

Abstract
Hydrogen sulfide (H2S) is an important biological signaling agent that exerts action on numerous (patho)physiological processes. Once generated, H2S can be oxidized to generate reductant-labile sulfane sulfur pools, which include hydrodisulfides/persulfides. Despite the importance of hydrodisulfides in H2S storage and signaling, little is known about the physical properties or chemical reactivity of these compounds. We report here the synthesis, isolation, and characterization (NMR, IR, Raman, HRMS, X-ray) of a small-molecule hydrodisulfide and highlight its reactivity with reductants, nucleophiles, electrophiles, acids, and bases. Our experimental results establish that hydrodisulfides release H2S upon reduction and that deprotonation results in disproportionation to the parent thiol and S0, thus providing a mechanism for transsulfuration in the sulfane sulfur pool.
Hydrogen sulfide (H2S) is now accepted as an important signaling molecule that exerts physiological functions in the cardiovascular, neuronal, and immune systems.1 Produced endogenously from cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), and cysteine amino transferase (CAT) in combination with 3-mercaptosulfurtransferase (3MST), H2S exists primarily as HS– under normal physiological conditions.1 In addition to this freely diffusible form, H2S oxidation can generate reductant-labile sulfane sulfur pools containing hydrodisulfides/persulfides (RSSH), hydropolysulfides (RS(Sn)SH), and polysulfides (RS(Sn)SR), all of which may be present in micromolar concentrations.2 In addition to their possible roles in H2S storage, the S-sulfhydration (or S-persulfidation) of protein Cys residues is postulated to be an important oxidative post-translational modification that contributes to H2S signaling and action.3 In addition to reductant-labile sulfane sulfur, sulfide can also be stored in acid-labile sulfur pools, such as iron sulfur cluster-containing proteins.4 Although, Kevil et al. recently reported methods to separate the free, acid-labile, and reductant-labile sulfide pools,5 there remains controversy surrounding the basic reactivity of sulfane sulfur, specifically persulfides. For example, although some reports suggest that reductant-labile sulfane sulfur is also acid-labile,6 it is more likely that acid-labile and reductant-labile sulfur form two chemically distinct pools.7
Because the chemical reactivity of hydrodisulfides is poorly established, the chemical underpinnings for detecting3a,8 and differentiating −SSH from −SH groups have been fraught with problems, often leading to variable results from different labeling strategies. A better functional understanding of the chemical reactivity of hydrodisulfides would not only contribute to the quickly evolving strategies for detection, but would also clarify the potential roles of hydrodisulfides in H2S chemistry and signaling. Based on this current need, we viewed that isolation of a discrete small-molecule hydrodisulfide model would allow for the reactivity of this important functional group to be established under controlled conditions. Motivated by these challenges, we report here the preparation, isolation, and full characterization of tritylhydrodisulfide (TrtSSH) and confirm that under hydrophobic conditions hydrodisulfides release H2S upon reduction, but not upon protonation or by nucleophilic attack.
To establish the spectroscopic and reactivity properties of hydrodisulfides, we chose to prepare tritylhydrodisulfide due to stability of the S-nitrosothiol analogue, TrtSNO, and the crystallinity of the trityl group. Although there are various reports of hydrodisulfides in the literature, to the best of our knowledge, none of these have been characterized completely.9 We viewed that isolation and characterization of the −SSH functional group would allow for other researchers to better identify and characterize similar compounds in the future. Additionally, studies focused on hydrodisulfide reactivity with different electrophiles have used hydrodisulfides generated in situ rather than isolated hydrodisulfides.10 Although the trityl group precludes water solubility, the investigation of hydrodisulfide reactivity in organic solution may provide insights into the chemistry of hydrodisulfides in hydrophobic environments in protein architectures, which are proposed to be involved in the activation of various enzymes. To prepare TrtSSH, we first treated TrtSH with AcSCl (3) in Et2O to generate TrtSSAc (4). Removal of the acetyl protecting group by acidic ethanolysis affords the desired hydrodisulfide product, TrtSSH (Scheme 1).
Scheme 1. Synthesis of TrtSSH.

With TrtSSH in hand, we fully characterized the compound and compared its chemical properties to those of TrtSH. Both the 1H and 13C{1H} NMR spectra of TrtSSH are similar to those of TrtSH, but the 1H NMR resonance of the sulfhydryl SSH is shifted upfield to 2.7 ppm, by comparison to 3.1 ppm in TrtSH (Figures S4, S5). Much of the infrared and Raman spectra of TrtSSH and TrtSH are also similar due to their related structures; however, the S–H stretch shifts to a lower wavenumber by 65 cm–1 for TrtSSH when compared to TrtSH (Figure 1b). Comparison of the Raman spectrum of TrtSSH with that of S8 showed similarities in the 200–500 cm–1 region corresponding sulfur–sulfur bond formation. HRMS analysis of TrtSSH confirmed its atomic composition, affording an m/z value of 307.0631 amu, which agrees well with the calculated value of 307.0621 amu for TrtSS–. To further confirm the molecular structure of TrtSSH, single crystals suitable for X-ray diffraction were grown from toluene/Et2O. TrtSSH crystallizes in P212121 with an S–S bond length of 2.0396(12) Å and a CSSH dihedral angle of 82.2°, both of which are within the ranges common for disulfides (Figure 1a). The terminal −SSH hydrogen was located from the residual density map, and inspection of the packing diagram shows that it does not form hydrogen bonds with other atoms in the solid state (Figure S28).
Figure 1.
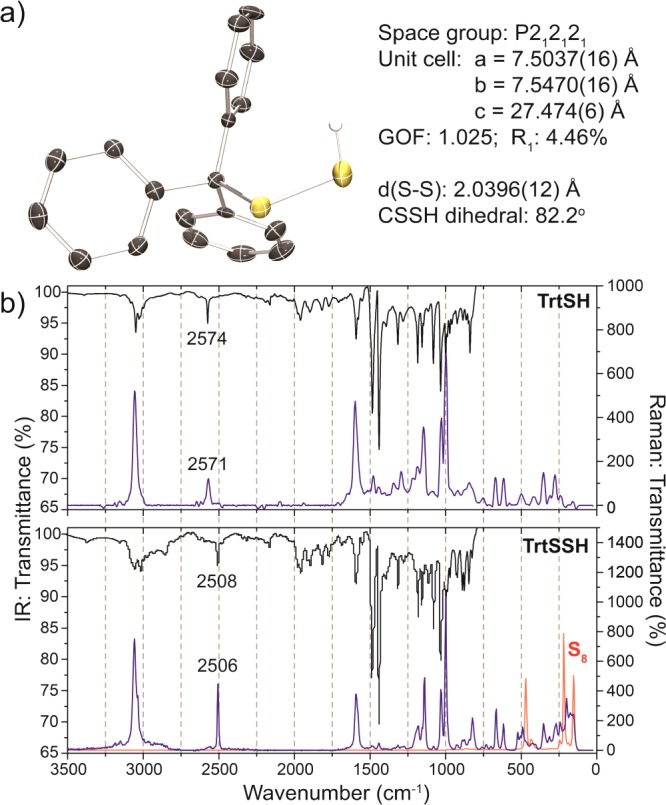
(a) X-ray crystal structure of TrtSSH. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms on the phenyl rings are omitted for clarity. Full crystallographic details are available in the Supporting Information. (b) Infrared (black) and Raman (blue) spectra of TrtSH (top) and TrtSSH (bottom). The Raman spectrum of S8 (red) is shown for comparison.
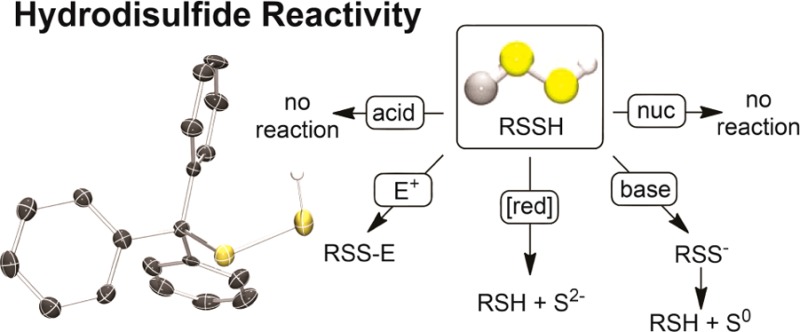
Having confirmed the molecular identity of TrtSSH and established its spectroscopic properties, we next investigated the reactivity of TrtSSH to determine under which conditions sulfide or other sulfur-containing products were released. Although reports have suggested that H2S can be released from hydrodisulfides upon protonation, reduction, and nucleophilic attack, we expected that only reduction would liberate sulfide because the sulfur oxidation state is higher in hydrodisulfides (either two S1– or one S0 and S2–) than in H2S (S2–). As expected, treatment of TrtSSH with reductants, such as PPh3 or [NBu4+][BH4–] (Figures S6, S7) resulted in clean conversion of TrtSSH to TrtSH with concurrent liberation of sulfide.11 These results confirm that hydrodisulfides are a source of reductant-labile sulfane sulfur (Table 1, entries 1–2). When TrtSSH was treated with sulfhydryl-containing species, such as TrtSH, BnSH, dithiopropane (DTP), or H2S, however, sulfide release did not occur, suggesting that the thiols were either insufficiently reducing or that proton transfer is a mechanistically important step (Table 1, entries 3–6). Treatment of TrtSSH with acid, either alone or in the presence of a thiol as a potential electron source, also failed to liberate sulfide (Table 1, entries 7–8). These results confirm that hydrodisulfides are a source of reductant-labile but not acid-labile sulfide.
Table 1. Reactivity of TrtSSH with Different Reactants Including Reductants, Nucleophiles, Electrophiles, Bases, and Acidsa.
| entry | reactant | completion | products |
|---|---|---|---|
| 1 | PPh3 | <2 min | Ph3PS, TrtSH |
| 2 | [NBu4+][BH4–] | <2 min | TrtSH, BH3, SH– |
| 3 | DTP | n.r. | – |
| 4 | TrtSH | n.r. | – |
| 5 | BnSH | n.r. | – |
| 6 | H2S | n.r. | – |
| 7 | AcOH | n.r. | – |
| 8 | AcOH + TrtSH | n.r. | – |
| 9 | [Na+][TrtS–] | <2 min | TrtSH, S8 |
| 10 | [NBu4+][CN–] | <2 min | TrtSH, S8 |
| 11 | [NBu4+][Cl–] | 1 h | TrtSH, S8 |
| 12 | [NBu4+][Br–] | 24 h | TrtSH, S8 |
| 13 | [NBu4+][I–] | >48 h | TrtSH, S8 |
| 14 | [NBu4+][OAc–] | <2 min | TrtSH, S8 |
| 15 | [NBu4+][TFA–] | 30 min | TrtSH, S8 |
| 16 | NEt3 | <2 min | TrtSH, S8 |
| 17 | 10% NEt3 | <2 min | TrtSH, S8 |
| 18 | DMEDA | <2 min | TrtSH, S8 |
| 19 | DMAP | <2 min | TrtSH, S8 |
| 20 | Pyridine | >48 h | TrtSH, S8 |
| 21 | NEMb | 24 h | TrtSS-NEM |
Experiments were performed in CD2Cl2 at room temperature with 18 mM TrtSSH and reactant. All products were confirmed by 1H or 31P{1H} NMR spectroscopy. BH3 formation was identified by trapping with THF and characterization by 11B{1H} NMR spectroscopy. S8 formation was confirmed and quantified by titration with PPh3 at the end of the reaction.
Reaction was incomplete due to slower reactivity of protonated TrtSSH, however addition of NEt3 increased the reaction rate. Abbreviations: DTP: dithiopropane; DMEDA: N,N′-dimethylethylenediamine; DMAP: dimethylaminopyridine; NEM: N-ethyl-maleimide.
To further understand the reactivity of thiols toward TrtSSH, we treated TrtSSH with TrtS–. Monitoring the reaction by 1H NMR spectroscopy revealed clean conversion to TrtSH and the formation of S0 (vide infra) (Table 1, entry 9). To clarify this reactivity, we investigated whether the TrtS– was acting as a nucleophile or as a base by treating TrtSSH with different nucleophiles (CN–, I–, Br–, Cl–, TFA–, and AcO–) and bases (NEt3, DMEDA, pyridine, and DMAP). Monitoring these reactions by 1H NMR spectroscopy (Table 1, entries 10–20) revealed that the rate of conversion of TrtSSH to TrtSH and S0 correlated with the basicity (CN– > AcO– > TFA– > Cl– > Br– > I–) rather than with anion nucleophilicity (Figures S15–S20). The same trends for reaction rate were maintained with amine bases: NEt3 ∼ DMEDA > DMAP > pyridine (Figures S21, S23–S25). Introduction of catalytic NEt3 (entry 17, Figure S22) resulted in the same reactivity, thus supporting a base-mediated disproportionation mechanism. We also wanted to verify the multitude of reports highlighting the nucleophilicity of hydrodisulfides. To verify the nucleophilicity of TrtSSH, we treated TrtSSH with N-ethylmaleimide (NEM), a common thiol labeling agent, which resulted in slow conversion to the NEM-labeled disulfide (Figure S26). The slow reaction rate is consistent with proton transfer being important in the observed reactivity. Adding a small amount of NEt3 to the reaction drastically increases the reaction rate, mimicking the observed reactivity of hydrodisulfides with electrophiles in water.
Having determined that deprotonation results in disproportionation of TrtSSH to TrtSH and S0, we wanted to further investigate the mechanistic details involved in this reaction. Monitoring the formation of TrtSH from the reaction between TrtSSH and different nucleophiles by 1H NMR spectroscopy resulted in clean conversion to the TrtSH product without the formation of any NMR-active intermediates (Figure 2a). After the completion of each reaction, titration with PPh3 and monitoring the formation of Ph3PS, which is generated by reaction of PPh3 with S0, by 31P{1H} NMR spectroscopy established that 1 equiv of S0 was formed for each equiv of TrtSSH (Figure 2b). Following the reaction of TrtSSH with [NBu4+][CN–] by UV–vis spectroscopy, however, showed the formation of a transient intermediate with λmax = 340 nm (Figure S27), which we attribute to TrtSS–. Disappearance of this band is accompanied by the appearance of a broad absorption band centered at 300 nm consistent with S8 formation. Taken together, these data suggest that TrtSSH deprotonation initially generates TrtSS–, which subsequently disproportionates into TrtS– and S0, thus providing a mechanism for transsulfuration in the sulfane sulfur pool without disruption of redox homeostasis.
Figure 2.
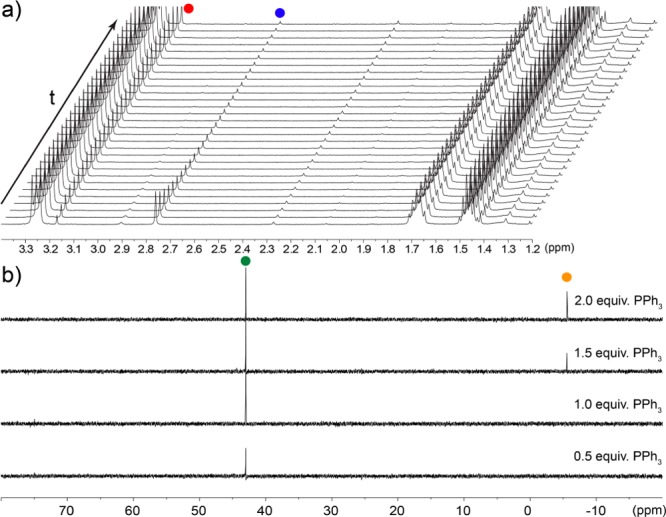
(a) 1H NMR spectra of the conversion of 18 mM TrtSSH (blue circle) to TrtSH (red circle) with 1 equiv of [NBu4+][TFA–] recorded every 60 s. (b) Addition of PPh3 (yellow circle) in 0.5 equiv. increments at the end of the reaction shows the clean formation of Ph3PS (green circle).
In summary, we have prepared, isolated, and fully characterized the stable hydrodisulfide TrtSSH and studied its reactivity toward reductants, nucleophiles, acids, and bases. The chemical reactivity data obtained from TrtSSH demonstrate that hydrodisulfides do not react with nucleophiles or acids in organic solution, but rather release sulfide upon reduction and disproportionate to release S0, likely as S8 under physiological conditions, upon deprotonation (Figure 3). These results clarify the innate reactivity of hydrodisulfides and confirm that hydrodisulfides are viable sources of reductant-labile sulfur. Additionally, deprotonation of hydrodisulfides may provide a mechanism for interconversion of different species within the reductant-labile sulfur pool, thereby allowing for facile conversion back to the parent thiol without disruption of thiol or redox homeostasis. We envision that these studies as well as future work using isolated hydrodisulfides will be invaluable in furthering our understanding of biological H2S signaling and storage by providing clear insight into the reactivity of hydrodisulfides under different reactive conditions.
Figure 3.
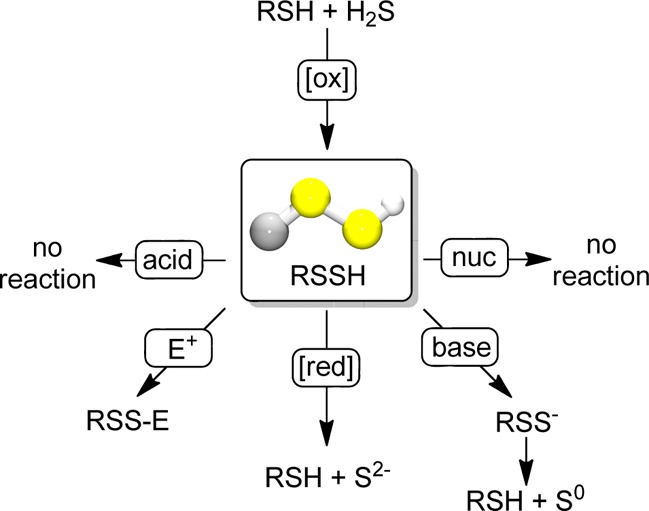
Reactivity of TrtSSH with acids, electrophiles (E+), reductants ([red]), bases, or nucleophiles (nuc).
Acknowledgments
We thank Milton Jackson, Jr. for assistance with Raman spectroscopy. This work was supported by the Oregon Medical Research Foundation and the NIGMS (R00GM092970). The NMR facilities at the UO are supported by NSF/ARRA (CHE-0923589). The Biomolecular Mass Spectrometry Core of the Environmental Health Sciences Core Center at Oregon State University is supported, in part, by the NIEHS (P30ES000210) and the NIH.
Supporting Information Available
Experimental details, NMR spectra, and UV–vis and crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wang R. Physiol. Rev. 2012, 92, 791–896. [DOI] [PubMed] [Google Scholar]
- Ida T.; Sawa T.; Ihara H.; Tsuchiya Y.; Watanabe Y.; Kumagai Y.; Suematsu M.; Motohashi H.; Fujii S.; Matsunaga T.; Yamamoto M.; Ono K.; Devarie-Baez N. O.; Xian M.; Fukuto J. M.; Akaike T. Proc. Natl. Acad. Sci. U.S.A. 2014, 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mustafa A. K.; Gadalla M. M.; Sen N.; Kim S.; Mu W. T.; Gazi S. K.; Barrow R. K.; Yang G. D.; Wang R.; Snyder S. H. Sci. Signal. 2009, 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vandiver M. S.; Paul B. D.; Xu R. S.; Karuppagounder S.; Rao F.; Snowman A. M.; Ko H. S.; Il Lee Y.; Dawson V. L.; Dawson T. M.; Sen N.; Snyder S. H. Nat. Commun. 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kessler D. FEMS Microbiol. Rev. 2006, 30, 825–840. [DOI] [PubMed] [Google Scholar]; d Kabil O.; Banerjee R. J. Biol. Chem. 2012, 287, 44561–44567. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kimura Y.; Mikami Y.; Osumi K.; Tsugane M.; Oka J.; Kimura H. FASEB J. 2013, 27, 2451–2457. [DOI] [PubMed] [Google Scholar]; f Paul B. D.; Snyder S. H. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [DOI] [PubMed] [Google Scholar]
- a Ubuka T. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2002, 781, 227–249. [DOI] [PubMed] [Google Scholar]; b Johnson D. C.; Dean D. R.; Smith A. D.; Johnson M. K. Annu. Rev. Biochem. 2005, 74, 247–281. [DOI] [PubMed] [Google Scholar]
- Shen X. G.; Peter E. A.; Bir S.; Wang R.; Kevil C. G. Free Radic. Biol. Med. 2012, 52, 2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M. D.; Abdel-Rehim M. S.; Furne J. Antioxid. Redox Signaling 2011, 15, 373–378. [DOI] [PubMed] [Google Scholar]
- Ishigami M.; Hiraki K.; Umemura K.; Ogasawara Y.; Ishii K.; Kimura H. Antioxid. Redox Signaling 2009, 11, 205–214. [DOI] [PubMed] [Google Scholar]
- a Krishnan N.; Fu C. X.; Pappin D. J.; Tonks N. K. Sci. Signaling 2011, 4, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang D. H.; Macinkovic I.; Devarie-Baez N. O.; Pan J.; Park C. M.; Carroll K. S.; Filipovic M. R.; Xian M. Angew. Chem., Int. Ed. 2014, 53, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nakabayashi T.; Tsurugi J.; Yabuta T. J. Org. Chem. 1964, 29, 1236–1238. [Google Scholar]; b Tsurugi J.; Nakabaya T.; Ishihara T. J. Org. Chem. 1965, 30, 2707–2710. [Google Scholar]; c Kawamura S.; Kitao T.; Nakabaya T.; Horii T.; Tsurugi J. J. Org. Chem. 1968, 33, 1179–1181. [Google Scholar]; d Tsurugi J.; Abe Y.; Nakabaya T.; Kawamura S.; Kitao T.; Niwa M. J. Org. Chem. 1970, 35, 3263–3266. [Google Scholar]; e Kawamura S.; Horii T.; Tsurugi J. Bull. Chem. Soc. Jpn. 1971, 44, 2878–2879. [Google Scholar]; f Franzi R.; Geoffroy M.; Bernardinelli G. Mol. Phys. 1984, 52, 947–954. [Google Scholar]; g Liu D. K.; Chang S. G. Can. J. Chem. 1987, 65, 770–774. [Google Scholar]; h Derbesy G.; Harpp D. N.; Rather B.; Carroll G. Sulfur Lett. 1992, 14, 199–204. [Google Scholar]; i Chatterji T.; Keerthi K.; Gates K. S. Bioorg. Med. Chem. Lett. 2005, 15, 3921–3924. [DOI] [PubMed] [Google Scholar]; j Heimer N. E.; Field L.; Waites J. A. J. Org. Chem. 1985, 50, 4164–4166. [Google Scholar]
- a Pan J.; Carroll K. S. ACS Chem. Biol. 2013, 8, 1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Francoleon N. E.; Carrington S. J.; Fukuto J. M. Arch. Biochem. Biophys. 2011, 516, 146–153. [DOI] [PubMed] [Google Scholar]
- PPh3 reduces S0 to S2– which is trapped as SPPh3. This sulfide can be liberated by hydrolysis in water. Direct reduction of the hydrodisulfide by PPh3 is more likely than deprotonation followed by reaction with S8 because compounds of similar or higher basicity than PPh3 react with the hydrodisulfide at a much slower rate
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.