

Abstract
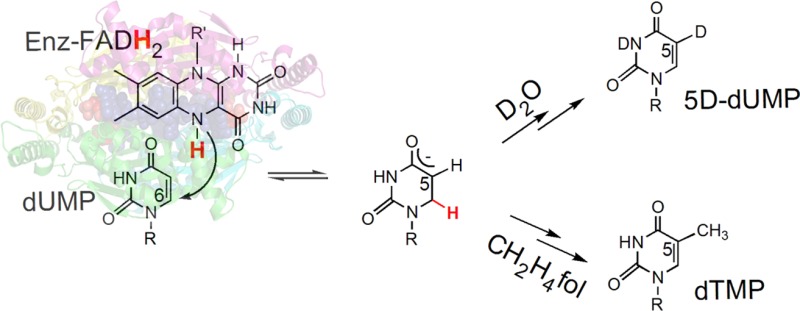
Thymidylate is a critical DNA nucleotide that has to be synthesized in cells de novo by all organisms. Flavin-dependent thymidylate synthase (FDTS) catalyzes the final step in this de novo production of thymidylate in many human pathogens, but it is absent from humans. The FDTS reaction proceeds via a chemical route that is different from its human enzyme analogue, making FDTS a potential antimicrobial target. The chemical mechanism of FDTS is still not understood, and the two most recently proposed mechanisms involve reaction intermediates that are unusual in pyrimidine biosynthesis and biology in general. These mechanisms differ in the relative timing of the reaction of the flavin with the substrate. The consequence of this difference is significant: the intermediates are cationic in one case and neutral in the other, an important consideration in the construction of mechanism-based enzyme inhibitors. Here we test these mechanisms via chemical trapping of reaction intermediates, stopped-flow, and substrate hydrogen isotope exchange techniques. Our findings suggest that an initial activation of the pyrimidine substrate by reduced flavin is required for catalysis, and a revised mechanism is proposed on the basis of previous and new data. These findings and the newly proposed mechanism add an important piece to the puzzle of the mechanism of FDTS and suggest a new class of intermediates that, in the future, may serve as targets for mechanism-based design of FDTS-specific inhibitors.
Thymidylate (2′-deoxythymidine-5′-monophosphate, or dTMP), an important DNA precursor, can be either scavenged by cells from thymidine in the environment, via thymidine kinase-catalyzed phosphorylation, or generated in cells de novo. The last committed step in the intracellular de novo biosynthesis of dTMP is catalyzed by the enzyme thymidylate synthase (TSase). TSase is encoded by thyA gene in eukaryotes and TYMS gene in mammals, while in many pathogenic bacteria and viruses this protein is the product of a completely different gene, thyX.1−3 TSase enzymes realize two chemical transformations: substitution of the C5 hydrogen of substrate 2′-deoxyuridine-5′-monophosphate (dUMP) with the methylene from the N5,N10-methylene-5,6,7,8-tetrahydrofolate (CH2H4fol) cofactor, and reduction of the transferred methylene by a hydride to form the C5 methyl of the product dTMP (Scheme 1). The thyA-encoded TSase uses CH2H4fol as a source of both the methylene and the reducing hydride.4,5 In thyX-encoded TSase, on the other hand, the hydride is supplied by the reduced flavin adenine dinucleotide prosthetic group (FADH2).6,7 Furthermore, the thyA- and thyX-encoded TSases share no sequence or structural resemblance and have been shown to catalyze dUMP→dTMP conversion by completely different chemical mechanisms.8 This structural and mechanistic divergence of the two enzymes provides an attractive direction for the design of drugs tailored to microbial thymidylate biosynthesis.
Scheme 1. Proposed Chemical Mechanisms for FDTS.
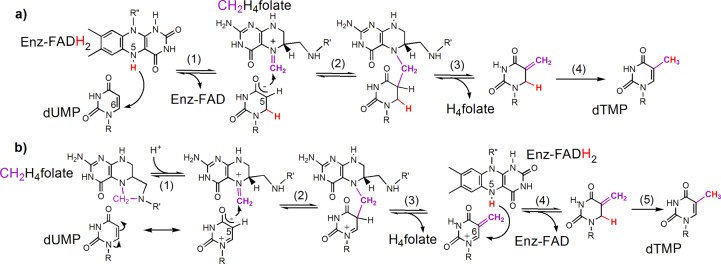
Adapted with permission from ref (11). R = 2′-deoxyribose-5′-phosphate; R′ = (p-aminobenzoyl)glutamate; R″ = adenosine-5′-pyrophosphate-ribityl.
The catalytic mechanism of thyA-encoded classical TSase enzymes has been studied for many years. In the chemical mechanism of classical TSase,4,5 the enzyme activates dUMP for subsequent reaction via a nucleophilic attack at C6 of the uracil by an active-site cysteine. This mechanistic feature is conserved in all thyA-encoded TSases, as well as in other uracil-methylating enzymes (e.g., rRNA- and tRNA-methyltransferases),8 and has been exploited by chemotherapeutic drugs targeting TSase (e.g., 5-fluorouracil).9 In contrast to thyA-encoded TSase, flavin-dependent TSase (FDTS) bypasses the need for an active-site nucleophile.10 Instead, FDTS has been proposed to accomplish substrate activation by either direct FADH2 reduction of dUMP to form reactive enolate (step 1 in Scheme 1a) or polarization of the uracil moiety in the active site to make the C5 nucleophilic (resonance form of dUMP in Scheme 1b).11 The possibility that the N5 of FADH2 nucleophilically activates dUMP has been eliminated by the observation that 5-deaza-FAD-FDTS is still active.10 As described below, hydrogen isotope exchange on dUMP supports substrate activation by the reduced flavin (Scheme 1a). However, the deuteration of the substrate and product but not the trapped intermediate in FDTS reactions in D2O cannot be explained by either of the mechanisms in Scheme 1; thus an alternative proposal is called for.
Direct hydride transfer from the N5 of reduced flavin to dUMP (Scheme 1a) was proposed to initiate the reaction based on the deuterium incorporation at C6 of dTMP, in the reactions of Thermatoga maritima FDTS in D2O conducted at sub-physiological temperatures.10 In all FDTS crystal structures in complex with both FAD and dUMP, the N5 of FAD is indeed in close proximity of the C6 of dUMP (ca. 3.4 Å), consistent with the postulated direct hydride transfer from the flavin. This chemistry is unusual in thymidylate biosynthesis and uridine methylation in general, but it is not without precedent in enzymology. For example, direct hydride addition from reduced flavin to an equivalent position of α,β-unsaturated substrates similar to dUMP occurs in reactions catalyzed by the old-yellow enzyme12 and dihydroorotate dehydrogenase.13 In the proposed mechanism in Scheme 1a, substrate reduction by FADH2 (i.e., flavin oxidation) takes place prior to the methylene transfer; consequently, the reaction intermediates along this path are reduced and non-aromatic in nature.
In an alternative mechanism (Scheme 1b), dUMP is activated for the reaction with CH2H4fol via electronic polarization of the uracil moiety in the enzyme’s active site upon binding. This dUMP polarization was proposed as the initial step on the basis of the disappearance of dUMP in a single-turnover experiment occurring before flavin oxidation (ref (11) and the green trace in Figure 1). To the best of our knowledge, no such addition of formaldehyde and/or Mannich amines (analogous to iminium CH2H4fol) to the C5 of uracil has been observed before without the aid of a nucleophile, either in enzymes or in solution; nevertheless, such chemistry does not violate any obvious chemical rules. Following elimination of H4fol from the dUMP-folate adduct (step 3 in Scheme 1b), a positively charged exocyclic methylene intermediate would be obtained. This intermediate could then be reduced by FADH2 at C6 to yield the same isomer proposed in Scheme 1a, accounting for the observed D6 in the product dTMP.10 In such a mechanism, flavin oxidation happens after the methylene transfer; as a consequence, the reaction intermediates are not reduced, in sharp contrast to the mechanism in Scheme 1a.
Figure 1.
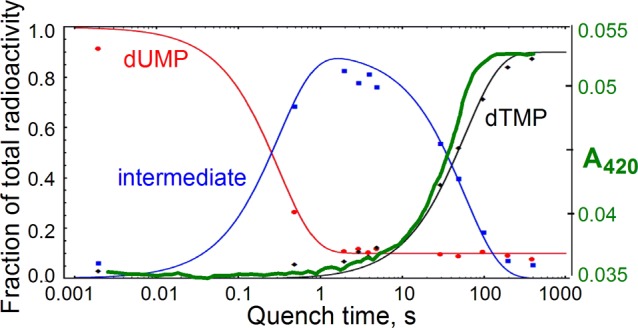
Single-turnover FDTS reaction kinetics overlaid with stopped-flow flavin absorbance trace (green, this work). Reduced flavin (FADH2) has no 420 nm absorbance, while oxidized flavin (FAD) does. Adapted with permission from ref (11).
Recently, we isolated and characterized a derivative of an intermediate(s) in FDTS-catalyzed thymidylate synthesis, in rapid acid-quenching experiments conducted at room temperature.11 This trapped species (5-hydroxymethyl-dUMP, or 5-HM-dUMP, Scheme 2) already contains the methylene of CH2H4fol. 5-HM-dUMP is consistent with either of the two proposed mechanisms for FDTS (Scheme 1) and does not distinguish between them. In the current work, in an attempt to differentiate between the mechanisms, we repeated acid-quenching experiments with FDTS reactions taking place in deuterated water (D2O). The reaction conditions were kept the same as in the quenching studies in H2O,11 except all reactants and buffers were exchanged into D2O by cycles of lyophilization and resuspension in heavy water (99.9% D). In D2O, all exchangeable hydrogens, including the N5 hydrogen of the reduced flavin to be transferred to the uracil moiety, are exchanged with their heavier isotopes. Thus, we anticipated that, if the hydride from the flavin is transferred to dUMP before the methylene (Scheme 1a), then a portion of acid-trapped 5-HM-dUMP would be deuterated, i.e., one mass unit heavier than in the reactions conducted in H2O (Scheme 2a). On the other hand, on the basis of the mechanism proposed in Scheme 1b, no effect on the mass of the trapped intermediate was expected (Scheme 2b). As shown in Figure 2, no deuterium enrichment is observed in 5-HM-dUMP isolated in the D2O experiment. Importantly, all dTMP product present in D2O reactions was singly deuterated, eliminating the possibility of protium contamination in the experiment and in accordance with previously reported deuterium incorporation into dTMP.10
Scheme 2. Acid Trapping of the Proposed Intermediates in the Reaction with Deuterium-Labeled Flavin (FADD2).

Formation of 5-HM-dUMP in (a) requires oxidation of the reduced intermediates at C6, i.e., loss of a hydron (H+ or D+) and two electrons. Due to an isotope effect on this nonenzymatic oxidation, the majority of 5-HM-dUMP is expected to be deuterated. Molecular oxygen has been proposed as the oxidant,11 since quenched reactions are exposed to oxygen during quenching.
Figure 2.

HRMS of 5-hydroxymethyl-dUMP isolated from the acid-quenched FDTS reactions in H2O and D2O.
The above observation appears to support the mechanism suggested in Scheme 1b; however, while analyzing acid-quenched FDTS reactions conducted in D2O by MS, we noticed that a significant portion (>60%) of unreacted substrate, dUMP, was singly deuterated (Figure 3b). Much longer incubations of the same reaction mixture with the oxidized FDTS yielded no trace of deuterated dUMP (Figure 3c). This observation is inconsistent with the polarization mechanism in Scheme 1b, which does not require reduced flavin. In stark contrast to classical TSase-catalyzed H/D exchange at C5 of dUMP, which is strongly dependent on CH2H4folate,14 the dUMP in FDTS reactions is deuterated to the same extent whether CH2H4folate is present or absent, depending only on the flavin being reduced (Figure 3c). The location of the incorporated deuterium was confirmed to be the C5 of uracil by the following series of observations: (i) incubation of reduced enzyme with 5D-dUMP in H2O resulted in loss of the deuterium label (Figure S1b); (ii) tritium of [5-3H]-dUMP was released into water upon incubation with reduced FDTS (Figure S2e) but not with oxidized FDTS or without the enzyme; and (iii) in the same experiment with [6-3H]-dUMP, all of the tritium remained on dUMP (Figure S2b), indicating that the reduced enzyme catalyzes the exchange of the C5 hydrogen and not that of C6. The choice of the reducing agent (sodium dithionite vs NADPH) had no effect on the observed dUMP deuteration (Figure 3b,d), ruling against dithionite decomposition products (e.g., thiosulfate)15 as uracil activators. The exchange on C5 of uracil generally requires Michael addition at C6 (Scheme S1), as demonstrated with a variety of nucleophiles in solution (see Supporting Information for references). Altogether, the above observations and controls are in line with the H/D exchange at C5 of dUMP being catalyzed by the attack of hydride from the enzyme-bound FADH2 on the C6 of dUMP, as proposed in step 1 of Scheme 1a. The possibility that substrate H/D exchange is enabled by a significant conformational change in FDTS upon reduction is not supported by any of the current crystal structures of FDTSs from several organisms and with various ligands and mutations, but it cannot be positively excluded.
Figure 3.

ESI-MS spectra of dUMP incubated in D2O with dithionite (a), dithionite-reduced FDTS (b), oxidized FDTS (c), and NADPH-reduced FDTS (d). All spectra were collected in the negative-ion mode.
Neither of the mechanisms in Scheme 1 can explain all of the findings described in this work. Specifically: when incubating the mixture in D2O, deuteration of the substrate dUMP at C5 occurs only with reduced enzyme-bound flavin; in D2O, the product dTMP is mono-deuterated at either C6 or C7, yet the acid-trapped intermediate is not deuterated, and the flavin is still reduced as this intermediate is trapped (Figure 1). A new mechanism is proposed in Scheme 3 that reconciles these seemingly contradictory observations. By this mechanism the H/D exchange at C5 of dUMP in D2O (after step 1) requires the flavin to be reduced, as observed experimentally (Figure 3). A reversible stereospecific hydride transfer from N5 of the reduced flavin to C6 of the uracil moiety, similar to the one proposed in Scheme 3 (steps 1 and 3), has been observed before in dihydrouridine synthase16,17 and dihydropyrimidine dehydrogenase.18 If steps 1–3 are fast and occur within the dead-time of the flow experiments (2 ms), this would also be in accordance with the presence of the methylene in the earliest intermediate trapped.11 We attempted to detect the presumed initial intermediate that follows the first hydride transfer from the flavin (step 1 in Scheme 3) using the substrate dUMP with and without the unreactive CH2H4folate analogue, folinic acid (see Supporting Information for experimental details and results). That step is reversible, and if at equilibrium a significant amount of oxidized flavin were to accumulate, its detection would have supported the mechanism in Scheme 3. As no oxidation of FADH2 was detected, this attempt cannot support or rule out the proposed mechanism. The unusual intermediates between steps 3 and 5 accumulate and are trapped by the acid in the quench-flow experiment.11 Step 5 (or 4b) seems to occur ∼20 s before step 6, in accordance with the delay observed between the stopped-flow kinetics and the formation of dTMP product (Figure 1). Because the oxidized FAD is formed only transiently and does not accumulate kinetically, the flavin is expected to appear spectrally reduced, in agreement with the stopped-flow kinetics in Figure 1. Finally, the intermediates forming between steps 3 and 5 would contain no deuterium in D2O and could readily undergo hydroxyl addition at the methylene carbon to yield non-deuterated 5-hydroxymethyl-dUMP observed in acid-quenched FDTS reactions (Figure 2). It is noteworthy that the bridged intermediate between steps 3 and 4 has recently been suggested by QM/MM calculations in classical thymidylate synthase.19
Scheme 3. Proposed Alternative Mechanism for FDTS That Agrees with Both Current and Past Findings.
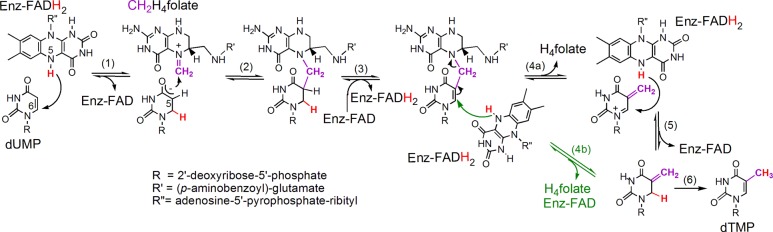
The hypothesis is that steps 1–3 occur within the dead-time of the flow experiments (2 ms), and that intermediates between steps 3 and 5 accumulate and are trapped by the acid in the quench-flow experiment.11 At this time it is not clear if the elimination of H4fol precedes the hydride transfer from the flavin (step 4a) or is concerted with it (the green arrows in step 4b). Note that FAD prosthetic group remains bound to the enzyme throughout the catalytic cycle, although its isoalloxazine ring fluctuates toward and away from the substrate as described in ref (21).
FDTS is a promising antibiotic drug target because, while it is absent in humans, it is present in ∼30% of all microorganisms, several of which are severe human pathogens. For example, all Rickettsia rely solely on FDTS for thymidylate, and the essentiality of FDTS has been recently illustrated in M. tuberculosis.20 FDTS is the only known uracil-methylating enzyme that does not employ an enzymatic nucleophile for uridylate activation,8 thus presenting a unique target for small-molecule inhibition. The current study sheds new light on the complex mechanism of the reaction catalyzed by this enzyme. Further investigation of the proposed mechanistic features is underway to build the platform for mechanism-based design of FDTS inhibitors. The mechanism proposed here is unique to FDTS, which lends confidence in high specificity of such inhibitors.
Acknowledgments
This work was supported by NIH R01 GM065368 and NSF CHE 1149023 to A.K., and by the Iowa Center for Biocatalysis and Bioprocessing to T.V.M., associated with NIH Predoctoral Training Program in Biotechnology, Grant T32 GM008365.
Supporting Information Available
Materials, stopped-flow, folinic acid assay and data, and substrate hydrogen isotope exchange procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Myllykallio H.; Lipowski G.; Leduc D.; Filee J.; Forterre P.; Liebl U. Science 2002, 297, 105–107. [DOI] [PubMed] [Google Scholar]
- Leduc D.; Graziani S.; Lipowski G.; Marchand C.; Le Marechal P.; Liebl U.; Myllykallio H. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7252–7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernyshev A.; Fleischmann T.; Kohen A. Appl. Microbiol. Biotechnol. 2007, 74, 282–289. [DOI] [PubMed] [Google Scholar]
- Carreras C. W.; Santi D. V. Annu. Rev. Biochem. 1995, 64, 721–762. [DOI] [PubMed] [Google Scholar]
- Finer-Moore J. S.; Santi D. V.; Stroud R. M. Biochemistry 2003, 42, 248–256. [DOI] [PubMed] [Google Scholar]
- Agrawal N.; Lesley S. A.; Kuhn P.; Kohen A. Biochemistry 2004, 43, 10295–10301. [DOI] [PubMed] [Google Scholar]
- Gattis S. G.; Palfey B. A. J. Am. Chem. Soc. 2005, 127, 832–833. [DOI] [PubMed] [Google Scholar]
- Mishanina T. V.; Koehn E. M.; Kohen A. Bioorg. Chem. 2011, 43, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touroutoglou N.; Pazdur R. Clin. Cancer Res. 1996, 2, 227–243. [PubMed] [Google Scholar]
- Koehn E. M.; Fleischmann T.; Conrad J. A.; Palfey B. A.; Lesley S. A.; Mathews I. I.; Kohen A. Nature 2009, 458, 919–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishanina T. V.; Koehn E. M.; Conrad J. A.; Palfey B. A.; Lesley S. A.; Kohen A. J. Am. Chem. Soc. 2012, 134, 4442–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown B. J.; Deng Z.; Karplus P. A.; Massey V. J. Biol. Chem. 1998, 273, 32753–32762. [DOI] [PubMed] [Google Scholar]
- Fagan R. L.; Nelson M. N.; Pagano P. M.; Palfey B. A. Biochemistry 2006, 45, 14926–14932. [DOI] [PubMed] [Google Scholar]
- Carreras C. W.; Climie S. C.; Santi D. V. Biochemistry 1992, 31, 6038–6044. [DOI] [PubMed] [Google Scholar]
- Burlamacchi L.; Guarini G.; Tiezzi E. Trans. Faraday Soc. 1969, 65, 496–502. [Google Scholar]
- Rider L. W.; Ottosen M. B.; Gattis S. G.; Palfey B. A. J. Biol. Chem. 2009, 284, 10324–10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Tanaka Y.; Yamashita K.; Suzuki T.; Nakamura A.; Hirano N.; Suzuki T.; Yao M.; Tanaka I. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 19593–19598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohkamp B.; Voevodskaya N.; Lindqvist Y.; Dobritzsch D. Biochim. Biophys. Acta 2010, 1804, 2198–2206. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Ferrer S.; Moliner V.; Kohen A. Biochemistry 2013, 52, 2348–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fivian-Hughes A. S.; Houghton J.; Davis E. O. Microbiology 2012, 158, 308–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehn E. M.; Perissinotti L. L.; Moghram S.; Prabhakar A.; Lesley S. A.; Mathews I. I.; Kohen A. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 15722–15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.