

Abstract

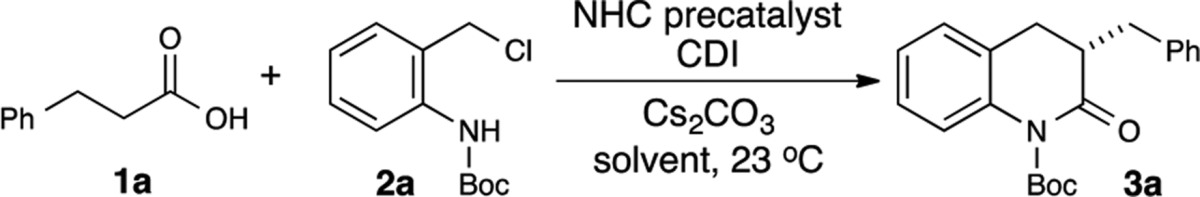
A convergent, catalytic asymmetric formal [4 + 2] annulation for the synthesis of dihydroquinolones has been developed. Carboxylic acids can be employed as precursors to NHC enolates through an in situ activation strategy. Simultaneous generation of a reactive aza-o-quinone methide under the basic conditions employed for NHC generation leads to a dual activation approach.
Dihydroquinolones are found in numerous natural products and therapeutically relevant compounds.1 Due to the prevalence of this structural motif in bioactive compounds and approved pharmaceuticals such as cilostazol, cartelolol, and pinolinone, as well as its utility as a synthetic intermediate for the synthesis of other bioactive compounds such as tetrahydroquinolines, the development of new asymmetric methods to fashion dihydroquinolone is a high value goal.2 Previous approaches for the preparation of enantioenriched quinolone derivatives mainly utilize transition metal catalysis.3 Within the area of organocatalysis, the Córdova group reported an asymmetric synthesis of 1,2-dihydroquinolidines using chiral amine catalysts,4 and the Lu group reported an asymmetric synthesis of 2-aryl-2,3-dihydro-4-quinolones using bifunctional thiourea catalysts.5 More recently, the groups of Akiyama6 and Gong7 independently reported a selective approach for the preparation of tetrahydroquinoline derivatives using chiral phosphoric acids.8 However, a convergent and efficient asymmetric approach for the synthesis of the dihydroquinolin-2-one scaffold is currently underdeveloped, and such a transformation could facilitate access to this privileged scaffold with various substitution patterns and high levels of enantioselectivity.
Organocatalysis has emerged as a powerful strategy to construct various hetero- and carbocyclic systems. N-Heterocyclic carbenes (NHCs) in particular demonstrate great versatility and selectivity to facilitate many transformations, including enolate processes and nontraditional umpolung reactivity.9 We have been engaged in enhancing NHC reactivity and selectivity through the integration of this Lewis base catalysis mode with other strategies, including Lewis acids,10,11 Lewis bases such as fluoride,12 and Brønsted acids.13,14 On the basis of this cooperative catalysis/activation concept, we envisioned that a “dual activation” approach could lead to the formation of dihydroquinolone derivatives. The dual activation classification would apply if a compatible Brønsted base could be leveraged to both provide NHC formation from the azolium salt precatalysts and promote in situ creation of an aza-o-quinone methide (aza-o-QM).15 This species could behave as an electrophile with an NHC-enolate to provide the desired dihydroquinolone in a convergent process (Figure 1A). However, given the potential high reactivity of the aza-o-QM, it was also entirely possible at the onset of these investigations that a nucleophilic NHC and/or base would simply undergo an unproductive addition reaction.
Figure 1.

Dual activation strategy.
At the onset, cinnamaldehyde and tert-butyl carbamate (Boc) protected 2-aminobenzyl chloride were employed as the nulcleophile and electrophile precursors, respectively (not shown). With cinnamaldehyde as the enolate progenitor, a mixture of several products was observed, with dimers formed through the enolate (not shown) and homoenolate (shown, Figure 1B) pathways predominating. This dimerization with enals as substrates for NHC-enolate or NHC-homoenolate reactions continues to be a significant challenge in reaction design in the area of carbene catalysis.16 We hypothesized that carboxylic acids could be activated in situ through the appropriate reagent combinations to react with a NHC and thus generate the desired NHC-enolate/acyl azolium. Once the carboxylic acid is activated, the acylation of the NHC and subsequent enolization could form the necessary NHC enolate, thereby eliminating the possibility of dimerization observed with enals. Carboxylic acids present two advantages: (1) they are readily available and more stable compared to enals, α-aryloxy acetalaldehydes,17 and α-functionalized alkyl aldehydes used previously in carbene-catalyzed enolate reactions,18 and (2) they are less prone to undergo enolate/aldol chemistry compared to ketones and activated esters. In situ activation of acids to access enolate reactivity has proven successful in organocatalysis,19 but this strategy has not seen successful implementation in carbene catalysis.20 A potential complication in this context with carbene catalysis would be the direct addition of the nucleophilic C2 position of the NHC to any in situ activation reagent. Recently, the Chi group has demonstrated the ability to access NHC enolates through the acylation of carbenes with saturated highly activated aryl esters.21a,11b,21b However, the use of more stable acid substrates directly in NHC catalysis has not been reported. Herein, we report an asymmetric annulation reaction for the synthesis of dihydroquinolone derivatives with carboxylic acids via in situ generated azolium enolates (Figure 1).22
To test our hypothesis, we screened several acid activating reagents (isobutyl chloroformate, SOCl2, 2,4,6-trichlorobenzoyl chloride, etc.) with hydrocinnamic acid and achiral NHC catalysts. Carbonyldiimidazole (CDI) in combination with an achiral 5,5-triazolium catalyst provided the desired dihydroquinolone in high isolated yield with minimal side products. After further screening with chiral triazolium catalysts (Table 1), we found that isobutyl-substituted catalyst D provided the desired product in moderate yield and enantioselectivity (entry 4). We hypothesized that the main reason for the depressed yield and enantioselectivity stemmed from decomposition of our in situ generated acyl imidazole and/or aza-o-quinone methide. By lowering the reaction temperature (4 °C), we were able to slightly improve both the yield and enantioselectivity (entry 5). Increasing the amount of CDI also improved the reaction yield (entry 6). The most drastic increase in yield was obtained when we added 60 mol % of imidazole, which presumably acts as a homogeneous scavenger of HCl to prevent the inactivation of our starting materials and catalyst in the reaction (entry 7). We turned our attention toward improving the enantioselectivity and screened various amino acid derived 5,5-triazolium catalysts (entries 8–16). The best results were obtained with benzyl-substituted catalyst G in terms of reactivity and enantioselectivity, with the desired product in 80% yield and 94:6 er (entry 10). With catalyst H, the desired product was obtained in slightly improved er, but decreased yield (entry 14).
Table 1. Optimization of Asymmetric Reaction Conditionsa.

Conditions: 1a (0.05 mmol, 1.0 equiv), 2a (2.0 equiv), triazolium (0.2 equiv), Cs2CO3 (2.5 equiv) at 23 °C for 15 h.
Determined by NMR analysis with 1,3,5-trimethoxybenzene as an internal standard.
Determined by HPLC analysis.
Reaction conducted at 4 °C.
60 mol % of imidazole was added.
With the optimized reaction conditions in hand, the scope of this formal [4 + 2] annulation was explored (Table 2). The reaction turned out to be tolerant of various starting acids. The desired dihydroquinolone derivatives were obtained in good yields (52–84%) with high enantioselectivities (up to 98:2 er) with alkyl (3l and 3m), benzyl (3a–3i), aryl (3j), and heteroalkyl (3k) substituted acids. The products derived from meta-substituted acids (3d and 3e) were obtained with slightly higher enantioselectivity; however, a noticeable electronic effect of the substituents was not observed. Gratifyingly, aliphatic carboxylic acids were suitable substrates under our reaction conditions. The isopropyl-substituted hydroquinolone was obtained in high yield (81%) and excellent enantioselectivity (98:2 er) by employing isovaleric acid (3l). Interestingly, this catalytic system could also provide high yield and enantioselectivity for acids of minimal size such as propanoic acid (3m). The tolerance of functionality and substitution on the aminobenzyl chloride 2 was also investigated. Both electron-withdrawing and electron-rich groups were tolerated at the C-4, C-5, and C-6 positions affording the desired products 3n–3t in good to high yields and high enantioselectivities. However, no product was isolated when using C-3 substituted aminobenzyl chlorides because many of these potential substrates were not stable under the reaction conditions.
Table 2. Substrate Scopea.


Reactions conducted on 0.3 mmol scale. Yields are of isolated product after column chromatography. Enantiomeric ratio was determined by HPLC analysis.
Reaction conducted on 5 mmol scale.
Absolute configuration was determined by X-ray crystallography.
Two different mechanisms are hypothesized for the formation of dihydroquinolone 3 from enol I-A (Scheme 1): (1) a concerted [4 + 2] process and (2) a formal [4 + 2] process involving a stepwise Michael addition and annulation. The addition of catalyst G to acyl imidazole 1 generated in situ and subsequent formal 1,2-H migration generates NHC-enolate I-A. The reaction between the intermediate I-A and Boc-aminobenzyl chloride 2 could proceed through either a concerted [4 + 2] pathway or a stepwise NHC-acylation process. In the [4 + 2] pathway, concerted addition of in situ generated aza-o-quinone methide 2 and NHC enolate I-A via II (via either endo or exo transition states) could provide the desired product 3.23 In the Michael addition/acylation pathway, carbon–carbon bond formation through the less sterically demanding transition state (III) and subsequent tautomerization forms acyl azolium intermediate IV, which then undergoes N-acylation to afford 3 and the NHC, now free to re-enter the catalytic cycle. The current model for asymmetric induction is based on the more favored NHC-enolate conformation I-A versus the higher energy possibility of I–B based on DFT calculations of these two rotamers (Figure 2).24−26
Scheme 1. Proposed Reaction Pathway.

Figure 2.

DFT model for asymmetric induction.
The enantioenriched dihydroquinolone product 3a was converted into enantioenriched tetrahydroquinoline derivative 4 and thioamide 5 to demonstrate the utility of this new transformation (Scheme 2). After removal of the Boc protecting group, the resulting N–H lactam was reduced using LiAlH4/AlCl3 to afford the desired tetrahydroquinoline derivative (4), which is another important scaffold due to its bioactive properties as antihypertensives, oxytocin antagonists, vasopressin antagonists, and calcium antagonists.27 The amide can also be converted into the corresponding thioamide (5) without racemization.28 This heteroatom conversion provides a platform for further known transformations (e.g., annulations and amidine formation) to access potentially bioactive architectures.29
Scheme 2. Synthetic Transformations.

Reagents: (a) TFA, CH2Cl2; (b) Lawesson’s reagent, CH2Cl2; (c) TFA, CH2Cl2; (d) LiAlH4, AlCl3, Et2O/DCM
In conclusion, the first highly efficient enantioselective NHC-catalyzed annulation reaction for the synthesis of dihydroquinolones has been developed. This transformation features a dual activation system focusing on combining a Lewis base (NHC) and in situ generated active nucleophiles and electrophiles. In addition, the utilization of unactivated carboxylic acids directly as substrates provides the desired dihydroquinolone derivatives and importantly avoids generation of dimerization side products observed when employing enals. The demonstration that activating strategies and highly reactive aza-o-quinone methides are compatible with nucleophilic carbene catalysis presents new opportunities to pursue in the context of asymmetric heterocyclic synthesis.
Acknowledgments
Financial support was generously provided by the NIH NIGMS (GM073072). We thank Michael Wang (NU) for assistance with X-ray crystallography.
Supporting Information Available
Experimental procedures and spectral data for new compounds, geometries, and energies of all structures discussed. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Katritzky A. R.; Rachwal S.; Rachwal B. Tetrahedron 1996, 52, 15031–15070. [Google Scholar]; b Tamura S. Y.; Goldman E. A.; Bergum P. W.; Semple J. E. Bioorg. Med. Chem. Lett. 1999, 9, 2573–2578. [DOI] [PubMed] [Google Scholar]; c Oshiro Y.; Sakurai Y.; Sato S.; Kurahashi N.; Tanaka T.; Kikuchi T.; Tottori K.; Uwahodo Y.; Miwa T.; Nishi T. J. Med. Chem. 2000, 43, 177–189. [DOI] [PubMed] [Google Scholar]; d Zhao H.; Thurkauf A.; Braun J.; Brodbeck R.; Kieltyka A. Bioorg. Med. Chem. Lett. 2000, 10, 2119–2122. [DOI] [PubMed] [Google Scholar]
- Horn J.; Li H. Y.; Marsden S. P.; Nelson A.; Shearer R. J.; Campbell A. J.; House D.; Weingarten G. G. Tetrahedron 2009, 65, 9002–9007. [Google Scholar]
- For selected examples, see:; a Shintani R.; Yamagami T.; Kimura T.; Hayashi T. Org. Lett. 2005, 7, 5317–5319. [DOI] [PubMed] [Google Scholar]; b Lei B.-L.; Ding C.-H.; Yang X.-F.; Wan X.-L.; Hou X.-L. J. Am. Chem. Soc. 2009, 131, 18250–18251. [DOI] [PubMed] [Google Scholar]; c Szöllösi G.; Makra Z.; Kovács L.; Fülöp F. Adv. Synth. Catal. 2013, 355, 1623–1629. [Google Scholar]
- Sundén H.; Rios R.; Ibrahem I.; Zhao G.-L.; Eriksson L.; Córdova A. Adv. Synth. Catal. 2007, 349, 827–832. [Google Scholar]
- Liu X.; Lu Y. Org. Lett. 2010, 12, 5592–5595. [DOI] [PubMed] [Google Scholar]
- Mori K.; Ehara K.; Kurihara K.; Akiyama T. J. Am. Chem. Soc. 2011, 133, 6166–6169. [DOI] [PubMed] [Google Scholar]
- Ren L.; Lei T.; Ye J.-X.; Gong L.-Z. Angew. Chem., Int. Ed. 2012, 51, 771–774. [DOI] [PubMed] [Google Scholar]
- For organocatalyzed asymmetric synthesis of 2-aryl-2,3-dihydro-4-quinolones, also see:; a Zheng H.; Liu Q.; Wen S.; Yang H.; Luo Y. Tetrahedron: Asymmetry 2013, 24, 875–882. [Google Scholar]; b Kanagaraj K.; Pitchumani K. J. Org. Chem. 2013, 78, 744–751. [DOI] [PubMed] [Google Scholar]
- a Enders D.; Niemeier O.; Henseler A. Chem. Rev. 2007, 107, 5606–5655. [DOI] [PubMed] [Google Scholar]; b Phillips E. M.; Chan A.; Scheidt K. A. Aldrichimica Acta 2009, 42, 55–66. [PMC free article] [PubMed] [Google Scholar]; c Vora H. U.; Sheeler P.; Rovis T. Adv. Synth. Catal. 2012, 354, 1617–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bugaut X.; Glorius F. Chem. Soc. Rev. 2012, 41, 3511–3522. [DOI] [PubMed] [Google Scholar]
- a Cardinal-David B.; Raup D. E. A.; Scheidt K. A. J. Am. Chem. Soc. 2010, 132, 5345–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Phillips E. M.; Riedrich M.; Scheidt K. A. J. Am. Chem. Soc. 2010, 132, 13179–13181. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Raup D. E. A.; Cardinal-David B.; Holte D.; Scheidt K. A. Nat. Chem. 2010, 2, 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Cohen D. T.; Cardinal-David B.; Scheidt K. A. Angew. Chem., Int. Ed. 2011, 50, 1678–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Dugal-Tessier J.; O’Bryan E. A.; Schroeder T. B. H.; Cohen D. T.; Scheidt K. A. Angew. Chem., Int. Ed. 2012, 51, 4963–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mo J.; Chen X.; Chi Y. R. J. Am. Chem. Soc. 2012, 134, 8810–8813. [DOI] [PubMed] [Google Scholar]; b Chen X.; Yang S.; Song B.-A.; Chi Y. R. Angew. Chem., Int. Ed. 2013, 52, 11134–11137. [DOI] [PubMed] [Google Scholar]
- Izquierdo J.; Orue A.; Scheidt K. A. J. Am. Chem. Soc. 2013, 135, 10634–10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker E. O.; Scheidt K. A. Angew. Chem., Int. Ed. 2013, 52, 13616–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhao X.; Dirocco D. A.; Rovis T. J. Am. Chem. Soc. 2011, 133, 12466–12469. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu G.; Wilkerson P. D.; Toth C. A.; Xu H. Org. Lett. 2012, 14, 858–861. [DOI] [PubMed] [Google Scholar]; c Fu Z.; Sun H.; Chen S.; Tiwari B.; Li G.; Chi Y. R. Chem. Commun. 2013, 49, 261–263. [DOI] [PubMed] [Google Scholar]
- a Steinhagen H.; Corey E. J. Angew. Chem., Int. Ed. 1999, 38, 1928–1931. [DOI] [PubMed] [Google Scholar]; b Avemaria F.; Vanderheiden S.; Bräse S. Tetrahedron 2003, 59, 6785–6796. [Google Scholar]; c Yang Q.-Q.; Xiao C.; Lu L.-Q.; An J.; Tan F.; Li B.-J.; Xiao W.-J. Angew. Chem., Int. Ed. 2012, 51, 9137–9140. [DOI] [PubMed] [Google Scholar]; d Yang Q.-Q.; Wang Q.; An J.; Chen J.-R.; Lu L.-Q.; Xiao W.-J. Chem.—Eur. J. 2013, 19, 8401–8404. [DOI] [PubMed] [Google Scholar]
- Cohen D. T.; Cardinal-David B.; Roberts J. M.; Sarjeant A. A.; Scheidt K. A. Org. Lett. 2011, 13, 1068–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kawanaka Y.; Phillips E. M.; Scheidt K. A. J. Am. Chem. Soc. 2009, 131, 18028–18029. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Phillips E. M.; Wadamoto M.; Roth H. S.; Ott A. W.; Scheidt K. A. Org. Lett. 2009, 11, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Reynolds N. T.; Alaniz J. R. d.; Rovis T. J. Am. Chem. Soc. 2004, 126, 9518–9519. [DOI] [PubMed] [Google Scholar]; b Show K. Y.-K.; Bode J. W. J. Am. Chem. Soc. 2004, 126, 8126–8127. [DOI] [PubMed] [Google Scholar]
- For a review of this area, see:Morrill L. C.; Smith A. D. Chem. Soc. Rev. 2014, 10.1039/C4CS00042K. [DOI] [Google Scholar]; For seminal and recent examples, see:; a Cortez G. S.; Tennyson R. L.; Romo D. J. Am. Chem. Soc. 2001, 123, 7945–7946. [DOI] [PubMed] [Google Scholar]; b Cortez G. S.; Oh S.-H.; Romo D. Synthesis 2001, 1731–1736. [Google Scholar]; c Ling K. B.; Smith A. D. Chem. Commun. 2011, 47, 373–375. [DOI] [PubMed] [Google Scholar]; d Belmessieri D.; Morrill L. C.; Simal C.; Slawin A. M. Z.; Smith A. D. J. Am. Chem. Soc. 2011, 133, 2714–2720. [DOI] [PubMed] [Google Scholar]; e Manoni F.; Cornaggia C.; Murray J.; Tallon S.; Connon S. J. Chem. Commun. 2012, 48, 6502–6504. [DOI] [PubMed] [Google Scholar]; f Liu G.; Shirley M. E.; Romo D. J. Org. Chem. 2012, 77, 2496–2500. [DOI] [PubMed] [Google Scholar]; g Taylor J. E.; Daniels D. S. B.; Smith A. D. Org. Lett. 2013, 15, 6058–6061. [DOI] [PubMed] [Google Scholar]; h Stark D. G.; Morrill L. C.; Yeh P.-P.; Slawin A. M. Z.; O’Riordan T. J. C.; Smith A. D. Angew. Chem., Int. Ed. 2013, 52, 11642–11646. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Davies A. T.; Taylor J. E.; Douglas J.; Collett C. J.; Morrill L. C.; Fallan C.; Slawin A. M. Z.; Churchill G.; Smith A. D. J. Org. Chem. 2013, 78, 9243–9257. [DOI] [PubMed] [Google Scholar]; j Smith S. R.; Douglas J.; Prevet H.; Shapland P.; Slawin A. M. Z.; Smith A. D. J. Org. Chem. 2014, 79, 1626–1639. [DOI] [PubMed] [Google Scholar]
- For NHC-catalyzed generation of acyl azolium species using α,β-unsaturated acid fluorides, see:; a Ryan S. J.; Candish L.; Lupton D. W. J. Am. Chem. Soc. 2009, 131, 14176–14177. [DOI] [PubMed] [Google Scholar]; b Ryan S.; Candish L.; Lupton D. W. Synlett 2011, 2275–2278. [Google Scholar]; c Ryan S. J.; Candish L.; Lupton D. W. J. Am. Chem. Soc. 2011, 133, 4694–4697. [DOI] [PubMed] [Google Scholar]; d Candish L.; Forsyth C. M.; Lupton D. W. Angew. Chem., Int. Ed. 2013, 52, 9149–9152. [DOI] [PubMed] [Google Scholar]; e Candish L.; Lupton D. W. J. Am. Chem. Soc. 2013, 135, 58–61. [DOI] [PubMed] [Google Scholar]
- a Hao L.; Du Y.; Lv H.; Chen X.; Jiang H.; Shao Y.; Chi Y. R. Org. Lett. 2012, 14, 2154–2157. [DOI] [PubMed] [Google Scholar]; b Xu J.; Jin Z.; Chi Y. R. Org. Lett. 2013, 15, 5028–5031. [DOI] [PubMed] [Google Scholar]
- Douglas J.; Churchill G.; Smith A. D. Synthesis 2012, 44, 2295–2309. [Google Scholar]
- Ryan S. J.; Candish L.; Lupton D. W. J. Org. Chem. 2011, 133, 4694–4697. [DOI] [PubMed] [Google Scholar]
- Computed with Schrödinger interface using Jaguar DFT with B3LYP/6-31G**.
- For a related model of asymmetric induction with NHC-enolates invoking a rotamer similar to I-A, see:Zhao X.; Ruhl K. E.; Rovis T. Angew. Chem., Int. Ed. 2012, 51, 12330–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketene generation during this reaction is unlikely given the known methods for their preparation with strong leaving groups (e.g., acid chlorides):; a Tidwell T. T. Acc. Chem. Res. 1990, 23, 273–279. [Google Scholar]; b Tidwell T. T.Ketenes; John Wiley and Sons, Inc.: New York, 1995. [Google Scholar]; c Tidwell T. T.Ketenes II; John Wiley and Sons, Inc.: New York, 2006. [Google Scholar]; d Allen A. D.; Tidwell T. T. Eur. J. Org. Chem. 2012, 1081–1096Additionally, we have not observed any ketene dimer products from alkanoic acid substrates (1, R1 = Me). [Google Scholar]
- Sridharan V.; Suryavanshi P. A.; Menendez J. C. Chem. Rev. 2011, 111, 7157–7259. [DOI] [PubMed] [Google Scholar]
- Koduri N. D.; Scott H.; Hileman B.; Cox J. D.; Coffin M.; Glicksberg L.; Hussaini S. R. Org. Lett. 2012, 14, 440–443. [DOI] [PubMed] [Google Scholar]
- a Guarna A.; Lombardi E.; Machetti F.; Occhiato E. G.; Scarpi D. J. Org. Chem. 2000, 65, 8093–8095. [DOI] [PubMed] [Google Scholar]; b Occhiato E. G.; Ferrali A.; Menchi G.; Guarna A.; Danza G.; Comerci A.; Mancina R.; Serio M.; Garotta G.; Cavalli A.; Vivo M. D.; Recanatini M. J. Med. Chem. 2004, 47, 3546–3560. [DOI] [PubMed] [Google Scholar]; c Delcros J.-G.; Tomasi S.; Duhieu S.; Foucault M.; Martin B.; Roch M. L.; Eifler-Lima V.; Renault J.; Uriac P. J. Med. Chem. 2006, 49, 232–245. [DOI] [PubMed] [Google Scholar]; d Lucas S.; Heim R.; Ries C.; Schewe K. E.; Birk B.; Hartmann R. W. J. Med. Chem. 2008, 51, 8077–8087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.