereas peptide 8e was formed preferably in the second metathesis reaction starting from 5d and 3e (Figure 1b).
In conclusion, we have demonstrated that selenopeptides can be transamidated by N-terminal cysteinyl peptides in water using mild experimental conditions. Besides the pH which is mildy acidic (pH 5.5), the conditions which enable the reversibility of the peptide bond to selenocysteine and the transamidation reactions are similar to those used for the NCL reaction. These conditions also catalyze the metathesis of selenopeptides. Further work is in progress to improve the kinetics of the metathesis reaction and minimize the hydrolysis rate, which nevertheless remained in our conditions within acceptable levels. These novel chemical properties for selenopeptides may open the possibility to use native peptide structures, i.e., with no modifications on the peptide backbone, in dynamic covalent chemistry approaches.
Supporting Information Available
Experimental procedures and characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kryukov G. V.; Castellano S.; Novoselov S. V.; Lobanov A. V.; Zehtab O.; Guigo R.; Gladyshev V. N. Science 2003, 300, 1439. [DOI] [PubMed] [Google Scholar]
- Reeves M. A.; Hoffmann P. R. Cell. Mol. Life Sci. 2009, 66, 2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. R.; Bar-Noy S.; Kwon J.; Levine R. L.; Stadtman T. C.; Rhee S. G. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromer S.; Johansson L.; Bauer H.; Arscott L. D.; Rauch S.; Ballou D. P.; Williams C. H. Jr.; Schirmer R. H.; Arner E. S. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroder L. J. Pept. Sci. 2005, 11, 187. [DOI] [PubMed] [Google Scholar]
- Muttenthaler M.; Andersson A.; de Araujo A. D.; Dekan Z.; Lewis R. J.; Alewood P. F. J. Med. Chem. 2010, 53, 8585. [DOI] [PubMed] [Google Scholar]
- Berry S. M.; Gieselman M. D.; Nilges M. J.; van der Donk W. A.; Lu Y. J. Am. Chem. Soc. 2002, 124, 2084. [DOI] [PubMed] [Google Scholar]
- Gowd K. H.; Yarotskyy V.; Elmslie K. S.; Skalicky J. J.; Olivera B. M.; Bulaj G. Biochemistry 2010, 49, 2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobli M.; Morgenstern D.; King G. F.; Alewood P. F.; Muttenthaler M. Angew. Chem., Int. Ed. 2011, 50, 11952. [DOI] [PubMed] [Google Scholar]
- Stephenson N. A.; Zhu J.; Gellman S. H.; Stahl S. S. J. Am. Chem. Soc. 2009, 131, 10003. [DOI] [PubMed] [Google Scholar]
- Johnson E. C.; Kent S. B. J. Am. Chem. Soc. 2006, 128, 6640. [DOI] [PubMed] [Google Scholar]
- Hadley E. B.; Witek A. M.; Freire F.; Peoples A. J.; Gellman S. H. Angew. Chem., Int. Ed. 2007, 46, 7056. [DOI] [PubMed] [Google Scholar]
- Price J. L.; Hadley E. B.; Steinkruger J. D.; Gellman S. H. Angew. Chem., Int. Ed. 2010, 49, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadon Z.; Samiappan M.; Shahar A.; Zarivach R.; Ashkenasy G. Angew. Chem., Int. Ed. 2013, 52, 9944. [DOI] [PubMed] [Google Scholar]
- Ruff Y.; Garavini V.; Giuseppone N. J. Am. Chem. Soc. 2014, 136, 6333. [DOI] [PubMed] [Google Scholar]
- Rowan S. J.; Cantrill S. J.; Cousins G. R.; Sanders J. K.; Stoddart J. F. Angew. Chem., Int. Ed. 2002, 41, 898. [DOI] [PubMed] [Google Scholar]
- Ramstrom O.; Lehn J. M. Nat. Rev. Drug Discovery 2002, 1, 26. [DOI] [PubMed] [Google Scholar]
- Corbett P. T.; Leclaire J.; Vial L.; West K. R.; Wietor J.-L.; Sanders J. K. M.; Otto S. Chem. Rev. 2006, 106, 3652. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Muir T. W.; Clark-Lewis I.; Kent S. B. Science 1994, 266, 776. [DOI] [PubMed] [Google Scholar]
- Durek T.; Alewood P. F. Angew. Chem., Int. Ed. 2011, 50, 12042. [DOI] [PubMed] [Google Scholar]
- Kang J.; Richardson J. P.; Macmillan D. Chem. Commun. 2009, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams A. L.; Cowper B.; Morgan R. E.; Premdjee B.; Caddick S.; Macmillan D. Angew. Chem., Int. Ed. 2013, 52, 13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams A. L.; Macmillan D. J. Pept. Sci. 2013, 19, 65. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Churchill M. J.; Ghadiri M. R.; Kent S. B. H. J. Am. Chem. Soc. 1997, 119, 4325. [Google Scholar]
- Ollivier N.; Dheur J.; Mhidia R.; Blanpain A.; Melnyk O. Org. Lett. 2010, 12, 5238. [DOI] [PubMed] [Google Scholar]
- Hoffmann F. W.; Ess R. J.; Simmons T. C.; Hanzel R. S. J. Am. Chem. Soc. 1956, 78, 6414. [Google Scholar]
- Walling C.; Rabinowitz R. J. Am. Chem. Soc. 1957, 79, 5326. [Google Scholar]
- Muttenthaler M.; Nevin S. T.; Grishin A. A.; Ngo S. T.; Choy P. T.; Daly N. L.; Hu S. H.; Armishaw C. J.; Wang C. I.; Lewis R. J.; Martin J. L.; Noakes P. G.; Craik D. J.; Adams D. J.; Alewood P. F. J. Am. Chem. Soc. 2010, 132, 3514. [DOI] [PubMed] [Google Scholar]
- Gorlatov S. N.; Stadtman T. C. Arch. Biochem. Biophys. 1999, 369, 133. [DOI] [PubMed] [Google Scholar]
- Quaderer R.; Sewing A.; Hilvert D. Helv. Chim. Acta 2001, 84, 1197. [Google Scholar]
- Gieselman M. D.; Xie L.; van der Donk W. A. Org. Lett. 2001, 3, 1331. [DOI] [PubMed] [Google Scholar]
- Metanis N.; Keinan E.; Dawson P. E. Angew. Chem., Int. Ed. 2010, 49, 7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malins L. R.; Payne R. J. Org. Lett. 2012, 14, 3142. [DOI] [PubMed] [Google Scholar]
- Rohde H.; Schmalisch J.; Harpaz Z.; Diezmann F.; Seitz O. ChemBioChem 2011, 12, 1396. [DOI] [PubMed] [Google Scholar]
- Forni L. G.; Monig J.; Mora-Arellano V. O.; Willson R. L. J. Chem. Soc., Perkin Trans 2 1983, 961. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.