

Abstract

A concise enantioselective synthesis of tetrahydrolipstatin (THL) and seven stereoisomers has been achieved. The synthesis of THL was accomplished in 10 steps and 31% overall yield from an achiral ynone. Key to the success of the approach is the use of a bimetallic [Lewis acid]+[Co(CO)4]− catalyst for a late-stage regioselective carbonylation of an enantiomerically pure cis-epoxide to a trans-β-lactone. The success of this route to THL and its stereoisomers also demonstrated the practicality of the carbonylation catalyst for complex molecule synthesis as well as its functional group compatibility.
Introduction
Tetrahydrolipstatin (THL, 1) is an over-the-counter anti-obesity drug that acts by inhibiting the absorption of dietary fats. THL is the saturated form of lipstatin (2), a natural product isolated from Streptomyces toxytricini in 1987.1 Due to its greater stability, THL was chosen over lipstatin for pharmaceutical development. Both THL and lipstatin contain an α-alkylated β,δ-dihydroxy acid, which exists in the β-lactone form (Figure 1). The β-lactone in THL and lipstatin is believed to ring-open and covalently bind to pancreatic lipase, which results in irreversible inhibition.1a,2 In addition, THL and related β-lactones have been found to inhibit the thioesterase domain of fatty acid synthase (FAS),3 the inhibition of which has been linked to anticancer activity.4 More recently, THL has been shown to inhibit the in vitro growth of Giardia duodenalis, the causative parasite of the gastrointestinal disease giardiasis.5
Figure 1.

Tetrahydrolipstatin (THL, 1), lipstatin (2), and analogues (3).
Since the first synthesis of THL by Schneider,6a there have been numerous total6−12 and formal syntheses of THL,13 which have involved a diverse range of approaches. In terms of how they derive absolute stereochemistry, these approaches can be classified into the following categories: (1) chiral auxiliary,6 (2) asymmetric aldol,7 (3) asymmetric allylation/crotylation,8 (4) asymmetric reductions,9 (5) asymmetric resolutions,10 (6) asymmetric oxidations,11 and (7) chiron approach.12 These routes include the elegant use of a chiral phosphate template,9c a tandem Mukaiyama aldol lactonization,7e,7i an anti-aldol segment via a non-aldol route,7h and a Prins cyclization for stereocontrol.10b
As part of a larger effort aimed at the use of catalysis for the asymmetric synthesis and structure–activity relationship studies of biologically active natural products,14 we became interested in the synthesis of THL (1) and related analogues 3 (Figure 1). We were particularly interested in the carbonylation of epoxides using bimetallic [Lewis acid]+[Co(CO)4]− catalysts, which has recently emerged as a reliable, direct route to β-lactones.15 We have had success using this type of carbonylation catalyst for the synthesis of terminal β-lactones en route to natural products.16 However, unsymmetrically disubstituted epoxides are prone to giving a mixture of regioisomeric β-lactones. Presumably this is because of an unselective SN2-ring opening reaction in the case of electronically or sterically unbiased substrates (Scheme 1).
Scheme 1. Regioselective Carbonylation of Protected cis-Epoxyhomoallylic Alcohols.
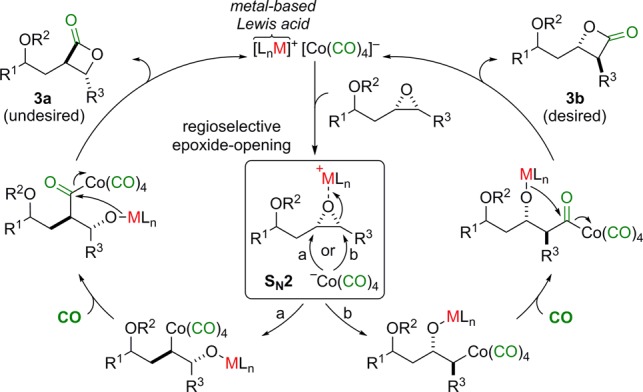
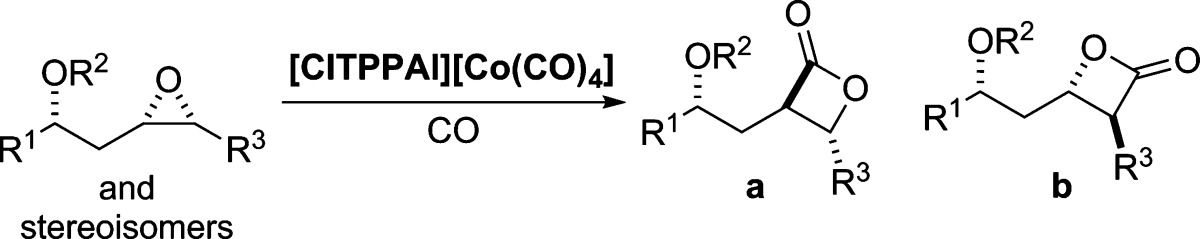
This problem has recently been addressed by the introduction of catalysts that can carbonylate racemic and enantioenriched trans-disubstituted epoxides to the corresponding cis-β-lactones with high and opposing regioselectivities.17 We hypothesized that it would be possible to obtain the desired β-lactone isomer 3b of THL and its analogues via regioselective carbonylation of protected cis-epoxyhomoallylic alcohols (Scheme 1). Herein, we disclose a new route for the synthesis of THL and seven stereoisomers using regioselective carbonylation of epoxides to form the β-lactone moiety. The fatty acid lipid portion of THL (4b) was prepared from protected cis-epoxyhomoallylic alcohol 5b with all the lipid stereogenic centers in place via a de novo asymmetric route (Scheme 2).
Scheme 2. Retrosynthetic Analysis of THL (1).

Retrosynthetic Analysis
Retrosynthetically, we envisioned preparing THL from its lipid core 4b and N-formyl-l-leucine (Scheme 2). Using a regioselective carbonylation reaction, the lipid portion of THL (4b) could be prepared either directly from cis-epoxyalcohol 5b or from 5a after alkylation of the terminal β-lactone 4a. Epoxides 5a,b could be prepared from a highly diastereoselective epoxidation of homoallylic alcohol 6a,b, which in turn could be prepared from 7a,b via asymmetric synthesis. Key to the success of this approach is the need for high regioselectivity in the carbonylation (5 to 4). Presumably, this could be accomplished with a high degree of confidence using terminal epoxide 5a.15a However, the subsequent alkylation of terminal β-lactones such as 4a is highly problematic.10a,18 Consequently, a greater degree of synthetic efficiency would result from a regioselective carbonylation of a 2,3-disubstituted epoxide with all the requisite lipid carbons in place, i.e. 5b to 4b. Regioselective carbonylation of epoxides had very little precedence in the synthesis of complex molecules prior to our endeavors.19
We anticipated that the precise choice of hydroxyl protecting group in 5b could be critical for the control of the carbonylation regioselectivity. At the outset, we chose a methoxymethyl group (MOM) for its potential to participate in chelated transition states. However, we were also interested in investigating the use of N-formyl-l-leucine, thus avoiding the need for a protecting group at this stage. This had the added advantage of testing the compatibility of the [Lewis acid]+[Co(CO)4]− carbonylation catalyst with the Lewis basic and Brønsted acidic formamide functional group as well as the rather epimerizable α-amino ester group.
Results and Discussion
Formal Synthesis of Tetrahydrolipstatin
To ensure success with this approach, we began our efforts with the synthesis of the diastereomeric terminal epoxides 10 and 12 (Scheme 3). The approach began with a Leighton allylation20 of dodecanal 7a to give homoallylic alcohol 6a. After Boc-protection of alcohol 6a, the resulting t-butylcarbonate 8 was treated with iodine to form cyclic carbonate 9. Hydrolysis of carbonate 9 led to in situ epoxidation to form 10, which was then protected as a MOM ether 5a (MOMCl/DIPEA). Alternatively, the stereochemistry at C5 in 10 was inverted using Mitsunobu conditions to give 12 after hydrolysis (PNBA/PPh3/DIAD, K2CO3/MeOH), which was also protected as a MOM ether 13. Epoxides 5a/13 underwent regioselective carbonylation to give β-hydroxy esters 11/14 when exposed to carbonylation conditions (CO, 10 mol % Co2(CO)8, 20 mol % 3-hydroxypyridine).21 The synthesis of β-hydroxy ester 14 constitutes a formal synthesis of THL (1) as 14 has been previously transformed into THL via the n-hexyl α-alkylation of a dianion generated from 14.12
Scheme 3. Formal Synthesis of THL (1).

Reagents and conditions: (a) (S,S)-Leighton, Sc(OTf)3 (2.5 mol %), CH2Cl2, −10 °C, 96%, >95% ee; (b) Boc2O, n-BuLi, THF, 0 °C, 93%; (c) I2, MeCN, −20 °C, 67%; (d) K2CO3, MeOH, rt, 99%, >95% dr; (e) MOMCl, DIPEA, CH2Cl2, 0 °C, 88%; (f) Co2(CO)8 (10 mol %), 3-hydroxypyridine (20 mol %), CO (900 psi), MeOH, THF, 60 °C, 89%; (g) PNBA, DIAD, PPh3, THF, 0 °C, 99%; (h) K2CO3, MeOH, 0 °C, 90%; (i) MOMCl, DIPEA, CH2Cl2, 0 °C, 95%; (j) Co2(CO)8 (10 mol %), 3-hydroxypyridine (20 mol %), CO (900 psi), MeOH, THF, 60 °C, 87%. DIAD = diisopropyl azodicarboxylate, PNBA = p-nitrobenzoic acid, MOMCl = chloromethyl methyl ether, DIPEA = N,N-diisopropylethylamine.
Asymmetric Synthesis of cis-Epoxyhomoallylic Alcohol 19
With access to a formal synthesis of THL, we turned our focus to potentially more efficient approaches to THL, which involved carbonylation of the more challenging 2,3-disubstituted cis-epoxide 5b. The synthesis of epoxide 5b required a practical asymmetric synthesis of epoxyhomoallylic alcohol 19 (Scheme 4). Our approach involved the novel construction of the Z-homoallylic alcohol functionality via a Noyori reduction/alkyne zipper/Lindlar reduction sequence.22 To establish the absolute stereochemistry for this route, we used a highly enantioselective (95% ee) Noyori asymmetric reduction23 of achiral ynone 7b, which could be prepared in one step from a known Weinreb amide. Using the alkyne zipper reaction,24 the internal 2-alkyne in 15 was isomerized into a terminal alkyne and then TBS protected to give 16. Alkylation of terminal alkyne 16 (n-BuLi then n-Hex-I, 88%) and TBAF-promoted deprotection of the TBS-ether provided the homopropargyl alcohol 17. Partial hydrogenation of alkyne 17 using Lindlar conditions25 (1 atm H2, Pd/CaCO3, quinoline, 96%) cleanly gave (Z)-olefin 6b. Finally, a highly diastereoselective hydroxy-directed epoxidation of 6b furnished 19 (t-BuOOH, 2 mol % VO(acac)2, 94%, 92% dr) via putative intermediate 18.26
Scheme 4. Synthesis of cis-Epoxyhomoallylic Alcohol 19.

Reagents and conditions: (a) (S,S)-Noyori (5 mol %), Et3N, HCO2H, rt, 83%, >95% ee; (b) H2N(CH2)3NH2, KH, 15 °C to rt, 80%; (c) TBSCl, imidazole, DMF, rt, 97%; (d) n-BuLi, HPMA, THF, −20 °C; C6H13I, −20 °C to rt, 88%; (e) TBAF, THF, rt, 96%; (f) Pd/CaCO3, quinoline, H2 (1 atm), MeOH, rt, 96%; (g) t-BuOOH, VO(acac)2 (2 mol %), CH2Cl2, 0 °C, 94%, 92% dr. KAPA = potassium 3-aminopropylamide, TBSCl = tert-butyldimethylsilyl chloride, HMPA = hexamethyl phosphoramide, TBAF = tetra-n-butylammonium fluoride.
Total Synthesis of Tetrahydrolipstatin
With the establishment of a practical and stereocontrolled synthesis of epoxide 19, we began our investigation of the regioselectivity of the carbonylation reaction (Scheme 5). This study began with the protection of the alcohol as a MOM ether 5b (MOMCl, 85%). To our delight, when the MOM-protected epoxide 5b was subjected to carbonylation (1 mol % [ClTPPAl][Co(CO)4], CO (900 psi)),15f a single regioisomer β-lactone 20 was formed, which was obtained in 81% isolated yield.
Scheme 5. Synthesis of THL (1): Carbonylation of MOM-Protected Epoxide 5b.

Reagents and conditions: (a) MOMCl, DIPEA, CH2Cl2, 0 °C, 85%; (b) [ClTPPAl][Co(CO)4] (1 mol %), CO (900 psi), THF, 50 °C, 81%; (c) BF3·OEt2, 1,2-ethanedithiol, CH2Cl2, 0 °C, 83%; (d) N-Cbz-l-leucine, DCC, DMAP, CH2Cl2, rt, 93%; (e) Pd/C (10% wt/wt, 10 mol %), H2 (1 atm), AcOCHO, rt, 80%. ClTPP = meso-tetra(4-chlorophenyl) porphyrinato, DCC = N,N′-dicyclohexylcarbodiimide, DMAP = 4-dimethylaminopyridine.
The synthesis of THL was easily finished via a four-step deprotection/acylation/deprotection/formylation procedure. Thus, the MOM group was removed with BF3 etherate to give 4b (83%). A DCC coupling of 4b with N-Cbz-l-leucine installed the amino acid side chain in 21. Finally, a two-step hydrogenolysis/formylation procedure both removed the Cbz group (1 atm H2, Pd/C in AcOCHO) and installed the N-formyl group to give THL (1) without any epimerization (vide infra).6f,7h The synthetic THL produced had spectral (1H, 13C NMR, IR) and optical properties (reported [α]D20 = −33 (c = 0.36, CHCl3);6b synthetic [α]D23 = −33.7 (c = 0.48, CHCl3)) consistent with what have been reported in the literature.
Alternative Approach to THL
To test the functional group compatibility of the carbonylation catalyst, we explored alternatives to the MOM protecting group. In this vein, we looked at the use of the N-formyl-l-leucine ester group (i.e., 22) (Scheme 6) as a replacement for the MOM ether. Along with testing the functional group compatibility of the carbonylation conditions, this substitution also had the advantage of reducing steps.
Scheme 6. Synthesis of THL (1): A Late-Stage Carbonylation of Epoxide.

Reagents and conditions: (a) N-formyl-l-leucine, DCC, DMAP, CH2Cl2, rt, 88%; (b) N-Cbz-l-leucine, DCC, DMAP, CH2Cl2, rt, 99%; (c) Pd/C (10% wt/wt, 10 mol %), H2 (1 atm), THF, rt; DCC, formic acid, CH2Cl2, rt, 79%; (d) [ClTPPAl][Co(CO)4] (2 mol %), CO (900 psi), THF, 50 °C, 80%.
We initially investigated the synthesis of epoxide 22 via the direct DCC coupling of N-formyl-l-leucine with 19. Unfortunately, we were not able to find conditions under which the coupling occurred without appreciable amounts of epimerization (i.e., 22 and 23 were isolated as a 6:4 mixture of epimers).6f,7h To circumvent the epimerization problem, we returned to the less epimerizable N-Cbz-protected l-leucine. Under the same DCC coupling procedure, N-Cbz-l-leucine coupled with 19 to afford ester 24 in 99% yield without any sign of epimerization.6f Exposure of 24 to the two-step hydrogenolysis/formylation conditions (1 atm H2, Pd/C then HCO2H/DCC) provided the desired epoxide 22 in 79% yield. Gratifyingly, under carbonylation conditions (2 mol % [ClTPPAl][Co(CO)4], CO (900 psi)), epoxide 22 with an N-formyl-l-leucine side chain was cleanly transformed into THL (1) as the single regioisomer, which was obtained in excellent yield (80%) without any sign of epimerization.
Preparation of Other THL Stereoisomers
The successful synthesis of THL via late-stage regioselective carbonylation of epoxide 22 prompted us to explore the scope of this approach. In particular, we were interested in exploring its utility for the synthesis of various stereoisomers of THL.27 To do this, we required access to a series of stereoisomeric epoxides with various groups at the C5 position. This was carried out from epoxyalcohol 19 and its enantiomer ent-19. Thus, the C5 diastereomer 26 with a MOM-protecting group was easily prepared from ent-19 by means of a three-step Mitsunobu/hydrolysis/protection sequence in a 65% overall yield (Scheme 7).
Scheme 7. Synthesis of Diastereomer 26.

Reagents and conditions: (a) PNBA, DIAD, PPh3, THF, 0 °C, 96%; (b) K2CO3, MeOH, 0 °C, 80%; (c) MOMCl, DIPEA, CH2Cl2, 0 °C, 84%.
By both invertive and retentive acylation chemistry, the three epoxides with an N-formyl leucine side chain (23, 28, and 30) were prepared from epoxide 19 (Scheme 8). To circumvent the problem associated with epimerization during the DCC coupling with N-formyl leucine, we resorted to the use of N-Cbz-d-leucine in the coupling to form 27, which could be readily converted into N-formyl amide 23 by hydrogenolysis followed by a DCC coupling with formic acid. In contrast to the DCC coupling with N-formyl leucine, the invertive Mitsunobu acylation of epoxide 19 with N-formyl leucine occurred with complete stereocontrol to give 28. Epoxide 30 was also made from 19 via ester 29 by means of a three-step Mitsunobu acylation and hydrogenolysis N-Cbz to N-formyl group exchange (1 atm H2, Pd/C then HCO2H/DCC).
Scheme 8. Synthesis of Stereoisomeric Epoxides from 19.

Reagents and conditions: (a) N-Cbz-d-leucine, DCC, DMAP, CH2Cl2, rt, 96%; (b) Pd/C (10% wt/wt), H2 (1 atm), THF, rt; DCC, formic acid, CH2Cl2, rt, 82%; (c) N-formyl-l-leucine, DIAD, PPh3, THF, 0 °C to rt, 82%; (d) N-Cbz-d-leucine, DIAD, PPh3, THF, 0 °C, 93%; (e) Pd/C (10% wt/wt), H2 (1 atm), THF, rt; DCC, formic acid, CH2Cl2, rt, 67%.
With related invertive and retentive acylation chemistry, three stereoisomeric epoxides (ent-24, ent-29, and 32) and their enantiomers were made with the N-Cbz leucine side chain from epoxide ent-19 (Scheme 9). To serve as a control group for the amide substitution, epoxide 31 with no amino-substitution was prepared by a Mitsunobu acylation with isohexanoic acid.
Scheme 9. Synthesis of Stereoisomeric Epoxides from ent-19.

Reagents and conditions: (a) isohexanoic acid, DIAD, PPh3, THF, 0 °C, 62%; (b) N-Cbz-d-leucine, DCC, DMAP, CH2Cl2, rt, 96%; (c) N-Cbz-l-leucine, DIAD, PPh3, THF, 0 °C, 98%; (d) N-Cbz-d-leucine, DIAD, PPh3, THF, 0 °C, 98%.
We next investigated the regioselectivity of the carbonylation with the various epoxide stereoisomers (Table 1). Exposure of the MOM-protected diastereomeric epoxide 26 to typical carbonylation conditions cleanly converted it into β-lactone 33 (entry 1, 88%) with complete regioselectivity. Similarly, clean conversion was found for the C2″ stereoisomeric epoxide 23, which reacted to give β-lactone 34 as a single regio- and stereoisomer (entry 2, 77%). In contrast to changes in stereochemistry at C2″, the inversion of the stereochemistry at C5 had a significant effect on the regioselectivity of the reaction (entries 3 and 4). Epoxide 28 with an N-formyl amide carbonylated to give β-lactones 35 in good yields of a 6:1 mixture of regioisomers. The C2″ epimeric epoxide 30 also carbonylated with lower regioselectivity to give β-lactones 36 in good yields of a 5:1 mixture of regioisomers. In addition to poor regioselectivities, epoxides 28 and 30 also required higher catalyst loadings.28
Table 1. Stereochemical Scope of Regioselective Carbonylationa.
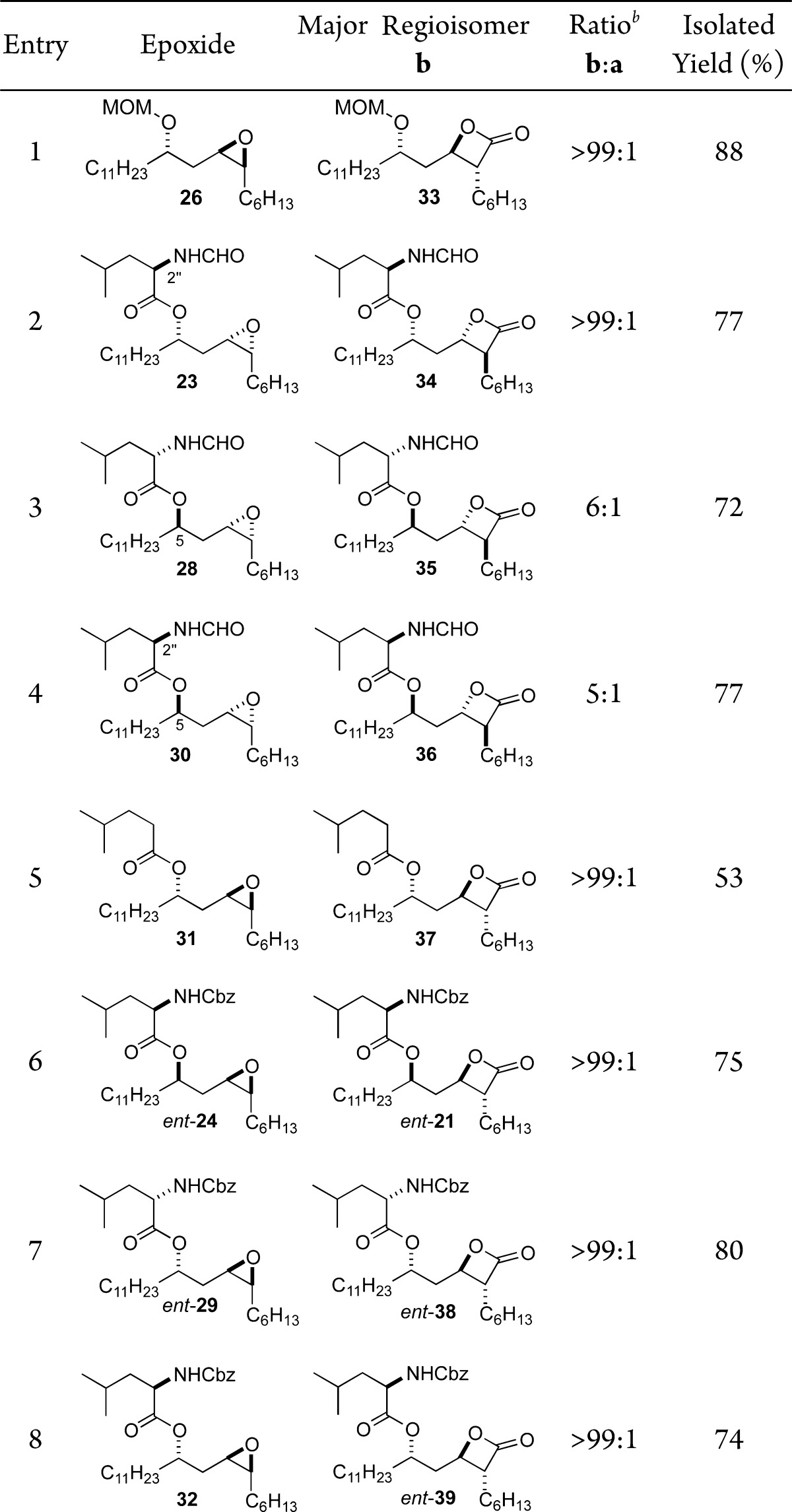
See Supporting Information for detailed reaction conditions for each entry.
Ratio determined by 1H NMR spectroscopy; for entries 1, 2, and 5–8 regioisomer a was not observed.
We hypothesized that the loss in regioselectivity for substrates 28 and 30 could be the result of hydrogen bonding interactions (Figure 2). This hydrogen bonding interaction in turn could lower the barrier for pathway a to the regioisomeric β-lactone. To test this hypothesis, we investigated the carbonylation of epoxides 31, ent-24, ent-29, and 32. When the N-formyl group was removed as in epoxide 31 (entry 5), the carbonylation occurred to give β-lactone 37 with complete control of regioselectivity. Interestingly, when the N-formyl group was replaced with the less acidic N-Cbz group, the high regioselectivity returned. Thus, epoxides ent-24, ent-29, and 32 carbonylated to form β-lactones ent-21, ent-38, and ent-39 as single regioisomers (entries 6–8; 75%, 80%, 74%, respectively).
Figure 2.

Rationale for loss of regiocontrol for epoxides 28 and 30.
In order to confirm the connectivity of unknown β-lactones 35, 36, 38, and 39, we prepared them independently from known β-lactone 4b (Scheme 10). The β-lactone 4b was converted into Cbz-protected leucine esters 38 and 39 via a Mitsunobu esterification. A hydrogenolysis/formylation reaction converted 38 and 39 into 36 and 35, respectively, which had identical 1H NMR spectra to the products prepared from the carbonylation reactions.
Scheme 10. Synthesis of β-Lactones 35 and 36 from Known β-Lactone 4b.

Reagents and conditions: (a) N-Cbz-l-leucine, DIAD, PPh3, THF, 0 °C, 72%; (b) Pd/C (10% wt/wt), H2 (1 atm), THF, rt; DCC, formic acid, CH2Cl2, rt, 63%; (c) N-Cbz-d-leucine, DIAD, PPh3, THF, 0 °C, 82%; (d) Pd/C (10% wt/wt), H2 (1 atm), THF, rt; DCC, formic acid, CH2Cl2, rt, 60%.
In support of the structure of the minor isomers formed from the carbonylation of epoxides 28 and 30 (entries 3 and 4; 35 and 36), we carried out a thermolytic decarboxylation reaction on the product mixture 35 and presumed regioisomer 35a (Scheme 11). Specifically, a 2:1 mixture of 35 and hypothesized 35a was heated to 230 °C in a sealed tube under argon for 1 hour producing trans-olefin 40 as the sole product. The identity of olefin 40 was confirmed by its synthesis from homoallylic alcohol 41, which in turn was made from terminal olefin 6a using cross metathesis.29 For comparison, cis-olefin 42 was prepared from 6b, which was readily distinguishable from its trans-isomer 40 by 13C NMR spectra.
Scheme 11. Evidence for Minor Regioisomer 35a by Thermolysis.

Reagents and conditions: (a) 230 °C, 60%; (b) 1-octene, Grubbs II, CH2Cl2, reflux, 59%, E/Z = 6:1; (c) N-formyl-l-leucine, DIAD, PPh3, THF, 0 °C, 69%; (d) N-formyl-l-leucine, DIAD, PPh3, THF, 0 °C, 87%.
Synthesis of ent-THL and Its Stereoisomers
With the route established above, we synthesized the enantiomer of THL (ent-1), as well as the stereoisomers ent-34, ent-35, and ent-36 (Scheme 12). The most direct route took advantage of the epimerization that occurred during the DCC coupling of epoxide ent-19 and N-formyl-l-leucine. Under these conditions, a mixture of ent-22 and ent-23 was afforded, which upon carbonylation were smoothly converted into a mixture of β-lactones ent-1 and ent-34. Preparative HPLC was used to purify the two isomers, ent-1 and ent-34. The remaining two enantiomers, ent-35 and ent-36, were prepared by an analogous route to the synthesis of their enantiomers (35 and 36, Scheme 10). This was accomplished by a two-step hydrogenolysis/formylation procedure, which replaced the N-Cbz group with an N-formyl group in β-lactones ent-38 and ent-39, cleanly providing ent-36 and ent-35, respectively.
Scheme 12. Synthesis of ent-THL (ent-1) and Its Stereoisomers.

Reagents and conditions: (a) N-formyl-l-leucine, DCC, DMAP, CH2Cl2, rt, 86%; (b) [ClTPPAl][Co(CO)4] (2 mol %), CO (900 psi), THF, 50 °C, 82%; (c) Pd/C (10% wt/wt, 20 mol %), H2 (1 atm), THF, rt; DCC, formic acid, CH2Cl2, rt, 70%; (d) Pd/C (10% wt/wt, 20 mol %), H2 (1 atm), THF, rt; DCC, formic acid, CH2Cl2, rt, 72%.
Conclusions
A concise enantioselective synthesis of THL has been achieved in 10 steps and 31% overall yield from achiral ynone 7b. The route is amenable for the production of THL and seven stereoisomers. In addition, the route demonstrated the versatility and regioselectivity of the bimetallic [Lewis acid]+[Co(CO)4]− catalyzed carbonylation of enantiomerically pure cis-epoxides to trans-β-lactones. Further application of bimetallic carbonylation catalysts for the synthesis and medicinal chemistry studies of natural products will be reported in due course.
Acknowledgments
This work is dedicated to the memory of Dr. Josephine (JoJo) O’Doherty. We are grateful to NIH (Grant GM090259), NSF (Grant CHE-1213596), and DOE (Grant DE-FG02-05ER15687) for financial support of this research.
Supporting Information Available
Detailed experimental procedures, full characterization data, and copies of spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ M.M., B.J.T. and Y.W. are co-first authors; the order is alphabetical.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Weibel E. K.; Hadváry P.; Hochuli E.; Kupfer E.; Lengsfeld H. J. Antibiot. 1987, 40, 1081–1085. [DOI] [PubMed] [Google Scholar]; b Hochuli E.; Kupfer E.; Maurer R.; Meister W.; Mercadal Y.; Schmidt K. J. Antibiot. 1987, 40, 1086–1091. [DOI] [PubMed] [Google Scholar]
- a Hadváry P.; Sidler W.; Meister W.; Vetter W.; Wolfer H. J. Biol. Chem. 1991, 266, 2021–2027. [PubMed] [Google Scholar]; b Stalder H.; Oesterhelt G.; Borgström B. Helv. Chim. Acta 1992, 75, 1593–1603. [Google Scholar]
- a Kridel S. J.; Axelrod F.; Rozenkrantz N.; Smith J. W. Cancer Res. 2004, 64, 2070–2075. [DOI] [PubMed] [Google Scholar]; b Little J. L.; Wheeler F. B.; Fels D. R.; Koumenis C.; Kridel S. J. Cancer Res. 2007, 67, 1262–1269. [DOI] [PubMed] [Google Scholar]; c Pemble C. W.; Johnson L. C.; Kridel S. J.; Lowther W. T. Nat. Struct. Mol. Biol. 2007, 14, 704–709. [DOI] [PubMed] [Google Scholar]
- a Menendez J. A.; Vellon L.; Lupu R. Ann. Oncol. 2005, 16, 1253–1267. [DOI] [PubMed] [Google Scholar]; b Dowling S.; Cox J.; Cenedella R. Lipids 2009, 44, 489–498. [DOI] [PubMed] [Google Scholar]; c Seguin F.; Carvalho M. A.; Bastos D. C.; Agostini M.; Zecchin K. G.; Alvarez-Flores M. P.; Chudzinski-Tavassi A. M.; Coletta R. D.; Graner E. Br. J. Cancer 2012, 107, 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J.; Seeber F.; Kolodziej H.; Ignatius R.; Laue M.; Aebischer T.; Klotz C. PLoS One 2013, 8, e71597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Barbier P.; Schneider F. Helv. Chim. Acta 1987, 70, 196–202. [Google Scholar]; b Barbier P.; Schneider F.; Widmer U. Helv. Chim. Acta 1987, 70, 1412–1418. [Google Scholar]; c Barbier P.; Schneider F. J. Org. Chem. 1988, 53, 1218–1221. [Google Scholar]; d Pons J.-M.; Kocieński P. Tetrahedron Lett. 1989, 30, 1833–1836. [Google Scholar]; e Fleming I.; Lawrence N. J. Tetrahedron Lett. 1990, 31, 3645–3648. [Google Scholar]; f Chadha N. K.; Batcho A. D.; Tang P. C.; Courtney L. F.; Cook C. M.; Wovkulich P. M.; Uskokovic M. R. J. Org. Chem. 1991, 56, 4714–4718. [Google Scholar]; g Fleming I.; Lawrence N. J. J. Chem. Soc., Perkin Trans. 1 1998, 2679–2686. [Google Scholar]; h Dirat O.; Kouklovsky C.; Langlois Y. Org. Lett. 1999, 1, 753–755. [DOI] [PubMed] [Google Scholar]; i Raghavan S.; Rathore K. Synlett 2009, 1285–1288. [Google Scholar]; j Raghavan S.; Rathore K. Tetrahedron 2009, 65, 10083–10092. [Google Scholar]; k Aubry S.; Aubert G.; Cresteil T.; Crich D. Org. Biomol. Chem. 2012, 10, 2629–2632. [DOI] [PubMed] [Google Scholar]
- a Case-Green S. C.; Davies S. G.; Hedgecock C. J. R. Synlett 1991, 781–782. [Google Scholar]; b Paterson I.; Doughty V. A. Tetrahedron Lett. 1999, 40, 393–394. [Google Scholar]; c Ghosh A. K.; Fidanze S. Org. Lett. 2000, 2, 2405–2407. [DOI] [PubMed] [Google Scholar]; d Yin J.; Yang X. B.; Chen Z. X.; Zhang Y. H. Chin. Chem. Lett. 2005, 16, 1448–1450. [Google Scholar]; e Ma G.; Zancanella M.; Oyola Y.; Richardson R. D.; Smith J. W.; Romo D. Org. Lett. 2006, 8, 4497–4500. [DOI] [PubMed] [Google Scholar]; f Kumaraswamy G.; Markondaiah B. Tetrahedron Lett. 2008, 49, 327–330. [Google Scholar]; g Case-Green S. C.; Davies S. G.; Roberts P. M.; Russell A. J.; Thomson J. E. Tetrahedron: Asymmetry 2008, 19, 2620–2631. [Google Scholar]; h Ghosh A. K.; Shurrush K.; Kulkarni S. J. Org. Chem. 2009, 74, 4508–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Xu Q. Y.; Yu J. H.; Hu W. H.; Yang L. P. Chin. J. Org. Chem. 2010, 30, 1175–1179. [Google Scholar]
- a Hanessian S.; Tehim A.; Chen P. J. Org. Chem. 1993, 58, 7768–7781. [Google Scholar]; b Ghosh A. K.; Liu C. Chem. Commun. 1999, 1743–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Thadani A. N.; Batey R. A. Tetrahedron Lett. 2003, 44, 8051–8055. [Google Scholar]
- a Pommier A.; Pons J.-M.; Kocienski P. J.; Wong L. Synthesis 1994, 1294–1300. [Google Scholar]; b Giese B.; Roth M. J. Braz. Chem. Soc. 1996, 7, 243–246. [Google Scholar]; c Venukadasula P. K. M.; Chegondi R.; Maitra S.; Hanson P. R. Org. Lett. 2010, 12, 1556–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Parsons P. J.; Cowell J. K. Synlett 2000, 107–109. [Google Scholar]; b Yadav J. S.; Reddy M. S.; Prasad A. R. Tetrahedron Lett. 2006, 47, 4995–4998. [Google Scholar]; c Shiina I.; Umezaki Y.; Kuroda N.; Iizumi T.; Nagai S.; Katoh T. J. Org. Chem. 2012, 77, 4885–4901. [DOI] [PubMed] [Google Scholar]; d Kim H.; Choi K.; Tae J. Synlett 2012, 1832–1834. [Google Scholar]
- a Bodkin J. A.; Humphries E. J.; McLeod M. D. Aust. J. Chem. 2003, 56, 795–803. [Google Scholar]; b Bodkin J. A.; Humphries E. J.; McLeod M. D. Tetrahedron Lett. 2003, 44, 2869–2872. [Google Scholar]; c Yadav J. S.; Rao K. V.; Reddy M. S.; Prasad A. R. Tetrahedron Lett. 2006, 47, 4393–4395. [Google Scholar]
- Yadav J. S.; Rao K. V.; Prasad A. R. Synthesis 2006, 3888–3894. [Google Scholar]
- a Landi J. J. Jr.; Garofalo L. M.; Ramig K. Tetrahedron Lett. 1993, 34, 277–280. [Google Scholar]; b Wedler C.; Costisella B.; Schick H. J. Org. Chem. 1999, 64, 5301–5303. [DOI] [PubMed] [Google Scholar]; c Sharma A.; Chattopadhyay S. J. Org. Chem. 1999, 64, 8059–8062. [DOI] [PubMed] [Google Scholar]; d Sato M.; Nakashima H.; Hanada K.; Hayashi M.; Honzumi M.; Taniguchi T.; Ogasawara K. Tetrahedron Lett. 2001, 42, 2833–2837. [Google Scholar]; e Polkowska J.; Łukaszewicz E.; Kiegiel J.; Jurczak J. Tetrahedron Lett. 2004, 45, 3873–3875. [Google Scholar]; f Tripathi D.; Kumar P. Tetrahedron: Asymmetry 2012, 23, 884–890. [Google Scholar]
- a Borisova S. A.; Guppi S. R.; Kim H. J.; Wu B.; Penn J. H.; Liu H.-w.; O’Doherty G. A. Org. Lett. 2010, 12, 5150–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shi P.; Silva M. C.; Wang H.-Y. L.; Wu B.; Akhmedov N. G.; Li M.; Beuning P. J.; O’Doherty G. A. ACS Med. Chem. Lett. 2012, 3, 1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hinds J. W.; McKenna S. B.; Sharif E. U.; Wang H.-Y. L.; Akhmedov N. G.; O’Doherty G. A. ChemMedChem. 2013, 8, 63–69. [DOI] [PubMed] [Google Scholar]; d Mrozowski R. M.; Vemula R.; Wu B.; Zhang Q.; Schroeder B. R.; Hilinski M. K.; Clark D. E.; Hecht S. M.; O’Doherty G. A.; Lannigan D. A. ACS Med. Chem. Lett. 2013, 4, 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Cuccarese M. F.; Wang Y.; Beuning P. J.; O’Doherty G. A. ACS Med. Chem. Lett. 2014, 5, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Getzler Y. D. Y. L.; Mahadevan V.; Lobkovsky E. B.; Coates G. W. J. Am. Chem. Soc. 2002, 124, 1174–1175. [DOI] [PubMed] [Google Scholar]; b Mahadevan V.; Getzler Y. D. Y. L.; Coates G. W. Angew. Chem., Int. Ed. 2002, 41, 2781–2784. [DOI] [PubMed] [Google Scholar]; c Schmidt J. A. R.; Lobkovsky E. B.; Coates G. W. J. Am. Chem. Soc. 2005, 127, 11426–11435. [DOI] [PubMed] [Google Scholar]; d Church T. L.; Getzler Y. D. Y. L.; Coates G. W. J. Am. Chem. Soc. 2006, 128, 10125–10133(mechanistic study). [DOI] [PubMed] [Google Scholar]; e Kramer J. W.; Lobkovsky E. B.; Coates G. W. Org. Lett. 2006, 8, 3709–3712. [DOI] [PubMed] [Google Scholar]; f Rowley J. M.; Lobkovsky E. B.; Coates G. W. J. Am. Chem. Soc. 2007, 129, 4948–4960. [DOI] [PubMed] [Google Scholar]; g Church T. L.; Getzler Y. D. Y. L.; Byrne C. M.; Coates G. W. Chem. Commun. 2007, 657–674(a review). [DOI] [PubMed] [Google Scholar]
- a Chen Q.; Mulzer M.; Shi P.; Beuning P. J.; Coates G. W.; O’Doherty G. A. Org. Lett. 2011, 13, 6592–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen Q.; Zhong Y.; O’Doherty G. A. Chem. Commun. 2013, 49, 6806–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mulzer M.; Whiting B. T.; Coates G. W. J. Am. Chem. Soc. 2013, 135, 10930–10933. [DOI] [PubMed] [Google Scholar]; For the regiodivergent carbonylation of racemic cis-disubstituted epoxides, see:; b Mulzer M.; Ellis W. C.; Lobkovsky E. B.; Coates G. W. Chem. Sci. 2014, 5, 1928–1933. [Google Scholar]
- a Mulzer J.; Kerkmann T. J. Am. Chem. Soc. 1980, 102, 3620–3622. [Google Scholar]; b Griesbeck A.; Seebach D. Helv. Chim. Acta 1987, 70, 1320–1325. [Google Scholar]; c Mead K. T.; Yang H.-L. Tetrahedron Lett. 1989, 30, 6829–6832. [Google Scholar]
- a Bates R. W.; Fernández-Moro R.; Ley S. V. Tetrahedron Lett. 1991, 32, 2651–2654. [Google Scholar]; b Bates R. W.; Fernández-Moro R.; Ley S. V. Tetrahedron 1991, 47, 9929–9938. [Google Scholar]; For other methods of carbonylation of epoxides to β-lactones, see:; c Annis G. D.; Ley S. V. J. Chem. Soc., Chem. Commun. 1977, 581–582. [Google Scholar]; d Kamiya Y.; Kawato K.; Ohta H. Chem. Lett. 1980, 9, 1549–1552. [Google Scholar]; e Annis G. D.; Ley S. V.; Self C. R.; Sivaramakrishnan R. J. Chem. Soc., Perkin Trans. 1 1981, 270–277. [Google Scholar]; f Shimizu I.; Maruyama T.; Makuta T.; Yamamoto A. Tetrahedron Lett. 1993, 34, 2135–2138. [Google Scholar]; g Lee J. T.; Thomas P. J.; Alper H. J. Org. Chem. 2001, 66, 5424–5426. [DOI] [PubMed] [Google Scholar]
- a Kubota K.; Leighton J. L. Angew. Chem., Int. Ed. 2003, 42, 946–948. [DOI] [PubMed] [Google Scholar]; For other applications of the Leighton reagent in natural product synthesis, see:; b Hoye T. R.; Eklov B. M.; Jeon J.; Khoroosi M. Org. Lett. 2006, 8, 3383–3386. [DOI] [PubMed] [Google Scholar]; c Li M.; O’Doherty G. A. Org. Lett. 2006, 8, 6087–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vintonyak V. V.; Maier M. E. Org. Lett. 2008, 10, 1239–1242. [DOI] [PubMed] [Google Scholar]; e Nicewicz D. A.; Satterfield A. D.; Schmitt D. C.; Johnson J. S. J. Am. Chem. Soc. 2008, 130, 17281–17283. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Schmidt B.; Hölter F. Chem.—Eur. J. 2009, 15, 11948–11953. [DOI] [PubMed] [Google Scholar]; g Wang Y.; O’Doherty G. A. J. Am. Chem. Soc. 2013, 135, 9334–9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinterding K.; Jacobsen E. N. J. Org. Chem. 1999, 64, 2164–2165. [Google Scholar]
- For the combination of the alkyne zipper and Noyori reduction in synthesis, see:; a Trost B. M.; Horne D. D.; Woltering M. J. Chem.—Eur. J. 2006, 12, 6607–6620. [DOI] [PubMed] [Google Scholar]; b Trost B. M.; Bartlett M. J. Org. Lett. 2012, 14, 1322–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Xing Y.; O’Doherty G. A. Org. Lett. 2009, 11, 1107–1110. [DOI] [PubMed] [Google Scholar]; d Xing Y.; Penn J. H.; O’Doherty G. A. Synthesis 2009, 2847–2854. [Google Scholar]
- Fujii A.; Hashiguchi S.; Uematsu N.; Ikariya T.; Noyori R. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar]
- Brown C. A.; Yamashita A. J. Am. Chem. Soc. 1975, 97, 891–892. [Google Scholar]
- a Lindlar H. Helv. Chim. Acta 1952, 35, 446–450. [Google Scholar]; b Lindlar H.; Dubuis R. Org. Synth. 1966, 46, 89–91. [Google Scholar]
- Mihelich E. D.; Daniels K.; Eickhoff D. J. J. Am. Chem. Soc. 1981, 103, 7690–7692. [Google Scholar]
- For THL congeners, see:; a Richardson R. D.; Ma G.; Oyola Y.; Zancanella M.; Knowles L. M.; Cieplak P.; Romo D.; Smith J. W. J. Med. Chem. 2008, 51, 5285–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ortar G.; Bisogno T.; Ligresti A.; Morera E.; Nalli M.; Di Marzo V. J. Med. Chem. 2008, 51, 6970–6979. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for detailed reaction conditions for each entry in Table 1.
- Scholl M.; Trnka T. M.; Morgan J. P.; Grubbs R. H. Tetrahedron Lett. 1999, 40, 2247–2250. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.