

Abstract
The bovine antibody (BLV1H12) which has an ultralong heavy chain complementarity determining region 3 (CDRH3) provides a novel scaffold for antibody engineering. By substituting the extended CDRH3 of BLV1H12 with modified CXCR4 binding peptides that adopt a β-hairpin conformation, we generated antibodies specifically targeting the ligand binding pocket of CXCR4 receptor. These engineered antibodies selectively bind to CXCR4 expressing cells with binding affinities in the low nanomolar range. In addition, they inhibit SDF-1-dependent signal transduction and cell migration in a transwell assay. Finally, we also demonstrate that a similar strategy can be applied to other CDRs and show that a CDRH2-peptide fusion binds CXCR4 with a Kd of 0.9 nM. This work illustrates the versatility of scaffold-based antibody engineering and could greatly expand the antibody functional repertoire in the future.
There is a considerable interest in the generation of antibodies that not only bind a cell surface receptor but also modulate receptor-mediated signal transduction. Typically the identification of such functional antibodies involves the generation of high-affinity binding antibodies against a target protein and subsequent screening of the resulting clones for agonist or antagonist activities.1−6 Recently, we showed that it is possible to fuse growth factors and cytokines directly into the heavy chain complementarity determining region 3 (CDRH3) of the antibody BLV1H12, which has a well-structured ultralong hypervariable region. The resulting fusion proteins express well and are stable, retain the effector function of the original signaling molecule, and have the favorable pharmacological properties of the antibody molecule.7,8 Here, we demonstrate that biologically active cyclic peptides can also be engineered into the CDR loops of this novel antibody framework and retain their biological activities.
As a model system to test the feasibility of this approach we chose the 16 residue cyclic peptide CVX15, which is an analogue of the horseshoe crab peptide polyphemusin and an antagonist of the chemokine receptor CXCR4.9,10 CXCR4 is a G protein coupled receptor (GPCR) of stromal cell-derived factor (SDF1/CXCL12) that is involved in a number of developmental and physiological processes including stem cell and lymphocyte migration.11 For example, the SDF-1/CXCR4 axis controls the trafficking of hematopoietic stem cells (HSCs) from bone marrow to the periphery.12 In addition, CXCR4 is a major receptor for HIV infection;13 and overexpression of CXCR4 is associated with the metastatic potential of various cancers.14 Thus, antagonists of CXCR4 are being developed for the treatment of metastatic cancer,15 HIV infection,16 and mobilization of HSCs for bone marrow transplants.17
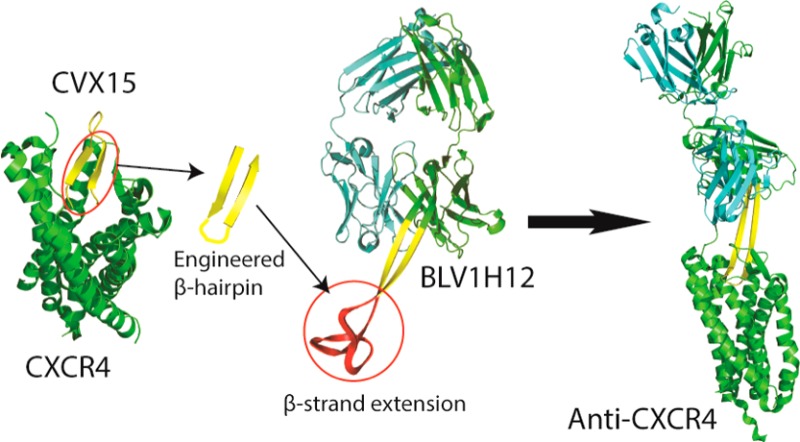
Recently, the X-ray crystal structure of a CVX15-CXCR4 complex revealed that the peptide is bound in a β-hairpin conformation with its N- and C-termini inserted into the transmembrane cavity of CXCR4 and its hairpin loop exposed to solvent10 (Figure 1A). The bound conformation of CVX15 suggests that it might make an excellent candidate to attempt to generate a novel CXCR4 antagonist antibody using antibody BLV1H12. This bovine antibody has an ultralong (61 residues) CDRH3 with an antiparallel β-sheet 20 Å in length, terminating in a disulfide cross-linked knob domain18 (Figure 1B). Replacement of this knob domain with the CVX15 hairpin peptide might be expected to afford an antibody with an extended CDR that can bind the ligand binding cavity of CXCR4. Schematic representations of three candidate antibody fusion proteins are shown in Figure 1C. Briefly, we first replaced the unnatural amino acids naphthylamine and citrulline of CVX15 by tryptophan and lysine based on sequence alignment with the peptide T22 from which CVX15 was derived.19 Next the N- and C-termini of the peptide were fused to sequences that promote β-turns: Gly-Arg (YRKCRGGRRWCYQK in bAb-AC1), Pro-Arg (bAb-AC2, YRKCRGPRRWCYQK), or Gly-Asn-Gly-Arg (bAb-AC3, YRKCRGGNGRRWCYQK).20−22 Based on the CVX15-CXCR4 complex structure, it was expected that such a β-turn linker would not affect the interaction of the peptide with CXCR4. Finally, the loop region of CVX15 that resides outside the binding pocket of CXCR4 was removed, and the resulting inverse hairpin sequence was substituted for the knob domain of BLV1H12. The final designs of the antibody-CVX fusion proteins are illustrated in Figure 1D. The three engineered antibodies were transiently expressed in FreeStyle 293 cells as a bovine-human chimera in which the Fc domain from human IgG1 was substituted for the bovine Fc. The antibodies were secreted into culture medium and purified by protein G column with yields of more than 5 mg/L (Figure S1).
Figure 1.
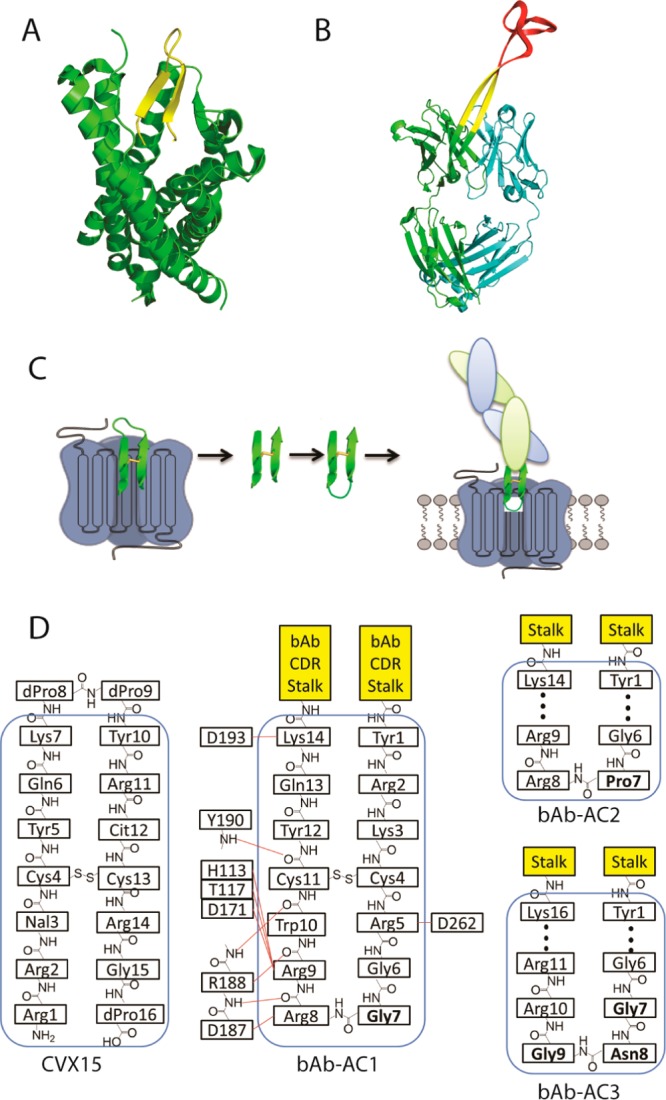
Antibody design. (A) Crystal structure of CXCR4 (green) in complex with a β-hairpin peptide antagonist CVX15 (yellow) (PDB code 3OE0). (B) Crystal structure of bovine antibody BLV1H12 (PDB code 4K3D) shows a disulfide cross-linked “knob” domain (red) grafted onto a solvent-exposed β-strand “stalk” (yellow). (C) A cartoon representation of the anti-CXCR4 antibody design. The loop region of the β-hairpin that resides outside the binding pocket of CXCR4 (blue) is removed, and the antiparallel β-strand region (green) is reconnected by selected β-turns to generate an inverted β-hairpin that is fused to the knob domain truncated bovine antibody scaffold. (D) A schematic representation of CVX15 and the engineered CDRs with β-turn promoting residues highlighted in bold. Potential interactions of bAb-AC1 with the CXCR4 ligand-binding pocket (blue box) are depicted on the basis of an analysis of the CXCR4-CVX15 complex.10
Next, we examined the binding of the engineered antibodies to CXCR4 by flow cytometry using human Jurkat cells, which highly express CXCR4.23 As shown in Figure 2A, all three antibodies (1 μg/mL) bind Jurkat cells, while the control antibody (BLV1H12) showed no detectable binding. To confirm that the observed binding is indeed mediated by CXCR4, flow cytometry experiments were performed using Chinese hamster ovary (CHO) cells (which have no detectable CXCR4 expression based on flow cytometry analysis of cells stained with a FITC labeled anti-CXCR4 antibody (clone 12G5)), with and without CXCR4 transfection (Figure S2). Incubation of CXCR4 transfected CHO cells with 1 μg/mL of the fusion antibodies resulted in a peak shift of 73.8%, 67.9%, and 67.4% for bAb-AC1, bAb-AC2, and bAb-AC3, respectively, in flow cytometry experiments. No peak shift was observed with nontransfected parental cells. In all cases, the control antibody showed no detectable binding. These results indicate that these engineered antibodies indeed bind specifically to CXCR4.
Figure 2.
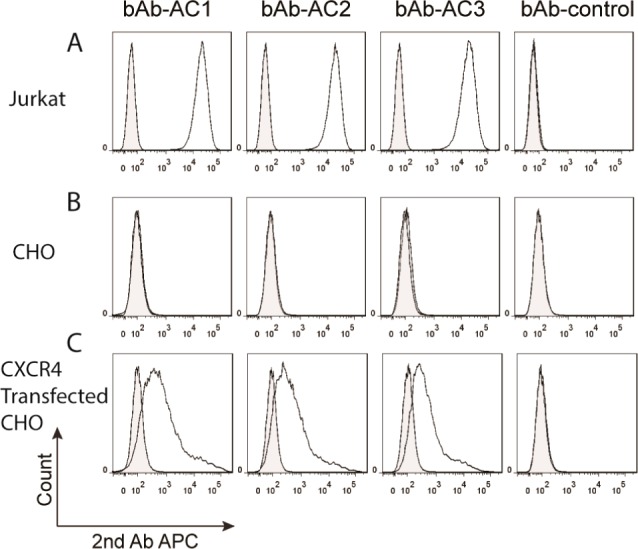
Flow cytometry analysis of interactions between CXCR4 and engineered antibodies. The engineered antibodies (A) bind to CXCR4 positive Jurkat cells, (B) do not bind to CXCR4 negative CHO cells, (C) but bind to CXCR4 transfected CHO cells. In all cases, the control antibody showed no peak shift by flow cytometry analysis. The shaded peaks are cells without antibody treatment.
To accurately determine the binding affinity between the engineered antibodies and CXCR4, we applied Tag-lite homogeneous time-resolved fluorescence (HTRF) (Cisbio Bioassays).24 Specific binding of fluorescently labeled SDF-1 to labeled SNAP-tag-CXCR4 results in a HTRF signal. The binding constant (Kd) between fluorescently labeled SDF-1 and the Tag-lite CXCR4 receptor was determined to be 14.2 ± 1.2 nM (Figure S3). A dose-dependent competition was observed between the engineered antibodies and 50 nM of labeled SDF-1 (Figure 3A). Assuming a competitive binding mode, the Kds of bAb-AC1, bAb-AC2, and bAb-AC3 to CXCR4 were calculated to be 2.1, 5.4, and 19.8 nM, respectively.25 These results indicate that bAb-AC1 with a more flexible glycine at i + 1 position of the hairpin turn binds the best to CXCR4, which is consistent with the flow cytometry analysis results. On the other hand, bAb-AC3, which has a β-turn promoting sequence (Asn-Gly) added at the end of the β-hairpin, has a decreased affinity compared to bAb-AC1 and bAb-AC2 that is probably due to spatial constraints within the CXCR4 ligand binding pocket.
Figure 3.
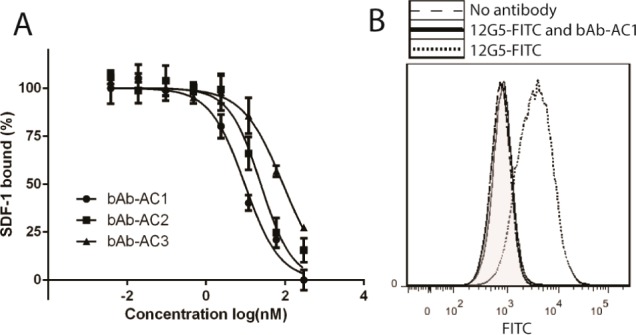
(A) Specific binding between bAb-AC1–3 and CXCR4 was determined by a Tag-lite HTRF binding assay. The binding affinities were calculated based on the Cheng–Prusoff equation to give Ki values of 2.1, 5.4, and 19.8 nM for bAb-AC1, bAb-AC2, and bAb-AC3, respectively. (B) Flow cytometry histogram demonstrating nearly complete inhibition of 12G5 binding to CXCR4 by a 3-fold excess of bAb-AC1.
Monoclonal antibody 12G5 is commonly used to assess CXCR4 expression as well as functionally inhibit the SDF1-CXCR4 interaction.26,27 The binding epitope of 12G5 includes extracellular loop (ECL) 2 as well as the N-terminus and ECL3.28 Because bAb-ACs are designed to bind the CXCR4 pocket, they should compete with binding of 12G5 to the receptor. To confirm this notion, a competitive binding assay was performed between 12G5 and bAb-AC1. A dose-dependent inhibition was observed for 12G5 binding to Jurkat cells by increasing concentrations of bAb-AC1 (Figure S4). Flow cytometry analysis (Figure 3B) indicated that a 3-fold excess of bAb-AC1 is sufficient to completely block the binding of 12G5 to CXCR4 on Jurkat cells.
Studies have shown that the CDR2 loop in the antibody VH domain is the most solvent exposed loop among all of the CDRs.29 An examination of the BLV1H12 structure suggests that the heavy chain CDR2 loop, which also connects two antiparallel β-strands in the canonical immunoglobulin fold, makes no direct contact with the rest of the antibody molecule. Thus, we hypothesized that an engineered CDRH2 with an extended antiparallel β-strand stalk can also be generated on the bovine antibody scaffold to afford a more solvent exposed antigen recognition domain. This design could be especially advantageous in the case of antibodies against certain GPCRs, as the ligand binding sites are often buried in the cell membrane. Therefore, a new antibody bAb-AC4 was designed by grafting the CDRH3 sequence from bAb-AC1 into the CDRH2 of the BLV1H12 scaffold (Figure S5). The truncated CDRH3 of the resulting antibody was capped with a GGGGS linker. bAb-AC4 was expressed in 293 cells with a much higher yield (17 mg/L) compared to bAb-AC1. This may be due to the fact that CDRH2 makes no direct contact with the rest of the antibody and therefore has less effect on heavy chain and light chain packing compared to the CDRH3 fusion. Binding between bAb-AC4 and CXCR4 was confirmed by both flow cytometry (Figure S6) and Tag-lite HTRF assay as described above (Figure S7) to give a Kd value of 0.92 nM against the receptor. This result indicates that the CDRH2 is indeed a viable alternative to CDRH3 for functional peptide grafting and suggests that it may be possible to simultaneously graft two polypeptide agonists or antagonists into two distinct CDRs of a single antibody fusion protein.
Next we tested if these engineered antibodies can block CXCR4-dependent intracellular signaling. Activation of CXCR4 by SDF1 can be measured by intracellular calcium flux, a secondary messenger involved in GPCR signaling. Ramos cells, a non-Hodgkin lymphoma cell line that highly express CXCR4, were loaded with Fluo-4 calcium indicators and incubated with 300 nM bAb-AC1, bAb-AC4, and the control antibody; SDF-1-mediated release of intracellular calcium was monitored by a fluorescence increase. As expected, bAb-AC1 significantly reduced calcium flux induced by 50 nM of SDF-1, whereas the same concentration of bAb-AC4 effectively blocks the calcium signaling post SDF-1 activation (Figures 4A and S8). These results indicate that these engineered antibodies are indeed CXCR4 antagonists.
Figure 4.
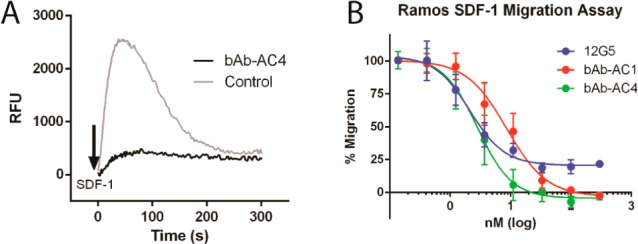
(A) 300 nM of bAb-AC4 efficiently blocks SDF-1-induced CXCR4 activation measured by intracellular calcium flux. (B) The antibodies bAb-AC1 and bAb-AC4 potently inhibit SDF-1-induced migration of Ramos cells in a dose-dependent manner with EC50 values of 8.6 and 3.1 nM, respectively. At saturating concentration, they are able to completely inhibit SDF-1-induced chemotaxis.
The physiological function of SDF-1 is to trigger the migration and recruitment of CXCR4 expressing cells. A chemotaxis assay was used to test if bAb-ACs can block SDF-1-dependent cell migration (Figure S9). Preincubation with the antibodies potently inhibits the migration of Ramos cells in a dose-dependent manner (Figure 4B) with EC50 values of 2.1, 8.6, and 3.1 nM for 12G5, bAb-AC1, and bAb-AC4, respectively. Interestingly, 30 nM of bAb-AC4 completely neutralizes SDF-1-induced migration of Ramos cells; while 12G5, even at its saturating concentration, cannot 100% block the migration (Figures 4B and S10). Studies have shown that the ECLs of CXCR4 exhibits considerable heterogeneity,27 which arises from post-translational modifications, including tyrosine sulfation, glycosylation, disulfide formation, etc.30 Thus, 12G5 recognizes only a subpopulation of CXCR4 molecules on the cell surface, resulting in an incomplete inhibition of chemotaxis.27 On the other hand, the conformational epitopes inside the ligand binding pocket of CXCR4 are likely more homogeneous, which makes the cavity targeting antibodies bAb-AC1 and bAb-AC4 more effective against SDF-1-induced cell migration.
In conclusion, we have demonstrated that by inserting a CXCR4 binding peptide that adopts a β-hairpin conformation into the ultralong CDRH3 of BLV1H12, one can generate potent antagonist antibodies against CXCR4. In addition, the elongated CDRs of these engineered antibodies can access the ligand binding pocket and effectively antagonize SDF1-dependent signal transduction and cell migration. Moreover, such CDR loop engineering is not limited to CDRH3 but can be applied to other CDRs. This result suggests that it may be possible to endow a single antibody molecule with two or more functions by grafting polypeptides into distinct CDRs. Thus, the work described here further expands the therapeutic potential of the antibody molecule.
Acknowledgments
We acknowledge Andrea Dashiell and Mishelle McClanahan-Shinn for their assistance in manuscript preparation. We thank Dr. Yong Zhang for helpful discussions. This work was supported by NIH R01 GM097206.
Supporting Information Available
Experimental details and supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wu A. L.; Kolumam G.; Stawicki S.; Chen Y.; Li J.; Zavala-Solorio J.; Phamluong K.; Feng B.; Li L.; Marsters S.; Kates L.; van Bruggen N.; Leabman M.; Wong A.; West D.; Stern H.; Luis E.; Kim H. S.; Yansura D.; Peterson A. S.; Filvaroff E.; Wu Y.; Sonoda J. Sci. Transl. Med. 2011, 3, 113ra126. [DOI] [PubMed] [Google Scholar]
- Zhang H. K.; Wilson I. A.; Lerner R. A. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 15728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide R. H.; Flaherty K. T.; Khalil M.; Stumacher M. S.; Bajor D. L.; Hutnick N. A.; Sullivan P.; Mahany J. J.; Gallagher M.; Kramer A.; Green S. J.; O’Dwyer P. J.; Running K. L.; Huhn R. D.; Antonia S. J. J. Clin. Oncol. 2007, 25, 876. [DOI] [PubMed] [Google Scholar]
- Kaplan-Lefko P. J.; Graves J. D.; Zoog S. J.; Pan Y.; Wall J.; Branstetter D. G.; Moriguchi J.; Coxon A.; Huard J. N.; Xu R.; Peach M. L.; Juan G.; Kaufman S.; Chen Q.; Bianchi A.; Kordich J. J.; Ma M.; Foltz I. N.; Gliniak B. C. Cancer Biol. Ther. 2010, 9, 618. [DOI] [PubMed] [Google Scholar]
- Camidge D. R. Expert Opin. Biol. Ther. 2008, 8, 1167. [DOI] [PubMed] [Google Scholar]
- Vitetta E. S.; Uhr J. W. Cancer Res. 1994, 54, 5301. [PubMed] [Google Scholar]
- Zhang Y.; Wang D.; de Lichtervelde L.; Sun S. B.; Smider V. V.; Schultz P. G.; Wang F. Angew. Chem., Int. Ed. Engl. 2013, 52, 8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Wang D.; Welzel G.; Wang Y.; Schultz P. G.; Wang F. ACS Chem. Biol. 2013, 8, 2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamura H.; Hori A.; Kanzaki N.; Hiramatsu K.; Mizumoto M.; Nakashima H.; Yamamoto N.; Otaka A.; Fujii N. FEBS Lett. 2003, 550, 79. [DOI] [PubMed] [Google Scholar]
- Wu B.; Chien E. Y. T.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C.; Hamel D. J.; Kuhn P.; Handel T. M.; Cherezov V.; Stevens R. C. Science 2010, 330, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C. Immunol. Rev. 2000, 177, 175. [DOI] [PubMed] [Google Scholar]
- Rettig M. P.; Ansstas G.; DiPersio J. F. Leukemia 2012, 26, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Broder C. C.; Kennedy P. E.; Berger E. A. Science 1996, 272, 872.8629022 [Google Scholar]
- Teicher B. A.; Fricker S. P. Clin. Cancer. Res. 2010, 16, 2927. [DOI] [PubMed] [Google Scholar]
- Kuhne M. R.; Mulvey T.; Belanger B.; Chen S.; Pan C.; Chong C.; Cao F.; Niekro W.; Kempe T.; Henning K. A.; Cohen L. J.; Korman A. J.; Cardarelli P. M. Clin. Cancer. Res. 2013, 19, 357. [DOI] [PubMed] [Google Scholar]
- Mosley C. A.; Wilson L. J.; Wiseman J. M.; Skudlarek J. W.; Liotta D. C. Expert Opin. Ther. Pat. 2009, 19, 23. [DOI] [PubMed] [Google Scholar]
- Pusic I.; DiPersio J. F. Curr. Opin. Hematol. 2010, 17, 319. [DOI] [PubMed] [Google Scholar]
- Wang F.; Ekiert D. C.; Ahmad I.; Yu W.; Zhang Y.; Bazirgan O.; Torkamani A.; Raudsepp T.; Mwangi W.; Criscitiello M. F.; Wilson I. A.; Schultz P. G.; Smider V. V. Cell 2013, 153, 1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda M.; Nakashima H.; Ueda T.; Naba H.; Ikoma R.; Otaka A.; Terakawa Y.; Tamamura H.; Ibuka T.; Murakami T.; et al. Biochem. Biophys. Res. Commun. 1992, 189, 845. [DOI] [PubMed] [Google Scholar]
- Guruprasad K.; Rajkumar S. J. Biosci. 2000, 25, 143. [PubMed] [Google Scholar]
- Hutchinson E. G.; Thornton J. M. Protein Sci. 1994, 3, 2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Alvarado M.; Blanco F. J.; Serrano L. Nat. Struct. Biol. 1996, 3, 604. [DOI] [PubMed] [Google Scholar]
- Hesselgesser J.; Liang M.; Hoxie J.; Greenberg M.; Brass L. F.; Orsini M. J.; Taub D.; Horuk R. J. Immunol. 1998, 160, 877. [PubMed] [Google Scholar]
- Zwier J. M.; Roux T.; Cottet M.; Durroux T.; Douzon S.; Bdioui S.; Gregor N.; Bourrier E.; Oueslati N.; Nicolas L.; Tinel N.; Boisseau C.; Yverneau P.; Charrier-Savournin F.; Fink M.; Trinquet E. J. Biomol. Screening 2010, 15, 1248. [DOI] [PubMed] [Google Scholar]
- Lazareno S.; Birdsall N. J. Br. J. Pharmacol. 1993, 109, 1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strizki J. M.; Turner J. D.; Collman R. G.; Hoxie J.; Gonzalez-Scarano F. J. Virol. 1997, 71, 5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribaud F.; Edwards T. G.; Sharron M.; Brelot A.; Heveker N.; Price K.; Mortari F.; Alizon M.; Tsang M.; Doms R. W. J. Virol. 2001, 75, 8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnec X.; Quan L.; Olson W. C.; Hazan U.; Dragic T. J. Virol. 2005, 79, 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori T.; Umetsu M.; Nakamshi T.; Tsumoto K.; Ohara S.; Abe H.; Naito M.; Asano R.; Adschiri T.; Kumagai I. Biochem. Biophys. Res. Commun. 2008, 365, 751. [DOI] [PubMed] [Google Scholar]
- Sloane A. J.; Raso V.; Dimitrov D. S.; Xiao X. D.; Deo S.; Muljadi N.; Restuccia D.; Turville S.; Kearney C.; Broder C. C.; Zoellner H.; Cunningham A. L.; Bendall L.; Lynch G. W. Immunol. Cell Biol. 2005, 83, 129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.