

Abstract
Context:
Chronic supraphysiological glucocorticoid therapy controls the androgen excess of 21-hydroxylase deficiency (21OHD) but contributes to the high prevalence of obesity, glucose intolerance, and reduced bone mass in these patients. Abiraterone acetate (AA) is a prodrug for abiraterone, a potent CYP17A1 inhibitor used to suppress androgens in the treatment of prostate cancer.
Objective:
The objective of the study was to test the hypothesis that AA added to physiological hydrocortisone and 9α-fludrocortisone acetate corrects androgen excess in women with 21OHD without causing hypertension or hypokalemia.
Design:
This was a phase 1 dose-escalation study.
Setting:
The study was conducted at university clinical research centers.
Participants:
We screened 14 women with classic 21OHD taking hydrocortisone 12.5–20 mg/d to enroll six participants with serum androstenedione greater than 345 ng/dL (>12 nmol/L).
Intervention:
AA was administered for 6 days at 100 or 250 mg every morning with 20 mg/d hydrocortisone and 9α-fludrocortisone acetate.
Main Outcome Measure:
The primary endpoint was normalization of mean predose androstenedione on days 6 and 7 (< 230 ng/dL [<8 nmol/L)] in greater than 80% of participants. Secondary end points included serum 17-hydroxyprogesterone and testosterone (T), electrolytes, plasma renin activity, and urine androsterone and etiocholanolone glucuronides.
Results:
With 100 mg/d AA, mean predose androstenedione fell from 764 to 254 ng/dL (26.7–8.9 nmol/L). At 250 mg/d AA, mean androstenedione normalized in five participants (83%) and decreased from 664 to 126 ng/dL (23.2–4.4 nmol/L), meeting the primary end point. Mean androstenedione declined further during day 6 to 66 and 38 ng/dL (2.3 and 1.3 nmol/L) at 100 and 250 mg/d, respectively. Serum T and urinary metabolites declined similarly. Abiraterone exposure was strongly negatively correlated with mean androstenedione. Hypertension and hypokalemia were not observed.
Conclusion:
AA 100–250 mg/d added to replacement hydrocortisone normalized several measures of androgen excess in women with classic 21OHD and elevated serum androstenedione.
Steroid 21-hydroxylase deficiency (21OHD) is among the most common genetic diseases and the most common form of congenital adrenal hyperplasia (1, 2). Most mutations causing 21OHD derive from gene conversions (3–5) from the CYP21A1P pseudogene to the CYP21A2 gene (6, 7), which encodes the cytochrome P450c21 (CYP21A2) enzyme. The incidence of classic or severe 21OHD is 1:16 000 worldwide (8), but nonclassic or mild 21OHD is at least 10 times more common (9), with an incidence of 1 in less than 300 in certain populations (10). The physiology of classic 21OHD results from cortisol and aldosterone deficiency, plus androgen excess. With the block in steroid 21-hydroxylation (Figure 1), the only remaining steroidogenic pathways in 21OHD involve the related enzyme steroid 17-hydroxylase/17,20-lyase [cytochrome P450c17 (CYP17A1)] (11). The flux of accumulating cortisol precursors via CYP17A1 generates the androgen excess characteristic of 21OHD.
Figure 1.
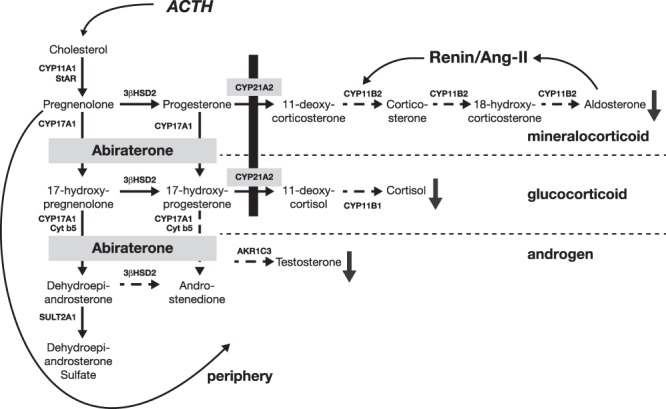
Steroidogenic pathways and disruption in 21OHD. Deficiency of CYP21A2 (black bar) impairs mineralocorticoid (aldosterone) and glucocorticoid (cortisol) production. Precursors accumulate and divert to pathways involving CYP17A1 to 19-carbon steroids dehydroepiandrosterone, androstenedione, and T (androgens). Addition of abiraterone acetate blocks CYP17A1-mediated pathways and lowers androgen production. AKR1C3, aldo-keto reductase family 1, member C3; Ang II, angiotensin II; 3βHSD2, 3β-hydroxysteroid dehydrogenase/isomerase type II; StAR, steroidogenic acute regulatory protein; SULT2A1, sulfotransferase family 2A, member 1.
The treatment of 21OHD consists of glucocorticoid and mineralocorticoid administration (12), which both replaces the hormone deficiencies and lowers ACTH and adrenal-derived androgen production. The androgen excess of poorly controlled 21OHD causes sexual precocity, compromised adult height, and impaired fertility as well as hirsutism, virilization, and male-pattern baldness in women (13, 14). Physiological doses of hydrocortisone (6–10 mg/m2 · d) correct the adrenal insufficiency; however, this minimal treatment often fails to normalize the androgen excess. To achieve good control of androgen excess, hydrocortisone is given in three or four divided doses at supraphysiological amounts up to 17 mg/m2 · d (15). Substitution of prednisolone or dexamethasone for hydrocortisone often improves control of androgen excess (12) but predisposes to iatrogenic Cushing syndrome with growth retardation, obesity, glucose intolerance, dermal atrophy, and bone loss (13, 14). The treatment of 21OHD is thus a difficult balance between the morbidities of androgen excess from undertreatment and glucocorticoid excess from overtreatment. No alternative nonsurgical strategies to lower the androgen excess of 21OHD currently exist.
Abiraterone acetate is a prodrug, which is metabolized to abiraterone, a potent active site-directed inhibitor of CYP17A1 (16). Abiraterone acetate added to medical castration suppresses circulating testosterone (T) and improves survival in castration-resistant prostate cancer (17, 18). CYP17A1 inhibition with abiraterone, however, also blocks cortisol synthesis and raises ACTH production as in genetic CYP17A1 deficiency (17-hydroxylase deficiency) (19, 20). By blocking CYP17A1-mediated pathways, abiraterone acetate therapy increases steroid flux via CYP21A2 to 11-deoxycorticosterone (DOC), a mineralocorticoid, which causes the hypertension and hypokalemia (21, 22) characteristic of genetic 17-hydroxylase deficiency. Consequently, abiraterone acetate is combined with prednisolone or prednisone to suppress ACTH and DOC production (22).
Because all androgen biosynthesis requires CYP17A1 activities, we reasoned that abiraterone acetate might control the androgen excess of 21OHD and obviate the need for supraphysiological glucocorticoids. Furthermore, because patients with 21OHD lack CYP21A2, they should not produce DOC during abiraterone acetate treatment. We therefore hypothesized that abiraterone acetate added to stable doses of physiological hydrocortisone and 9α-fludrocortisone acetate will control the androgen excess in 21OHD without causing hypertension or hypokalemia, and we tested this hypothesis in a phase 1 study of adult women with inadequately controlled classic 21OHD.
Participants and Methods
Study oversight and conduct
Academic and sponsor-employed investigators designed the study and wrote the protocol. The first author wrote the manuscript, and the sponsor provided funding for editorial assistance. All the authors then completed and approved the manuscript. The authors assume responsibility for the completeness and integrity of the data and the fidelity of the study to the protocol and analysis plan. Data were analyzed by statisticians employed by the sponsor.
The review boards of all participating institutions approved the study, which was conducted according to the principles of the Declaration of Helsinki, the International Conference on Harmonisation, and the guidelines for good clinical practice. All participants provided written informed consent.
Study design, participants, and safety measures
In this phase 1 nonrandomized, open-label, multiple-dose, intrasubject, sequential dose-escalation study, we screened 14 adult women with genotype-proven classic 21OHD, lacking wild-type or nonclassic 21OHD alleles. The six enrolled participants met all entry criteria, including a serum androstenedione concentration greater than 1.5 times above the upper limit of normal for adult women [345 ng/dL (12 nmol/L)]. The study consisted of a 40-day screening period with a qualifying period of up to 25 days and up to three treatment periods of escalating dosage. The primary end point was serum androstenedione, mean of days 6 and 7, within the normal range for at least 80% of participants. If the primary end point was not met, another treatment period at the next higher dose began after a rest period of at least 7 days. Participants maintained study-defined replacement doses of hydrocortisone (20 mg/d in two to three divided doses not later than 8:00 pm) and 9α-fludrocortisone acetate (∼0.1 mg/d, not later than 10:00 pm) for at least 6 days prior to the qualifying androstenedione draw, through all treatment periods and rest periods, and until study day 8 of the last treatment period. The Supplemental Material file contains all inclusion and exclusion criteria; CYP21A2 genotypes, medications, and screening androstenedione values; baseline characteristics; safety measures and adverse events (AEs); and a study flow scheme (Supplemental Tables 1–4 and Supplemental Figure 1, respectively).
The study team maintained a log of abiraterone acetate dispensed and administered and inventoried drug supplies throughout the study. Diary cards were provided to each subject to record at-home administration of abiraterone acetate, hydrocortisone, and 9α-fludrocortisone acetate. Participants returned the unused study drug and empty glass vials for accountability. Participants and investigators documented all medications taken during the study and until 30 days after completion. Drugs known to induce and/or inhibit CYP3A4 were not permitted during the study. An end-of-study visit was conducted on study day 8 of the last treatment period.
Safety analyses included documentation of all AEs, particularly hypertension, hypokalemia, and liver or renal dysfunction. Other laboratory tests obtained during the study included hemoglobin, white blood cell count (with differential at screening), platelet count, serum chemistry panel, liver function tests, thyroid function tests, urinalysis and urine sediment, and serum or urine pregnancy test for women with an intact uterus. Hepatitis A, B, and C serologies and electrocardiogram were obtained at screening. Vital signs were obtained at screening and at all visits during the treatment periods. A complete physical examination was conducted at screening and on days 1 and 8 of the treatment periods. A follow-up telephone call was conducted approximately 30 days after the last dose of abiraterone acetate to collect data on AEs and any concomitant medications used to treat AEs.
Intervention
All participants received continuous oral contraceptive (OC) therapy containing estrogen and progestin plus a nonhormonal form of contraception to minimize androgen contribution from ovarian sources. Abiraterone acetate was formulated by Janssen as a 25-mg/mL suspension with half the bioavailability of tablets in 10- or 20-mL single-use glass bottles. Abiraterone acetate suspension was administered as a single first-morning dose on an empty stomach with more than 240 mL water from study days 1 to 6 of each treatment period. No food or medication was consumed for 1 hour after the dose, and hydrocortisone and 9α-fludrocortisone acetate were taken 1 hour after each abiraterone acetate dose. A starting abiraterone acetate dose of 100 mg/d was chosen based on data from prostate cancer trials (23), and additional 250- and 500-mg/d abiraterone acetate treatment periods were planned if the primary end point was not met in more than 80% of participants.
Outcome measures, pharmacodynamic end points, and pharmacokinetics
Blood and urine samples were collected before the abiraterone acetate dose on study days 1 and 6; on day 6 at 2, 4, 6, and 8 hours after the dose; and at 24 hours (day 7) and 48 hours (day 8) after the final abiraterone acetate dose. Secondary end points included serum concentrations of 17-hydroxyprogesterone (17OHP), T, and potassium; plasma renin activity; urinary excretion of androsterone and etiocholanolone glucuronides (uAn+Et); and abiraterone pharmacokinetic assessments, blood pressure, and safety profiles. Steroids were measured by liquid chromatography-tandem mass spectrometry at Esoterix/Laboratory Corporation of America. Esoterix also performed the CYP21A2 genotyping.
Blood samples for pharmacokinetics were collected predose on study day 6 and at 0.25, 0.5, 1, 2, 4, 6, 8, 24, and 48 hours after the dose. Plasma concentrations of abiraterone were determined using a validated liquid chromatography-tandem mass spectrometry assay under the supervision of Janssen's bioanalytical department. Some participants also had optional predose blood samples for pharmacokinetics on days 3 and 5.
Statistical analyses
Descriptive statistical methods were used to analyze the pharmacokinetics, pharmacodynamics, and safety end points. Frequencies and percentages of subjects together with the associated exact two-sided 95% binomial confidence intervals for the percentages were provided for the primary pharmacodynamic end point. Mean and mean changes from baseline in both primary and secondary end points were also summarized overall and by dose level with subject number, mean, SD, median, and range.
Results
Participants and screening
While taking hydrocortisone in two to three divided doses of 8 mg/m2 · d or greater up to 20 mg total per day, 8 of 14 participants with genotype-proven classic 21OHD were excluded due to qualifying serum androstenedione less than 1.5 times the upper limit of the normal range prior to the first-morning hydrocortisone dose [<345 ng/dL (<12 nmol/L); Supplemental Tables 2 and 3]. All six participants completed all treatment periods while taking hydrocortisone of 10 mg, 5 mg, 5 mg with meals, or 10 mg twice daily, plus stable, clinically indicated doses of 9α-fludrocortisone acetate. No increases in hydrocortisone dosage for acute illness immediately prior to and during treatment periods occurred during the trial. The mean change in serum androstenedione from the qualifying draw to day 1 up to 10 days later was −8%, demonstrating minimal influence of concomitant hydrocortisone and OC therapy on the primary end point during the time frame of each treatment period.
Outcomes
Predose serum mean androstenedione was chosen as the primary end point because androstenedione is a 19-carbon adrenal androgen precursor, which is characteristically elevated in 21OHD when disease control is inadequate (12, 24). Additional measures of androgen production included serum T and uAn+Et, which are the major androgen metabolites in urine (25). At the initial abiraterone acetate dose of 100 mg/d, serum androstenedione fell sharply to less than 40% of baseline values in all participants (Figure 2A). Serum T and uAn+Et declined in parallel with serum androstenedione (Figure 2, B and C). Serum mean androstenedione normalized in only three participants (50%), which triggered a second treatment period at a higher dose, per study protocol. At 250 mg/d abiraterone acetate, mean androstenedione normalized in five participants (83%), and serum androstenedione fell to normal [<230 ng/dL (< 8 nmol/L)] in all six participants on day 7 (Figure 2D). Serum T and uAn+Et also declined in parallel at 250 mg/d abiraterone acetate (Figure 2, E and F). The primary end point was thus met in more than 80% of participants, and no further dose escalations were conducted.
Figure 2.

Serum androstenedione (A and D), serum T (B and E), and the sum of urine androsterone + etiocholanolone glucuronides (C and F; Andro + Etio, micrograms per gram creatinine) during 100 mg/d (A–C) and 250 mg/d (D–F) abiraterone acetate treatment periods. Concentrations are shown at left and percentage changes at right. Dotted line in left graph of panels A and D indicate upper limit of normal (230 ng/dL = 8 nmol/L). To convert to SI units, multiply androstenedione by 0.0349 (nanomoles per liter), T by 0.0347 (nanomoles per liter), and androsterone + etiocholanolone glucuronides by 0.000242 (nanomoles per micromole of creatinine). Each participant is consistently shown by the same color and symbol in all figures.
After the final abiraterone acetate dose on day 6, serum androstenedione and T declined sharply to nadirs of 20–97 ng/dL and 9–40 ng/dL (0.7–3.4 and 0.31–0.14 nmol/L), respectively, during the 100 mg/d abiraterone acetate treatment period and to <10–65 ng/dL and <3–37 ng/dL (<0.3–2.3 and <0.1–1.3 nmol/L), respectively, during the 250 mg/d abiraterone acetate treatment period. Serum androstenedione and T were similar on days 6 and 7 and then remained well below baseline values at day 8, indicating that androgens had reached steady state in 1 week and demonstrating a sustained duration of action of at least 48 hours (Figure 2). In two participants, serum androstenedione was within the normal range at the start of the 250 mg/d treatment period, after 10–12 months of consistent hydrocortisone dosing. These two participants had normalized mean androstenedione during the 100 mg/d abiraterone acetate treatment period 10–12 months earlier. In all but one participant with low baseline uAn+Et for the 250 mg/d abiraterone acetate treatment period, the uAn+Et also declined further during day 6 and remained below baseline at day 8. The fall in serum androstenedione and T slightly preceded the decline in uAn+Et (Figure 2).
The predose serum 17OHP values, in contrast, mostly rose back to baseline on day 6, 24 hours after the prior abiraterone acetate dose. After the day 6 abiraterone acetate dose, however, serum 17OHP declined precipitously, often to less than 10% of baseline values at 2–8 hours after the final dose, and then returned to near baseline on days 7 and 8 (Figure 3). The mean serum T on days 6 and 7 and mean androstenedione values correlated inversely with exposure to abiraterone (Figure 4; r = −0.72 to −0.73 for all data, r = −0.91 to −0.98 for the 250 mg/d abiraterone acetate treatment period). Although the numbers of data points are small, this correlation is consistent with active drug exposure being the major contributor to the androgen declines during the treatment periods.
Figure 3.
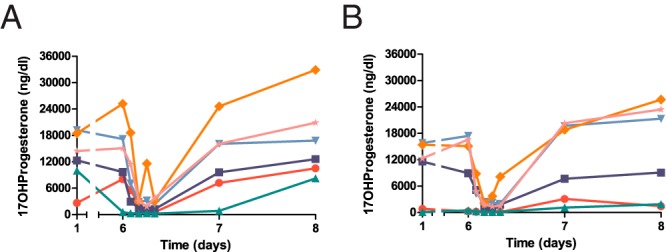
Serum 17-hydroxyprogesterone (17OHProgesterone) concentrations during 100 mg/d (A) or 250 mg/d (B) abiraterone acetate treatment periods. To convert to SI units, multiply by 0.0303 (nanomoles per liter).
Figure 4.
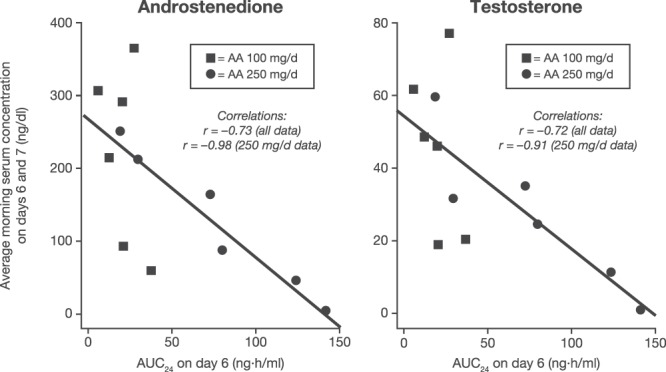
Pharmacokinetic/pharmacodynamic correlation during abiraterone acetate therapy. Mean serum concentrations of androstenedione (left panel) and T (right panel) are graphed as a function of area under the curve (AUC) over 24 hours (AUC24) for plasma abiraterone concentration on day 6 of treatment periods (squares, 100 mg/d; circles, 250 mg/d treatment periods). Lines are least squares fits to the data, with curve-fitting statistics shown in insets. AA, abiraterone acetate.
Adverse events
No participants developed hypertension or hypokalemia during the study. Plasma renin activity was not suppressed at the end of the treatment periods (Supplemental Table 4). These findings are consistent with lack of mineralocorticoid accumulation during abiraterone acetate therapy in this cohort of women with classic 21OHD. No serum transaminase or creatinine elevations were observed. No deaths, serious AEs, or AEs leading to discontinuation of abiraterone acetate were reported during this study, and the abiraterone acetate suspension was generally well tolerated.
Discussion
Our data provide compelling proof of concept that CYP17A1 inhibition is a feasible method to control androgen excess in classic 21OHD without using supraphysiological doses of hydrocortisone or more potent synthetic glucocorticoids. Given the challenges of conducting phase 1 studies with stringent entry criteria for rare diseases, this trial included a limited number of participants, a single treatment regimen, and two cycles of 6 days each. Nevertheless, abiraterone acetate at approximately 5%–15% of the dose equivalent used to treat prostate cancer markedly, consistently, and reproducibly lowered serum and urine androgens in all six adult women with classic 21OHD and elevated serum androgens without causing hypertension, hypokalemia, or renin suppression. Abiraterone acetate therapy afforded large fractional reductions in androgens for all participants, regardless of baseline values, as long as drug exposure was adequate (Figures 2–4). The study terminated early after meeting the primary end point during the 250 mg/d abiraterone acetate dose period, and both serum androstenedione and T fell well within the normal range in all participants within 6 hours of the final dose at both 100 mg/d and 250 mg/d [T < 55 ng/dL (< 1.9 nmol/L), Figure 2]. We cannot exclude a potential study effect from improved compliance with concurrent therapies, but the small differences between the qualifying and baseline androstenedione values for the first study period suggest that this contribution is small relative to the effect of abiraterone acetate (Supplemental Table 2).
Abiraterone acetate does not replace traditional therapies but rather allows for lower glucocorticoid dosing and maintains normal serum androgens despite lessened ACTH suppression. With reduced hydrocortisone doses, long-term exposure to higher circulating ACTH might predispose to the development of adrenal rest tumors, even in women who have had bilateral adrenalectomies (26), or of adrenal adenomas and myelolipomas (13, 14, 27, 28). Although adherence to conventional therapy for 21OHD reduces these neoplastic consequences (27), the side effects of standard regimens pose significant disincentives to compliance. Addition of abiraterone acetate, which allows lower glucocorticoid dosing and thus mitigates these untoward consequences, might facilitate compliance to well-tolerated glucocorticoid replacement regimens. We emphasize that 8 of 14 women screened did not qualify due to normal serum androstenedione, attesting to the efficacy of divided-dose hydrocortisone in many adults with 21OHD if taken consistently. These findings affirm that consistent glucocorticoid dosing remains the cornerstone of treatment for 21OHD into adulthood.
This study permits analysis of abiraterone action in patients with high androgen production and provides additional insight into abiraterone pharmacology. First, the absence of hypertension and hypokalemia or plasma renin activity suppression in our cohort with 21OHD supports the theory that 21-hydroxylated DOC causes these AEs associated with abiraterone (21, 22). The long-term consequences of accumulated upstream steroids under our treatment paradigm are unknown, but high progesterone typically impairs menses and might inhibit ovulation (29). The extraadrenal 21-hydroxylation of progesterone to DOC via hepatic cytochrome P450 enzymes is well documented (30, 31) but appears to be minor in our cohort. Second, a comparison of participants treated with hydrocortisone in two vs three divided doses illustrates the interplay of hydrocortisone and abiraterone action. In participants receiving 10 mg hydrocortisone 1 hour after abiraterone acetate plus 5 mg 3–5 and 8–10 hours later, serum androstenedione and T, as well as uAn+Et, declined progressively to plateau values during the 8 hours of sampling on day 6 after the final abiraterone acetate dose. In contrast, most participants taking hydrocortisone 10 mg twice daily with the doses more than 8 hours apart experienced an escape from androgen suppression during the 8 hours after the final abiraterone acetate dose (Figure 5). Abiraterone is a tight-binding CYP17A1 inhibitor with slow dissociation (16, 32, 33), rendering its inhibition functionally irreversible. Without a second dose of hydrocortisone less than 6 hours after abiraterone acetate, the rise in ACTH appears to induce the synthesis of new CYP17A1 protein, thus escaping inhibition. This analysis underscores that abiraterone acetate works in concert with hydrocortisone to control androgen excess and cannot substitute for glucocorticoid therapy in classic 21OHD.
Figure 5.

Comparison of androstenedione and T changes for participants taking three divided doses of hydrocortisone 20 mg/d (ie, three times a day, 10–5–5) (left) or two divided doses of hydrocortisone 20 mg/d (ie, twice a day, 10–0–10) (right) plus abiraterone acetate at 100 mg/d (A and B) or 250 mg/d (C and D). Two participants changed from three times a day to twice a day therapy between the 100 and 250 mg/d abiraterone acetate treatment periods. AA, abiraterone acetate.
The observed discrepancy in time courses of 17OHP and androgen changes reflects the simultaneous inhibition of sequential steps in steroidogenesis (17-hydroxylase and 17,20-lyase), both of which CYP17A1 catalyzes and abiraterone inhibits (11). The intermediate 17-hydroxylated products, reflected in serum 17OHP, fall rapidly, but with the subsequent accumulation of 17-deoxy precursors, 17OHP rises back to baseline values prior to the next dose. Because the 17-hydroxylated intermediates are not increased during multidose therapy, the inhibition of 17,20-lyase activity prevails and lowers androgen production. Similar changes in serum steroids and their urinary metabolites were observed in phase 2 prostate cancer trials using abiraterone acetate without glucocorticoid cotreatment (21). These changes also resonate with the known limitations of using 17OHP values alone to gauge control and to guide therapy in 21OHD, particularly for adults (12, 34).
Basal urinary androsterone to etiocholanolone glucuronide ratios, typically less than 1 in women (35), ranged from 1.1 to 2.3 in our cohort (Table 1). The urinary androsterone to etiocholanolone glucuronide ratio rises as steroid flux via the 5α-reduced or backdoor pathway to dihydrotestosterone (36) increases, because androsterone is a direct product of the steroidogenic tissue (37, 38). A previous study of patients with classic 21OHD found urinary androsterone to etiocholanolone glucuronide ratios near 1 in young adults and children older than 1 year of age (36). In contrast to that study, our participants were older, and their androgen excess control was suboptimal. The urinary androsterone to etiocholanolone glucuronide ratio fell to less than 1 during the 250 mg/d abiraterone acetate treatment period in three participants, two of whom had the lowest androgen values. Our data suggest that flux via the backdoor pathway increases in adults with 21OHD as control of androgen excess deteriorates.
Table 1.
Urinary Androsterone to Etiocholanolone Glucuronide Ratios
| Participant | Abiraterone Acetate 100 mg/d Treatment Period |
Abiraterone Acetate 250 mg/d TreatmentPeriod |
||
|---|---|---|---|---|
| Basal | Mean (Range) | Basal | Mean (Range) | |
| 200102 | 1.5 | 1.9 (1.5–2.2) | 1.2 | 1.0 (0.3–1.7)a |
| 200105 | 1.9 | 2.0 (1.8–2.1) | 1.5 | 1.3 (1.2–1.4) |
| 200106 | 1.4 | 1.7 (1.5–1.9) | 1.1 | 1.0 (0.7–1.3)a |
| 200111 | 1.9 | 2.2 (2.0–2.9) | 1.9 | 1.7 (1.3–2.0) |
| 200114 | 2.3 | 2.4 (2.0–2.7) | 1.5 | 1.6 (1.4–1.8) |
| 200115 | 1.5 | 1.4 (1.1–1.5) | 1.1 | 0.9 (0.7–1.3) |
Patients had two or more low androsterone glucuronide values on day 6, less than 250 μg/g creatinine.
In summary, CYP17A1 inhibition using abiraterone acetate, added to physiological hydrocortisone, 9α-fludrocortisone acetate, and OC therapy, ameliorates the androgen excess of women with classic 21OHD, with an excellent short-term safety profile. Our data encourage extended trials of this design, using modern, potent inhibitors of androgen biosynthesis and action, in selected adult women with classic and nonclassic 21OHD and in prepubertal children with poorly controlled classic 21OHD.
Acknowledgments
We thank the Nursing and Laboratory Staff of the Michigan Clinical Research Unit for their expert assistance with conducting the trial. We also thank the Janssen Research and Development Clinical Operations Team, including Ray Lekich, Shawn Kern, and Charlyn Logan, for monitoring, coordination, and regulatory activities, and we thank Dr D. Walt Chandler and Esoterix-Lab Corp Clinical Trials for supervising the steroid analyses and for discussions about the methodologies and results. We also thank Dr Italo Poggesi (Janssen Research and Development) for incorporating the pharmacokinetic/pharmacodynamic modeling into the trial design. We also thank Dr David Ehrmann (University of Chicago), who participated in the trial design and initiation. We also thank Dr Ira Mills (PAREXEL) for his assistance in the preparation of the manuscript.
This study is registered with ClinicalTrials.gov, with the number of NCT01495910.
This work was supported by Janssen Research and Development and Grant UL1-RR024986 from the National Institutes of Health to the Nursing and Laboratory Staff of the Michigan Clinical Research Unit. E.O.B. received support from Grant T32DK007245 from the National Institutes of Health.
This work was presented in abstract form at the 94th and 95th Annual Meetings of The Endocrine Society in June 2012 and June 2013.
Disclosure Summary: R.J.A., E.O.B., A.Y.C., G.D.H., C.R., and D.M. have received research grant support (September 2012 through January 2014) from Janssen Research and Development. G.W., M.G., X.S.X., J.W.S., J.J., and M.K.Y. are employed by Janssen Research and Development and own stock in Johnson & Johnson.
Footnotes
- AE
- adverse event
- CYP17A1
- cytochrome P450c17
- CYP21A2
- cytochrome P450c21
- DOC
- 11-deoxycorticosterone
- 21OHD
- 21-hydroxylase deficiency
- OC
- oral contraceptive
- 17OHP
- 17-hydroxyprogesterone
- uAn+Et
- urinary excretion of androsterone and etiocholanolone glucuronides.
References
- 1. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245–291 [DOI] [PubMed] [Google Scholar]
- 2. Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776–788 [DOI] [PubMed] [Google Scholar]
- 3. Higashi Y, Tanae A, Inoue H, Fujii-Kuriyama Y. Evidence for frequent gene conversions in the steroid 21-hydroxylase (P-450c21) gene: implications for steroid 21-hydroxylase deficiency. Am J Hum Genet. 1988;42:17–25 [PMC free article] [PubMed] [Google Scholar]
- 4. Donohoue PA, Van Dop C, McLean RH, White PC, Jospe N, Migeon CJ. Gene conversion in salt-losing congenital adrenal hyperplasia with absent complement C4B protein. J Clin Endocrinol Metab. 1986;62:995–1002 [DOI] [PubMed] [Google Scholar]
- 5. Morel Y, David M, Forest MG, et al. Gene conversions and rearrangements cause discordance between inheritance of forms of 21-hydroxylase deficiency and HLA types. J Clin Endocrinol Metab. 1989;68:592–599 [DOI] [PubMed] [Google Scholar]
- 6. White PC, Grossberger D, Onufer BJ, et al. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proc Natl Acad Sci USA. 1985;82:1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Higashi Y, Yoshioka H, Yamane M, Gotoh O, Fujii-Kuriyama Y. Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and genuine gene. Proc Natl Acad Sci USA. 1986;83:2841–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Therrell BLJ, Berenbaum SA, Manter-Kapanke V, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101:583–590 [DOI] [PubMed] [Google Scholar]
- 9. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet. 1985;37:650–667 [PMC free article] [PubMed] [Google Scholar]
- 10. Speiser PW, New MI, Tannin GM, Pickering D, Yang SY, White PC. Genotype of Yupik Eskimos with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Genet. 1992;88:647–648 [DOI] [PubMed] [Google Scholar]
- 11. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95:4133–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arlt W, Willis DS, Wild SH, et al. Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab. 2010;95:5110–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finkielstain GP, Kim MS, Sinaii N, et al. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97:4429–4438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonfig W, Pozza SB, Schmidt H, Pagel P, Knorr D, Schwarz HP. Hydrocortisone dosing during puberty in patients with classical congenital adrenal hyperplasia: an evidence-based recommendation. J Clin Endocrinol Metab. 2009;94:3882–3888 [DOI] [PubMed] [Google Scholar]
- 16. Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017α (17α-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem. 1995;38:2463–2471 [DOI] [PubMed] [Google Scholar]
- 17. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Costa-Santos M, Kater CE, Auchus RJ. Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab. 2004;89:49–60 [DOI] [PubMed] [Google Scholar]
- 20. Auchus RJ. Genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17-hydroxylase/17,20-lyase and isolated 17,20-lyase deficiency. In: New MI, ed. Genetic Steroid Disorders. Waltham, MA: Elsevier; 2014:111–123 [DOI] [PubMed] [Google Scholar]
- 21. Attard G, Reid AHM, Auchus RJ, et al. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J Clin Endocrinol Metab. 2012;97:507–516 [DOI] [PubMed] [Google Scholar]
- 22. Auchus ML, Auchus RJ. Human steroid biosynthesis for the oncologist. J Invest Med. 2012;60:495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Attard G, Reid AH, A'Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Verma S, Vanryzin C, Sinaii N, et al. A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 2010;72:441–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krone N, Hughes BA, Lavery GG, Stewart PM, Arlt W, Shackleton CH. Gas chromatography/mass spectrometry (GC/MS) remains a pre-eminent discovery tool in clinical steroid investigations even in the era of fast liquid chromatography tandem mass spectrometry (LC/MS/MS). J Steroid Biochem Mol Biol. 2010;121:496–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crocker MK, Barak S, Millo CM, et al. Use of PET/CT with cosyntropin stimulation to identify and localize adrenal rest tissue following adrenalectomy in a woman with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97:E2084–E2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reisch N, Scherr M, Flade L, et al. Total adrenal volume but not testicular adrenal rest tumor volume is associated with hormonal control in patients with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95:2065–2072 [DOI] [PubMed] [Google Scholar]
- 28. Nermoen I, Rorvik J, Holmedal SH, et al. High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21-hydroxylase deficiency. Clin Endocrinol (Oxf). 2011;75:753–759 [DOI] [PubMed] [Google Scholar]
- 29. Casteràs A, De Silva P, Rumsby G, Conway GS. Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clin Endocrinol (Oxf). 2009;70:833–837 [DOI] [PubMed] [Google Scholar]
- 30. Casey ML, MacDonald PC. Extra-adrenal formation of a mineralocorticoid: Deoxycorticosterone and deoxycorticosterone sulfate biosynthesis and metabolism. Endocr Rev. 1982;3:396–403 [DOI] [PubMed] [Google Scholar]
- 31. Gomes LG, Huang N, Agrawal V, Mendonça BB, Bachega TA, Miller WL. Extraadrenal 21-hydroxylation by CYP2C19 and CYP3A4: effect on 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2009;94:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482:116–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garrido M, Peng HM, Yoshimoto FK, Upadhyay SK, Bratoeff E, Auchus RJ. A-Ring modified steroidal azoles retain similar potent and slowly reversible CYP17A1 inhibition as abiraterone. J Steroid Biochem Mol Biol. 2014;143C:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Auchus RJ, Arlt W. Approach to the patient: the adult with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2013;98:2645–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Callies F, Arlt W, Siekmann L, Hubler D, Bidlingmaier F, Allolio B. Influence of oral dehydroepiandrosterone (DHEA) on urinary steroid metabolites in males and females. Steroids. 2000;65:98–102 [DOI] [PubMed] [Google Scholar]
- 36. Kamrath C, Hochberg Z, Hartmann MF, Remer T, Wudy SA. Increased activation of the alternative “backdoor” pathway in patients with 21-hydroxylase deficiency: evidence from urinary steroid hormone analysis. J Clin Endocrinol Metab. 2012;97:E367–E375 [DOI] [PubMed] [Google Scholar]
- 37. Wilson JD, Auchus RJ, Leihy MW, et al. 5α-Androstane-3α,17β-diol is formed in tammar wallaby pouch young testes by a pathway involving 5α-pregnane-3α,17α-diol-20-one as a key intermediate. Endocrinology. 2003;144:575–580 [DOI] [PubMed] [Google Scholar]
- 38. Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab. 2004;15:432–438 [DOI] [PubMed] [Google Scholar]