

Abstract
Down-regulation of expression of trmD, encoding the enzyme tRNA (guanosine-1)-methyltransferase, has shown that this gene is essential for growth of Streptococcus pneumoniae. The S. pneumoniae trmD gene has been isolated and expressed in Escherichia coli by using a His-tagged T7 expression vector. Recombinant protein has been purified, and its catalytic and physical properties have been characterized. The native enzyme displays a molecular mass of approximately 65,000 Da, suggesting that streptococcal TrmD is a dimer of two identical subunits. In fact, this characteristic can be extended to several other TrmD orthologs, including E. coli TrmD. Kinetic studies show that the streptococcal enzyme utilizes a sequential mechanism. Binding of tRNA by gel mobility shift assays gives a dissociation constant of 22 nM for one of its substrates, 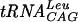 . Other heterologous nonsubstrate tRNA species, like
. Other heterologous nonsubstrate tRNA species, like 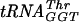 , tRNAPhe, and
, tRNAPhe, and 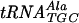 , bind the enzyme with similar affinities, suggesting that tRNA specificity is achieved via a postbinding event(s).
, bind the enzyme with similar affinities, suggesting that tRNA specificity is achieved via a postbinding event(s).
Proteins that methylate tRNA at position G37 are ubiquitous in nature, and no cellular form has been found which does not possess this important activity. In bacteria, the enzyme tRNA (guanosine-1)-methyltransferase (1MGT or TrmD) modifies the subset of tRNAs that have a G at position 36 and recognize codons beginning with C, including those for leucine, proline, and arginine. It has been shown that methylation at G37 prevents frameshifting (7), and in strains of Salmonella enterica serovar Typhimurium lacking this activity, frameshifting occurs at selected proline codons (32, 33). The deficiency of m1G in these strains also appears to have a more global effect on cell physiology as evidenced by changes in carbon source metabolism and sensitivity to amino acid analogs (24). In particular, lack of m1G is found to affect metabolism of thiamine and pantothenate (4, 6). TrmD activity has been shown to be important for growth of the gram-negative bacteria S. enterica serovar Typhimurium (5) and Escherichia coli (30), and recent studies suggest that TrmD is essential for growth of the gram-positive organisms Streptococcus pneumoniae (36) and Bacillus subtilis (22).
It has been shown that the entire tRNA structure is required for maximal catalytic activity of E. coli TrmD (18). For example, a derivative of 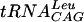 with the D and T loops deleted is a very poor substrate for this enzyme. Subsequent studies have shown that the sequence G36pG37 is the key recognition element in tRNA (19) and that G37 must be in the correct position in space relative to important interactions elsewhere in the molecule (19). Enzymatic and chemical probing experiments have shown that the enzyme makes contacts in key sites in the anticodon loop (13).
with the D and T loops deleted is a very poor substrate for this enzyme. Subsequent studies have shown that the sequence G36pG37 is the key recognition element in tRNA (19) and that G37 must be in the correct position in space relative to important interactions elsewhere in the molecule (19). Enzymatic and chemical probing experiments have shown that the enzyme makes contacts in key sites in the anticodon loop (13).
Although tRNA-modifying enzymes from E. coli have been well studied, little is known concerning modification enzymes from gram-positive bacteria. It is not clear, for instance, if gram-negative and gram-positive TrmD orthologs will display similar catalytic and physical properties.
In this study we show that S. pneumoniae TrmD carries out a function that is essential for growth of this key gram-positive pathogen. This discovery, together with the ubiquitous and highly conserved nature of the enzyme in eubacteria and the absence of a closely related eukaryotic homolog, indicates that TrmD can be a valuable novel antibacterial target. Consequently, S. pneumoniae TrmD has been isolated, and the enzyme has been biochemically characterized by a novel and efficient assay. The catalytic and physical properties of this enzyme have been determined and compared to those of other orthologs. A method for measuring the specificity of tRNA binding for TrmD has also been developed.
MATERIALS AND METHODS
Materials, strains, and growth conditions.
All chemicals were analytical grade and were used without further purification. Radiolabeled methyl donor (S-adenosyl-l-[methyl-3H]methionine [AdoMet]) and phosphodiesterase YSi-scintillation proximity assay (SPA) beads were from Amersham Pharmacia Biotech. Buffer reagents were from Sigma, and restriction enzymes were from Promega.
S. pneumoniae 0100993 (NCIMB 40794; GlaxoSmithKline strain collection), a virulent capsular serotype 3 strain, was grown at 37°C in Todd-Hewitt broth (Difco Laboratories) containing 0.5% yeast extract. S. pneumoniae R6 wild-type and mutant strains were maintained on Todd-Hewitt broth plus 0.5% yeast extract, AGCH medium (23), or blood agar plates. Media were supplemented with erythromycin (1 μg/ml), chloramphenicol (2.5 μg/ml), or fucose (0.01 to 1% [wt/vol]) where appropriate. E. coli strains were grown in Luria broth (LB) at 37°C.
TrmD alignments.
Translated open reading frames and complete DNA sequences were searched using the program BLASTP (2). Individual protein data sets were aligned using the program CLUSTALW v1.8 with default settings (37).
Regulation of trmD expression in S. pneumoniae.
Methodology is illustrated in Fig. 2. DNA fragments were amplified by PCR using the high-fidelity DNA polymerase enzyme Pwo and standard protocols (Roche Applied Science). Where appropriate, restriction sites were incorporated into primer ends in order to facilitate subsequent cloning. PCR products and fragments released by plasmid restriction digestion were gel purified by standard methods (Qiagen). S. pneumoniae R6 and intermediate strains were transformed according to the method described by Havarstein and colleagues (16).
FIG. 2.

Strategy for construction of S. pneumoniae TrmD regulatable strain.
The fucose kinase promoter (Pfcsk) (9), regions flanking the tex gene, and trmD together with its putative ribosome binding site were amplified from S. pneumoniae R6 genomic DNA. Primers used for amplification of trmD were 5′GTGGGGATCCTAGAAGGGTTAGACGATG3′ and 5′CATCGAATTCTCCTTTATTCTTTGTTTTC3′. The chloramphenicol resistance determinant (cat) with its promoter sequence was isolated from plasmid pC194 (20).
The region upstream of the tex gene (approximately 500 bp), cat in the reverse orientation, Pfcsk, trmD, and the region downstream of the tex gene (approximately 500 bp) were sequentially cloned into pBluescript II (Stratagene). DNA sequencing confirmed identity of inserts and fidelity of PCR. The tex integration cassette was released by digestion with NgoMIV/KpnI. S. pneumoniae R6 was transformed with this DNA fragment, and transformants were selected on chloramphenicol. Insertion of cat-Pfcsk-trmD within the tex gene by allelic replacement mutagenesis was confirmed by diagnostic PCR.
The native gene was then replaced by the erythromycin resistance marker ermAM (27) by a precise allelic replacement method that minimizes polarity effects (42). Transformants were recovered on erythromycin and fucose, and genomic organization was confirmed by diagnostic PCR.
S. pneumoniae growth studies.
Inoculum for growth studies was prepared by dilution (1/50) of exponential-phase cultures in AGCH medium plus fucose and static incubation at 37°C until an optical density at 650 nm (OD650) of 0.3 (1-cm path length) was reached. This growth cycle was repeated, and the final exponential-phase cultures were used as inoculum for growth studies. Growth studies were carried out in flat-bottomed 96-well plates (Nunc) with the SpectraMax spectrophotometer (Molecular Devices). Final culture volume was 250 μl in each well including a 1/50 (vol/vol) inoculum. Plates were incubated for 20 h at 37°C with absorbance readings (650 nm) taken each hour after agitation for 1 min.
Cloning of S. pneumoniae trmD.
Homologs of E. coli TrmD were identified in both the TIGR (htpp://www.tigr.org) and GlaxoSmithKline S. pneumoniae 0100993 genome database through BLAST analysis. PCR amplification and double-strand sequencing confirmed the S. pneumoniae sequence. To clone trmD, PCR forward and reverse primers were designed with an NdeI site (underlined) flanking the 5′ end of the gene (5′GGGAATTCCATATGAAGATTGATATTTTAACCCTA3′) and a BamHI site (underlined) at the 3′ end (5′CGCCGCGGATCCTTATTCTTTGTTTTCTTTGATTTC3′). DNA was amplified from S. pneumoniae 0100993 with high-fidelity Pfu DNA polymerase (Stratagene) as described by the manufacturer. The PCR product was digested with NdeI and BamHI and ligated to pET-15b vector (Novagen) cut with both restriction enzymes and treated with calf intestinal alkaline phosphatase (GIBCO-BRL). DNA fragments were purified after each manipulation by using the Qiaquick PCR purification kit (Qiagen). The ligation mixture was used to electroporate DH10B (GIBCO-BRL) electrocompetent cells according to the manufacturer's conditions. Cells were plated onto LB plates supplemented with ampicillin (100 μg/ml), IPTG (isopropyl-β-d-thiogalactopyranoside; 0.1 mM), and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; 2%) and incubated at 37°C overnight. Plasmids prepared from different bacterial colonies were checked for the correct insert by DNA restriction analysis and DNA sequencing. The correct plasmid, pET-TrmD, was introduced into BL21(DE3) (Novagen) cells for overexpression purposes.
Overexpression and purification of S. pneumoniae TrmD.
One liter of LB supplemented with ampicillin (50 μg/ml) was inoculated with 20 ml of an overnight culture of BL21(DE3)/pET-TrmD and grown at 37°C to an OD600 of 0.7 (1-cm path length) and then cooled down to 18°C and induced with 1.0 mM IPTG for 24 h at 18°C. Cells were pelleted and resuspended in 45 ml of 50 mM NaPO4 (pH 7.5)-10 mM imidazole-5 mM β-mercaptoethanol-50 mM NaCl-0.05 mg of lysozyme/ml. Cells were lysed using a French press at 20,000 lb/in2. After centrifugation (12,000 × g, 30 min), the lysate was loaded onto a 5-ml Qiagen Ni-nitrilotriacetic acid agarose affinity column. After a 100-ml wash with 50 mM phosphate buffer, pH 7.5, containing 10 mM imidazole and 500 mM NaCl, TrmD was eluted with 50 mM phosphate buffer, pH 7.5, containing 250 mM imidazole and 50 mM NaCl. Next, the enzyme was bound onto a 12-ml Bio-Rad (Uno) MonoQ fast protein liquid chromatography column and then eluted with an ascending linear salt gradient from 50 mM to 1.2 M NaCl in Tris-HCl, pH 8.0. Eluted enzyme was dialyzed against Tris-HCl (pH 7.5)-100 mM NaCl-10% (vol/vol) glycerol. Enzyme was stored at either −80 or −20°C in the presence of 50% glycerol and was stable to repeated thawing.
Protein native size determination.
A TSK high-pressure liquid chromatography gel filtration column (Tohaus) was calibrated using gel filtration molecular weight markers from Sigma (MW-GF-200). The enzyme was dialyzed against 50 mM Tris-HCl (pH 7.5)-100 mM KCl and run at 1 ml/min with the same buffer through the column. A Beckman Gold high-pressure liquid chromatography system was employed.
Preparation of tRNA transcripts.
Plasmid pUTL4 encoding wild-type E. coli 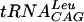 and pUTL66 encoding the
and pUTL66 encoding the 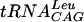 minihelix have been previously described (19). Plasmids encoding wild-type yeast tRNAAsp (29) as well as mutants G36 and A37 were a gift from Catherine Florentz. All these pTFMa constructs differ from the true wild type in that the first base pair, U1-A72, has been replaced by G1-C72 for improving in vitro transcription. In vitro transcription was done according to standard protocols (29) on BstNI-linearized plasmids. Transcripts were size purified on short denaturing polyacrylamide gels, electroeluted, precipitated, dissolved in water, and stored at −20°C. Quantitation was made by OD assuming that 1 A260 unit corresponds to a concentration of 40 μg/ml.
minihelix have been previously described (19). Plasmids encoding wild-type yeast tRNAAsp (29) as well as mutants G36 and A37 were a gift from Catherine Florentz. All these pTFMa constructs differ from the true wild type in that the first base pair, U1-A72, has been replaced by G1-C72 for improving in vitro transcription. In vitro transcription was done according to standard protocols (29) on BstNI-linearized plasmids. Transcripts were size purified on short denaturing polyacrylamide gels, electroeluted, precipitated, dissolved in water, and stored at −20°C. Quantitation was made by OD assuming that 1 A260 unit corresponds to a concentration of 40 μg/ml.
Fully modified native E. coli 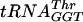 ,
, 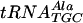 , and tRNAPhe were purchased from Plenum Scientific Research Inc.
, and tRNAPhe were purchased from Plenum Scientific Research Inc.
TrmD methylation assays.
Enzyme activity and kinetic properties of the purified protein were determined essentially as previously described (17) but using an SPA format. This is a homogenous radiometric assay technique applicable to a wide range of molecular interactions (8). Low-energy β particles, such as those emitted from 3H-labeled compounds, can stimulate scintillant-containing microspheres to emit light, which can be detected. Due to the short path length of these β emitters the microspheres can be stimulated only if the radiolabeled product is in direct contact with the surface of the bead, in this case by precipitation of tRNA with acid pH (26). The β emission from [3H]AdoMet present in free solution is absorbed by the solvent and does not stimulate the beads.
All the reactions were carried out in 96-well Optiplate plates (Packard Bioscience). The reaction mixture (50 μl) contained 0.1 M Tris-HCl (pH 8.5), 0.5 mM dithiothreitol (DTT), 0.1 mM EDTA, 6 mM MgCl2, 24 mM NH4Cl, 0.5 mg of bovine serum albumin (BSA)/ml, 1 μg of 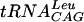 , 0.8 μCi of [3H]AdoMet, and 50 nM S. pneumoniae TrmD. The level of tRNA methylated was estimated by scintillation proximity; after the above mixture was incubated for 15 min at 25°C, 100 μl of 0.3 M sodium citrate, pH 2.0, containing 5 mg of YSi-SPA beads/ml was added to stop the reaction. Radioactivity was measured using a Packard TopCount NXT instrument (Packard Bioscience). The velocities are expressed as counts per minute. All the reagents were prepared using diethyl pyrocarbonate-treated water. All data fitting was carried out with nonlinear least-squares regression using the commercial software package Graphit 4.0 (Erithacus Software Limited). The assay mixture was later optimized to contain 0.1 M HEPES [pH 7.5], 2 mM DTT, 0.1 mM EDTA, 10 mM MgCl2, 1 μg of
, 0.8 μCi of [3H]AdoMet, and 50 nM S. pneumoniae TrmD. The level of tRNA methylated was estimated by scintillation proximity; after the above mixture was incubated for 15 min at 25°C, 100 μl of 0.3 M sodium citrate, pH 2.0, containing 5 mg of YSi-SPA beads/ml was added to stop the reaction. Radioactivity was measured using a Packard TopCount NXT instrument (Packard Bioscience). The velocities are expressed as counts per minute. All the reagents were prepared using diethyl pyrocarbonate-treated water. All data fitting was carried out with nonlinear least-squares regression using the commercial software package Graphit 4.0 (Erithacus Software Limited). The assay mixture was later optimized to contain 0.1 M HEPES [pH 7.5], 2 mM DTT, 0.1 mM EDTA, 10 mM MgCl2, 1 μg of 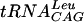 , 0.8 μCi of [3H]AdoMet, and 50 nM S. pneumoniae TrmD.
, 0.8 μCi of [3H]AdoMet, and 50 nM S. pneumoniae TrmD.
In experiments testing substrate specificity of the streptococcal enzyme by using selected tRNA substrates, reaction mixtures contained 0.2 mg of transcript/ml, 0.08 mg of S. pneumoniae TrmD enzyme/ml, and 50 μM AdoMet (isotopic dilution of unlabeled and tritiated AdoMet at 0.55 Ci/mmol). Reactions were started by addition of AdoMet, and reaction mixtures were incubated at 37°C. Samples were removed at times ranging from 4 to 8 min and spotted onto 3MM paper squares, and these were dropped into cold 5% trichloracetic acid. Papers were washed three times for 10 min each in cold trichloracetic acid, twice for 2 min each in cold ethanol, and dried. They were counted using an aqueous scintillation mixture.
TrmD binding assays. (i) RNA labeling and purification.
The 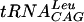 transcript's 5′ γ-phosphate was removed using a method previously described (10) with the exception of replacing calf intestinal phosphatase with bacterial alkaline phosphatase (Sigma). After phenol-chloroform treatment, ethanol precipitation, and centrifugation, the pellet was resuspended in 50 mM Tris-HCl, pH 7.5, to a concentration of 0.5 mg/ml. Water (0.5 μl) was added to 3.6 μl of tRNA, heated to 95°C for 2 min, and cooled for 5 min on ice to partially denature the tRNA. To this, 1.4 μl of 10× T4 polynucleotide kinase buffer (New England Biolabs), 7 μl of [γ-32P]ATP (0.55 μCi/μl) (Perkin-Elmer Life Sciences Inc.), and 1.5 μl of T4 polynucleotide kinase (10 U/μl) (New England Biolabs) were added for a total volume of 14 μl. The reaction was allowed to proceed at 37°C for 45 min, and then 14 μl of RNA loading buffer was added and the mixture was heated to 60°C for 5 min, loaded onto an 0.75-mm-thick RNA denaturing gel, and run for 2 h at 200 V. The labeled
transcript's 5′ γ-phosphate was removed using a method previously described (10) with the exception of replacing calf intestinal phosphatase with bacterial alkaline phosphatase (Sigma). After phenol-chloroform treatment, ethanol precipitation, and centrifugation, the pellet was resuspended in 50 mM Tris-HCl, pH 7.5, to a concentration of 0.5 mg/ml. Water (0.5 μl) was added to 3.6 μl of tRNA, heated to 95°C for 2 min, and cooled for 5 min on ice to partially denature the tRNA. To this, 1.4 μl of 10× T4 polynucleotide kinase buffer (New England Biolabs), 7 μl of [γ-32P]ATP (0.55 μCi/μl) (Perkin-Elmer Life Sciences Inc.), and 1.5 μl of T4 polynucleotide kinase (10 U/μl) (New England Biolabs) were added for a total volume of 14 μl. The reaction was allowed to proceed at 37°C for 45 min, and then 14 μl of RNA loading buffer was added and the mixture was heated to 60°C for 5 min, loaded onto an 0.75-mm-thick RNA denaturing gel, and run for 2 h at 200 V. The labeled 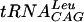 transcript was visualized by exposure and development of Kodak X-ray film according to the manufacturer's instructions. The band corresponding to labeled
transcript was visualized by exposure and development of Kodak X-ray film according to the manufacturer's instructions. The band corresponding to labeled 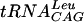 was excised and soaked overnight in Maxam-Gilbert buffer (0.5 M ammonium acetate, 10 mM magnesium acetate, 0.1 M EDTA, and 0.1% [wt/vol] sodium dodecyl sulfate) rotating in the cold room. Following ethanol precipitation, labeled RNA was washed with ice-cold 70% ethanol and resuspended in deionized water. Radiospecific activity was determined by spotting 1 μl on 4-mm2-sized filter paper, drying it, immersing it in 5 ml of scintillation fluid, and counting activity in a Packard Tri-Carb 1500 liquid scintillation counter.
was excised and soaked overnight in Maxam-Gilbert buffer (0.5 M ammonium acetate, 10 mM magnesium acetate, 0.1 M EDTA, and 0.1% [wt/vol] sodium dodecyl sulfate) rotating in the cold room. Following ethanol precipitation, labeled RNA was washed with ice-cold 70% ethanol and resuspended in deionized water. Radiospecific activity was determined by spotting 1 μl on 4-mm2-sized filter paper, drying it, immersing it in 5 ml of scintillation fluid, and counting activity in a Packard Tri-Carb 1500 liquid scintillation counter.
(ii) Gel retardation assay.
Approximately 5 fmol of 5′ γ-phosphate-labeled 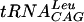 transcript (∼20,000 cpm, as determined above) was added to increasing amounts of S. pneumoniae TrmD in 50 mM Tris-HCl (pH 8.0)-100 mM KCl-5 mM MgCl2-5 mM β-mercaptoethanol-10% glycerol and incubated for 15 min at 37°C to determine a concentration of enzyme that would bind >97% of the tRNA. The Kd was then determined by taking this concentration of enzyme and radiolabeled tRNA and adding increasing quantities of nonlabeled tRNA under the same reaction conditions. Four types of tRNA were used: the
transcript (∼20,000 cpm, as determined above) was added to increasing amounts of S. pneumoniae TrmD in 50 mM Tris-HCl (pH 8.0)-100 mM KCl-5 mM MgCl2-5 mM β-mercaptoethanol-10% glycerol and incubated for 15 min at 37°C to determine a concentration of enzyme that would bind >97% of the tRNA. The Kd was then determined by taking this concentration of enzyme and radiolabeled tRNA and adding increasing quantities of nonlabeled tRNA under the same reaction conditions. Four types of tRNA were used: the 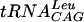 transcript and the fully modified
transcript and the fully modified 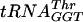 ,
,  , and tRNAPhe. Bound versus free tRNA concentrations were determined using a gel retardation assay as described previously (35). An 0.75-mm-thick native gel was made using 5% acrylamide-bis (39:1). The samples, with increasing concentrations of nonlabeled RNA, were run in 0.5× Tris-borate running buffer at 200 V in the cold room for 2 h. The gels were dried onto Whatman 3MM CHR paper with a Bio-Rad model 383 gel drier at 90°C for 2 h and exposed on a 20- by 25-cm Imaging Screen-K (Kodak) overnight. The screen was scanned by a Bio-Rad Personal Molecular Imager FX system, and bands were analyzed with Quantity One software.
, and tRNAPhe. Bound versus free tRNA concentrations were determined using a gel retardation assay as described previously (35). An 0.75-mm-thick native gel was made using 5% acrylamide-bis (39:1). The samples, with increasing concentrations of nonlabeled RNA, were run in 0.5× Tris-borate running buffer at 200 V in the cold room for 2 h. The gels were dried onto Whatman 3MM CHR paper with a Bio-Rad model 383 gel drier at 90°C for 2 h and exposed on a 20- by 25-cm Imaging Screen-K (Kodak) overnight. The screen was scanned by a Bio-Rad Personal Molecular Imager FX system, and bands were analyzed with Quantity One software.
RESULTS
Bioinformatic analysis of S. pneumoniae trmD.
In the majority of bacteria, particularly gram-negative organisms, trmD is the third gene in an operon that encodes ribosomal protein S16, RimM (a 16S rRNA-processing protein), and ribosomal protein L19 (Fig. 1A). Although TrmD is transcriptionally coupled, lower levels of TrmD than of the other products of the operon are produced, and it appears that this is due to poor translation of the trmD open reading frame (39, 40). Unlike the case with many other eubacteria, the S. pneumoniae trmD gene is not found in the same operon with ribosomal protein genes but instead appears to be translationally coupled to rimM in a putative five-gene operon in which the other three open reading frames have no known function.
FIG. 1.

The trmD operon structure and alignment of trmD products from different bacteria. (A) Chromosomal organization of the trmD-containing region in different bacteria. Patterns representing the different genes are illustrated in the figure. Gene names have been chosen from different genome annotations (accession numbers in parentheses): E. coli rpsP (P02372), rimM (P21504), trmD (P07020), and rplS (P02420); B. subtilis ylqC (O31738) and ylqD (D69880); S. pneumoniae spr0684 (AAK99488.1), spr0685 (AAK99489.1), and spr0688 (AAK99492.1); and T. maritima tm1570 (Q9X1Q6). Solid lines indicate intergenic regions. (B) Multiple sequence alignments of orthologous TrmD sequences from E. coli (NP_417098), H. influenzae (NP_438371), Pseudomonas aeruginosa (NP_252432), B. subtilis (NP_389485), S. pneumoniae (NP_358281), S. aureus (NP_371764), and T. maritima (NP_229369). According to the percent conservation, columns of residues are shaded as dark (100%), medium (80% or higher), or light (60% to 80%) or not at all.
Bioinformatic analysis shows that TrmD is a highly conserved protein, present in all bacteria for which genome sequence is available. In fact, there is a remarkable 50% identity between gram-positive and gram-negative TrmD proteins over their entire length, with this percentage being much higher in certain functional domains (Fig. 1B).
Essentiality testing of S. pneumoniae TrmD.
As part of a genome-scale analysis of essentiality in S. pneumoniae, Thanassi and colleagues (36) could not obtain mutants with a plasmid insertion in trmD, although the insertion event itself did not have a polarity effect on a downstream essential gene. Such a result suggests that TrmD is essential for growth in S. pneumoniae, but the conclusion is obviously based on the lack of recovery of mutants on a plate. In order to undoubtedly prove the essential nature of this protein, a strain in which the expression of trmD could be regulated was constructed. The trmD gene was placed under the control of an S. pneumoniae regulatable promoter that responds to the presence of fucose (9), following the specific strategy outlined in Fig. 2. In the first step, trmD was positioned downstream of the fucose kinase promoter, along with a chloramphenicol resistance determinant, within a nonessential single gene operon (unpublished results) on the S. pneumoniae chromosome. The second step involved precise allelic replacement of the native trmD gene with an erythromycin resistance cassette in such a way that expression of downstream genes was ensured and polarity effects were minimized (42). The resulting strain contains a single copy of trmD which is ectopically expressed in response to fucose. Western blot studies demonstrated that the level of TrmD in the regulatable strain is significantly lower than that in the parent strain, even under conditions of maximal induction (results not shown). Titration of protein levels with inducer fucose in the regulatable strain could not be investigated due to the lack of sensitivity of the S. pneumoniae TrmD antibodies.
In contrast to the control or the intermediate strain, the S. pneumoniae trmD regulatable strain was dependent on fucose for growth (Fig. 3), indicating that TrmD function is required for viability of this organism. This confirms the suggestion that TrmD is essential in S. pneumoniae under normal growth conditions (36).
FIG. 3.

Growth of different S. pneumoniae strains in the presence of various concentrations of fucose. Growth was monitored over 20 h. For each individual graph, the x axis represents time and the y axis represents OD650. The shape of the growth curves is expected and reflects a rapid cell lysis event at stationary phase in AGCH medium.
Cloning, expression, and purification of S. pneumoniae TrmD.
It was already known that the kinetic properties of native and His-tagged E. coli TrmD were essentially identical (34). Therefore, in order to biochemically characterize S. pneumoniae TrmD, the gene was amplified by PCR and cloned into the expression vector pET-15b so that the overexpressed protein contained six extra histidines at the N-terminal end. TrmD production was induced with average yields of 40 mg of >95% pure protein from 1 liter of culture. The identity of the protein was confirmed by N-terminal amino acid sequencing.
TrmD is dimeric in bacteria.
Initial reports on E. coli TrmD indicated that the protein was monomeric with a subunit molecular mass of approximately 32,000 Da (17). Our gel size-exclusion chromatography experiments gave a molecular mass for the streptococcal enzyme of 69,000 Da, a value consistent with a dimeric configuration. In order to investigate whether this is a special characteristic of the streptococcal enzyme, we carried out similar analyses with purified native protein orthologs from Staphylococcus aureus, Thermotoga maritima, and E. coli. Molecular masses of 67,000, 53,000, and 63,000 Da, respectively, were obtained. The molecular mass of S. pneumoniae TrmD was also determined to be approximately 65,000 Da by dynamic light scattering. Therefore, it appears that TrmD proteins are dimeric rather than monomeric as previously reported.
Optimization of the TrmD methylation assay.
With use of the purified enzyme, the effect of incubation time and enzyme concentration was determined using the SPA conditions described in Materials and Methods. The data indicated that the reaction was linear for an incubation time of up to 60 min and for TrmD concentrations up to 100 nM (data not shown). The remaining experiments were performed with 50 nM enzyme and an incubation time of 15 min. Each factor was tested independently, and the assay conditions were modified for the subsequent factors in the order in which they are described in the text. The effect of the nature and concentration of the reducing agent, the divalent cation, and the monovalent cation was first evaluated. Accordingly, the analysis of a number of reducing agents (DTT, β-mercaptoethanol, and [tris(2-carboxyethyl)phosphine HCl] [TCEP]) indicated that the activity was optimal with 2 mM DTT (Fig. 4A). The effect of divalent cations (Mg2+, Mn2+, Ca2+, and Co2+) was then evaluated for concentrations ranging from 0 to 100 mM. Results showed that the activity was optimal in the presence of 10 mM Mg2+ (Fig. 4B) with a ranking order of activity of Mg2+ > Mn2+ > Ca2+. No activity was observed in the absence of divalent cation or in the presence of Co2+. Similarly, the effect of monovalent cations was evaluated for concentrations ranging from 0 to 5 mM. S. pneumoniae TrmD activity increased only 10% in the presence of a monovalent cation, indicating the nonessential nature of this factor (data not shown) while BSA and casein had a significant adverse effect on the enzyme activity (data not shown). The remaining experiments were carried out in the presence of 2 mM DTT and 10 mM MgCl2 and in the absence of monovalent cation and BSA.
FIG. 4.

Effects of reducing agents, cations, and pH on S. pneumoniae TrmD activity. (A) Effect of the reducing agent. The assay conditions were 100 mM Tris-HCl, pH 8.5, containing 10 mM MgCl2, 0.1 mM EDTA, 1 μg of 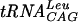 , 0.8 μCi of [3H]AdoMet, and various concentrations of reducing agent: DTT (•), β-mercaptoethanol (○), and TCEP (▪). (B) Effect of the cation. The assay conditions were 100 mM Tris-HCl, pH 8.5, containing 2 mM DTT, 0.1 mM EDTA, 1 μg of
, 0.8 μCi of [3H]AdoMet, and various concentrations of reducing agent: DTT (•), β-mercaptoethanol (○), and TCEP (▪). (B) Effect of the cation. The assay conditions were 100 mM Tris-HCl, pH 8.5, containing 2 mM DTT, 0.1 mM EDTA, 1 μg of 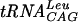 , 0.8 μCi of [3H]AdoMet, and various concentrations of divalent cations: Mg2+ (•), Mn2+ (○), Ca2+ (▪), and Co2+ (□). (C) Effect of pH. The assay conditions were 100 mM buffer containing 2 mM DTT, 10 mM MgCl2, 0.1 mM EDTA, 1 μg of
, 0.8 μCi of [3H]AdoMet, and various concentrations of divalent cations: Mg2+ (•), Mn2+ (○), Ca2+ (▪), and Co2+ (□). (C) Effect of pH. The assay conditions were 100 mM buffer containing 2 mM DTT, 10 mM MgCl2, 0.1 mM EDTA, 1 μg of 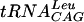 , and 0.8 μCi of [3H]AdoMet. The buffers were MES for pHs between 5.6 and 6.7, HEPES for pHs between 6.8 and 8.2, Tris-HCl for pHs between 7.5 and 9, and CHES for pHs between 9.2 and 9.8. Velocity is expressed in counts per minute. In all cases, each point represents the average of duplicate experiments.
, and 0.8 μCi of [3H]AdoMet. The buffers were MES for pHs between 5.6 and 6.7, HEPES for pHs between 6.8 and 8.2, Tris-HCl for pHs between 7.5 and 9, and CHES for pHs between 9.2 and 9.8. Velocity is expressed in counts per minute. In all cases, each point represents the average of duplicate experiments.
The effect of pH on enzyme activity was examined using the buffers MES (morpholineethanesulfonic acid), HEPES, Tris, and CHES [2-(cyclohexylamino)ethanesulfonic acid] at a concentration of 100 mM. The ionic strength was held constant at 50 mM by adding various concentrations of NaCl (see http://www.bi.umist.ac.uk/users/mjfrbn/Buffers/Makebuf.asp). S. pneumoniae TrmD was found to have an optimum activity between pH 7 and 8 (Fig. 4C). The effect of NaCl was reevaluated in 100 mM HEPES, pH 7.5, but no significant change was observed at NaCl concentrations between 0 and 5 mM (data not shown). Therefore, the remaining experiments were carried out in 100 mM HEPES at pH 7.5 and in the absence of NaCl.
Kinetic properties of S. pneumoniae TrmD.
Enzyme assays were performed in SPA format in a four-by-six array with various concentrations of 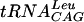 (0.38 to 3.05 μM), cold AdoMet (0.2 to 22.4 μM), and 0.8 μCi of [3H]AdoMet. Absolute initial velocities were calculated from raw initial velocities taking into account the isotopic dilution of the mixture by the following equation:
(0.38 to 3.05 μM), cold AdoMet (0.2 to 22.4 μM), and 0.8 μCi of [3H]AdoMet. Absolute initial velocities were calculated from raw initial velocities taking into account the isotopic dilution of the mixture by the following equation:
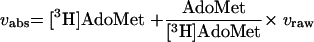 |
where [3H]AdoMet and AdoMet represent the concentrations of tritiated and cold AdoMet, respectively. Absolute initial velocities were then fitted to the steady-state equation:
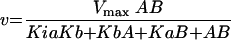 |
where A and B represent the concentrations of 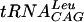 and cold AdoMet, respectively. The double-reciprocal plots of the kinetic data are linear and intersect, which is indicative of a ternary mechanism (Fig. 5). The velocities obtained in counts per minute were then normalized to molarity by running the reaction to completion (2 h) with 0.1 μM
and cold AdoMet, respectively. The double-reciprocal plots of the kinetic data are linear and intersect, which is indicative of a ternary mechanism (Fig. 5). The velocities obtained in counts per minute were then normalized to molarity by running the reaction to completion (2 h) with 0.1 μM  in the presence of [3H]AdoMet. The kinetic parameters are summarized in Table 1. Although the Km for AdoMet was observed to be at least 10 times lower than that of the E. coli enzyme, the Km for
in the presence of [3H]AdoMet. The kinetic parameters are summarized in Table 1. Although the Km for AdoMet was observed to be at least 10 times lower than that of the E. coli enzyme, the Km for 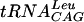 was comparable for the two enzymes (19, 34).
was comparable for the two enzymes (19, 34).
FIG. 5.

Double-reciprocal plots of substrate concentration against the initial velocities. The assay conditions were 100 mM HEPES, pH 7.5, containing 2 mM DTT, 10 mM MgCl2, and 0.1 mM EDTA. The concentration of 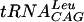 was varied from 0.38 to 3.05 μM, while the concentration of cold AdoMet was held constant at 0.2 (○), 0.47 (•), 1 (□), 2.6 (▪), 7.6 (▵), or 22.4 (▴) μM. Velocity is expressed in counts per minute. Each point represents the average of duplicate experiments.
was varied from 0.38 to 3.05 μM, while the concentration of cold AdoMet was held constant at 0.2 (○), 0.47 (•), 1 (□), 2.6 (▪), 7.6 (▵), or 22.4 (▴) μM. Velocity is expressed in counts per minute. Each point represents the average of duplicate experiments.
TABLE 1.
Kinetic parameters of S. pneumoniae TrmD
| Substrate | Km (μM) | kcat (min−1) | kcat/Km (mM−1 · min−1) |
|---|---|---|---|
| AdoMet | 0.7 ± 0.3 | 15 ± 2 | 2.1 × 104 |
| tRNACAGLeu | 4.3 ± 1.7 | 15 ± 2 | 3.3 × 103 |
tRNA substrate specificity.
The activity of S. pneumoniae TrmD has been tested with a number of tRNA substrates in order to determine the structural elements of tRNA required for recognition. The streptococcal TrmD requires the complete tRNA structure since it cannot modify the 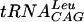 minihelix lacking the D, T, and extra loops of complete tRNA. In addition, and consistent with a requirement for G at positions 36 and 37 in the tRNA, the enzyme methylates yeast tRNAAsp (G36G37) with kinetic values that are indistinguishable from those obtained with substrate
minihelix lacking the D, T, and extra loops of complete tRNA. In addition, and consistent with a requirement for G at positions 36 and 37 in the tRNA, the enzyme methylates yeast tRNAAsp (G36G37) with kinetic values that are indistinguishable from those obtained with substrate 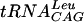 but does not methylate either tRNAAsp (C36G37) or tRNAAsp (C36A37).
but does not methylate either tRNAAsp (C36G37) or tRNAAsp (C36A37).
tRNA binding.
Direct tRNA-TrmD interactions were examined using a gel mobility shift assay. End-labeled tRNA species were mixed with purified streptococcal enzyme, and complexes were separated from free tRNA by native gel electrophoresis as shown in Fig. 6. S. pneumoniae TrmD clearly binds 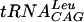 , one of its substrates, under these conditions. Since the number of enzyme molecules competent to bind tRNA cannot be determined in this assay, cold tRNA was used as a competitor for labeled tRNA binding (Fig. 6B). The slope of the Scatchard-type plot indicates a Kd of approximately 22 nM for the
, one of its substrates, under these conditions. Since the number of enzyme molecules competent to bind tRNA cannot be determined in this assay, cold tRNA was used as a competitor for labeled tRNA binding (Fig. 6B). The slope of the Scatchard-type plot indicates a Kd of approximately 22 nM for the 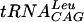 -TrmD interaction. To determine if S. pneumoniae TrmD can discriminate between substrate and nonsubstrate tRNA species at the level of binding, three other nonsubstrate tRNA molecules were tested by this method. All tRNAs show similar Kd values (Fig. 6), indicating that the streptococcal enzyme does not discriminate at the level of tRNA binding under these conditions. In addition, tRNA binding is unaffected by the presence of inhibitory concentrations (up to 100 μM [data not shown]) of sinefungin, a potent AdoMet analog (41).
-TrmD interaction. To determine if S. pneumoniae TrmD can discriminate between substrate and nonsubstrate tRNA species at the level of binding, three other nonsubstrate tRNA molecules were tested by this method. All tRNAs show similar Kd values (Fig. 6), indicating that the streptococcal enzyme does not discriminate at the level of tRNA binding under these conditions. In addition, tRNA binding is unaffected by the presence of inhibitory concentrations (up to 100 μM [data not shown]) of sinefungin, a potent AdoMet analog (41).
FIG. 6.

Scatchard analysis of TrmD-tRNA binding. (A) Binding of labeled synthetic 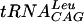 to increasing concentrations of TrmD protein: 0 (lane 1), 1 (lane 2), 2 (lane 3), 3 (lane 4), and 4 (lane 5) μM. (B) Competition of labeled
to increasing concentrations of TrmD protein: 0 (lane 1), 1 (lane 2), 2 (lane 3), 3 (lane 4), and 4 (lane 5) μM. (B) Competition of labeled 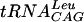 binding with increasing concentrations of unlabeled
binding with increasing concentrations of unlabeled 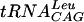 : 0 (lane 1), 5 (lane 2), 10 (lane 3), 20 (lane 4), and 40 (lane 5) nM. Similar concentrations were needed to determine Kd values of fully modified tRNAPhe,
: 0 (lane 1), 5 (lane 2), 10 (lane 3), 20 (lane 4), and 40 (lane 5) nM. Similar concentrations were needed to determine Kd values of fully modified tRNAPhe, 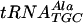 , and
, and 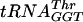 .
.
DISCUSSION
Base modification of tRNA in bacteria is a complex process involving about 45 enzymes (3). The importance of tRNA modification in bacteria is indicated by the large amount of genetic material which is dedicated to this process, approximately 1% of the genome in E. coli (3), and reports that show its involvement in normal cell growth (12, 28, 31). The subject of this work, tRNA (guanosine-1)-methyltransferase, has been shown in previous studies to be required for optimal growth of the gram-negative organisms E. coli and S. enterica serovar Typhimurium (5, 6, 30), and more recent global gene disruption studies suggest that the gene encoding this enzyme is essential for growth of the gram-positive organisms S. pneumoniae and B. subtilis (22, 36). It has also been reported that the TRM5 enzyme, which carries out a similar function in Saccharomyces cerevisiae, is essential for normal growth in that eukaryotic system (5). Using technology allowing selective regulation of gene expression in the important respiratory tract pathogen S. pneumoniae, we have clearly shown for the first time that the trmD gene is absolutely essential for bacterial growth, at least in this gram-positive organism. The essential nature of TrmD is not altogether surprising given the role of this enzyme in prevention of frameshifting (7, 15, 32, 33). Production of aberrant proteins as a result of frameshifting would be expected to have a lethal effect on a bacterial cell.
Certain domains of TrmD are remarkably well conserved among a diverse group of bacteria, and the streptococcal enzyme displays no unusual deviation from this pattern (Fig. 1B). Particularly striking is the very strong conservation of primary sequence between residues 130 and 147 (E. coli TrmD numbering), which we now know constitutes part of the catalytic center of the enzyme (1, 11). Other regions are less conserved, like the carboxy-terminal domain, residues 174 to terminus, which is involved in tRNA binding (unpublished observations).
Characterization of the physicochemical and kinetic properties of S. pneumoniae TrmD, and comparison with those of other orthologs, has allowed us to define the general properties of the bacterial TrmD enzyme.
The molecular masses of the S. pneumoniae and E. coli enzyme subunits are very similar (approximately 30 kDa), and it can be seen from the highly conserved primary sequences that this is true for many other bacterial orthologs. Here we report that a dimer configuration is a common feature of diverse bacterial enzymes including TrmD from gram-positive and gram-negative eubacteria as well as from the extreme thermophile T. maritima. Further evidence for the dimeric structure of TrmD comes from the solution of the E. coli and Haemophilus influenzae TrmD crystal structures, which clearly shows two subunits forming the catalytic site of the enzyme (1, 11).
The kinetic properties of the S. pneumoniae and E. coli enzymes are also similar. Both display sequential mechanisms and similar Km and Vmax values for tRNA (34). Surprisingly, the Km values for AdoMet are substantially different, 0.7 versus 15 μM (19), respectively. This may reflect different in vivo levels of this important metabolite in gram-positive bacteria. Interestingly, the kcat/Km values are similar, indicating that the two enzymes have comparable efficiency.
We have shown that the activity of the streptococcal enzyme is dependent upon the presence of Mg2+ cations. So far only one methyltransferase of any kind has been shown to contain Mg2+ as an integral part of the active site. This enzyme, catechol-O-methyltransferase, contains a single Mg2+ ion which is critically involved in substrate binding and deprotonation (38). It is tempting to predict a similar role for Mg2+ in the activity of TrmD. Perhaps these cations are required for binding crucial elements of tRNA, the target nucleotide or its neighbor, or in deprotonation of N1G37.
As is the case with the E. coli enzyme, S. pneumoniae TrmD requires the complete tRNA molecule for methylation and modifies G at position 37 in a tRNA molecule containing G36G37. However, the streptococcal enzyme displays essentially no activity with a simple anticodon loop and stem structure whereas there is low but measurable activity with the E. coli enzyme (19). This may reflect subtle differences in how productive complexes are achieved.
In contrast to E. coli TrmD, the streptococcal enzyme forms very stable complexes with tRNA, which can be detected in gel mobility shift assays, a property that allows the study of the specificity of the TrmD-tRNA interactions. Our experiments show clearly that the initial binding of tRNA to the enzyme is relatively nonspecific with regard to the identity of the tRNA isoacceptor, with TrmD binding all tRNA species with more or less equal affinity in the presence or absence of AdoMet. In E. coli it has been estimated that about 10% of bulk tRNA species are substrates for the TrmD protein (14), given that only 7 out of the 49 isoacceptors contain the G36G37 identity element. The relatively high Km value associated with both the S. pneumoniae and E. coli enzymes is consistent with the idea that in vivo bacterial TrmD makes many nonproductive binding efforts, effectively browsing the available tRNA molecules until postbinding “recognition” leads to subsequent catalytic steps. Therefore, only if the identity element G36pG37 is present in the tRNA does AdoMet-dependent methylation proceed. It may be that AdoMet binds TrmD independently, so that the TrmD-AdoMet complex would be always prepared to methylate a specific substrate tRNA which enters a precatalytic complex. Nonsubstrate tRNA species would simply dissociate without being methylated.
The fact that the E. coli enzyme does not make complexes with tRNA stable enough to survive gel electrophoresis may reflect differences in “on” or “off” rates for tRNA between the two proteins. We have been able to determine, using plasmon surface resonance, that the Kd of E. coli TrmD for tRNA is similar to that of the streptococcal enzyme (approximately 30 nM) (unpublished results), so very subtle differences in the structure of these two proteins might be affecting their affinity for tRNA. Further studies are necessary in order to determine the structural basis for the kinetic differences in tRNA-protein interaction.
Finally, there is general agreement that existing and emerging resistance to standard antibacterial agents necessitates the development of novel classes of antibiotics (21, 25). In this regard, TrmD is a suitable novel antibacterial target for several reasons. The essential nature of TrmD in the key respiratory tract pathogen S. pneumoniae, together with the similarity of the S. pneumoniae enzyme to other eubacterial orthologs in terms of primary sequence conservation, quaternary structure, kinetic characteristics, and substrate specificity, is a strong indication that the function of TrmD is critical for growth and/or survival of a broad spectrum of bacterial pathogens. The eukaryotic enzymes which carry out an equivalent function, although related in evolutionary terms, have diverged considerably and are significantly different from bacterial TrmD (5). Any specific inhibitor of the bacterial TrmD activity would therefore be expected to have antibacterial activity, low cytotoxicity, no preexisting bacterial resistance, and no particular propensity for high rates of resistance development in the clinic. Opportunities for high-throughput screen development are realistic given the type of assays described in this study. TrmD, therefore, represents a novel and attractive target for antibacterial drug discovery.
Acknowledgments
We are grateful to Joshua West and Howard Kallender for help with purification of S. pneumoniae TrmD, to Pan Chan for help with strain construction, and to GlaxoSmithKline's DNA and protein sequencing groups for confirmation of identity of constructs and proteins.
This work was supported in part by a grant from the National Science Foundation to W.M.H.
REFERENCES
- 1.Ahn, H. J., H. W. Kim, H. J. Yoon, B. I. Lee, S. W. Suh, and J. K. Yang. 2003. Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. EMBO J. 22:2593-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Björk, G. R. 1996. Stable RNA modification, p. 861-886. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, Jr., B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
- 4.Björk, G. R., J. M. Durand, T. G. Hagervall, R. Leipuviene, H. K. Lundgren, K. Nilsson, P. Chen, Q. Qian, and J. Urbonavicius. 1999. Transfer RNA modification: influence on translational frameshifting and metabolism. FEBS Lett. 452:47-51. [DOI] [PubMed] [Google Scholar]
- 5.Björk, G. R., K. Jacobsson, K. Nilsson, M. J. Johansson, A. S. Byström, and O. P. Persson. 2001. A primordial tRNA modification required for the evolution of life? EMBO J. 20:231-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Björk, G. R., and K. Nilsson. 2003. 1-Methylguanosine-deficient tRNA of Salmonella enterica serovar Typhimurium affects thiamine metabolism. J. Bacteriol. 185:750-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Björk, G. R., P. M. Wikström, and A. S. Byström. 1989. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244:986-989. [DOI] [PubMed] [Google Scholar]
- 8.Bosworth, N., and P. Towers. 1989. Scintillation proximity assay. Nature 341:167-168. [DOI] [PubMed] [Google Scholar]
- 9.Chan, P. F., K. M. O'Dwyer, L. M. Palmer, J. D. Ambrad, K. A. Ingraham, C. So, M. A. Lonetto, S. Biswas, M. Rosenberg, D. J. Holmes, and M. Zalacain. 2003. Characterization of a novel fucose-regulated promoter (PfcsK) suitable for gene essentiality and antibacterial mode-of-action studies in Streptococcus pneumoniae. J. Bacteriol. 185:2051-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke, P. 1999. Labeling and purification of RNA synthesized by in vitro transcription. Methods Mol. Biol. 118:1-10. [DOI] [PubMed] [Google Scholar]
- 11.Elkins, P. A., J. M. Watts, M. Zalacain, A. van Thiel, P. R. Vitazka, M. Redlak, C. Andraos-Selim, F. Rastinejad, and W. M. Holmes. 2003. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J. Mol. Biol. 333:931-949. [DOI] [PubMed] [Google Scholar]
- 12.Elseviers, D., L. A. Petrullo, and P. J. Gallagher. 1984. Novel E. coli mutants deficient in biosynthesis of 5-methylaminomethyl-2-thiouridine. Nucleic Acids Res. 12:3521-3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabryszuk, J., and W. M. Holmes. 1997. tRNA recognition for modification: solution probing of tRNA complexed with Escherichia coli tRNA (guanosine-1) methyltransferase. RNA 3:1327-1336. [PMC free article] [PubMed] [Google Scholar]
- 14.Grosjean, H., and B. Benne. 1998. Modification and editing of RNA. ASM Press, Washington, D.C.
- 15.Hagervall, T. G., T. M. Tuohy, J. F. Atkins, and G. R. Björk. 1993. Deficiency of 1-methylguanosine in tRNA from Salmonella typhimurium induces frameshifting by quadruplet translocation. J. Mol. Biol. 232:756-765. [DOI] [PubMed] [Google Scholar]
- 16.Havarstein, L. S., G. Coomaraswamy, and D. A. Morrison. 1995. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 92:11140-11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hjalmarsson, K. J., A. S. Byström, and G. R. Björk. 1983. Purification and characterization of transfer RNA (guanine-1)methyltransferase from Escherichia coli. J. Biol. Chem. 258:1343-1351. [PubMed] [Google Scholar]
- 18.Holmes, W. M., C. Andraos-Selim, and M. Redlak. 1995. tRNA-m1G methyltransferase interactions: touching bases with structure. Biochimie 77:62-65. [DOI] [PubMed] [Google Scholar]
- 19.Holmes, W. M., C. Andraos-Selim, I. Roberts, and S. Z. Wahab. 1992. Structural requirements for tRNA methylation. Action of Escherichia coli tRNA(guanosine-1)methyltransferase on tRNA(1Leu) structural variants. J. Biol. Chem. 267:13440-13445. [PubMed] [Google Scholar]
- 20.Horinouchi, S., and B. Weisblum. 1982. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 150:815-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isaacson, R. 2002. Genomics and the prospects for the discovery of new targets for antibacterial and antifungal agents. Curr. Pharm. Des. 8:1091-1098. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi, K., S. D. Ehrlich, A. Albertini, G. Amati, K. K. Andersen, M. Arnaud, K. Asai, S. Ashikaga, S. Aymerich, P. Bessieres, F. Boland, S. C. Brignell, S. Bron, K. Bunai, J. Chapuis, L. C. Christiansen, A. Danchin, M. Debarbouille, E. Dervyn, E. Deuerling, K. Devine, S. K. Devine, O. Dreesen, J. Errington, S. Fillinger, S. J. Foster, Y. Fujita, A. Galizzi, R. Gardan, C. Eschevins, T. Fukushima, K. Haga, C. R. Harwood, M. Hecker, D. Hosoya, M. F. Hullo, H. Kakeshita, D. Karamata, Y. Kasahara, F. Kawamura, K. Koga, P. Koski, R. Kuwana, D. Imamura, M. Ishimaru, S. Ishikawa, I. Ishio, D. Le Coq, A. Masson, C. Mauel, R. Meima, R. P. Mellado, A. Moir, S. Moriya, E. Nagakawa, H. Nanamiya, S. Nakai, P. Nygaard, M. Ogura, T. Ohanan, M. O'Reilly, M. O'Rourke, Z. Pragai, H. M. Pooley, G. Rapoport, J. P. Rawlins, L. A. Rivas, C. Rivolta, A. Sadaie, Y. Sadaie, M. Sarvas, T. Sato, H. H. Saxild, E. Scanlan, W. Schumann, J. F. Seegers, J. Sekiguchi, A. Sekowska, S. J. Seror, M. Simon, P. Stragier, R. Studer, H. Takamatsu, T. Tanaka, M. Takeuchi, H. B. Thomaides, V. Vagner, J. M. Van Dijl, K. Watabe, A. Wipat, H. Yamamoto, M. Yamamoto, Y. Yamamoto, K. Yamane, K. Yata, K. Yoshida, H. Yoshikawa, U. Zuber, and N. Ogasawara. 2003. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. USA 100:4678-4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacks, S. A., and R. D. Hotchkiss. 1960. A study of the genetic material determining an enzyme activity in pneumococcus. Biochim. Biophys. Acta 39:508-517. [DOI] [PubMed] [Google Scholar]
- 24.Li, J. N., and G. R. Björk. 1995. 1-Methylguanosine deficiency of tRNA influences cognate codon interaction and metabolism in Salmonella typhimurium. J. Bacteriol. 177:6593-6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livermore, D. 2003. Bacterial resistance: origins, epidemiology, and impact. Clin. Infect. Dis. 36(Suppl. 1):S11-S23. [DOI] [PubMed] [Google Scholar]
- 26.Macarron, R., L. Mensah, C. Cid, C. Carranza, N. Benson, A. J. Pope, and E. Diez. 2000. A homogeneous method to measure aminoacyl-tRNA synthetase aminoacylation activity using scintillation proximity assay technology. Anal. Biochem. 284:183-190. [DOI] [PubMed] [Google Scholar]
- 27.Martin, B., G. Alloing, V. Mejean, and J. P. Claverys. 1987. Constitutive expression of erythromycin resistance mediated by the ermAM determinant of plasmid pAM beta 1 results from deletion of 5′ leader peptide sequences. Plasmid 18:250-253. [DOI] [PubMed] [Google Scholar]
- 28.Palmer, D. T., P. H. Blum, and S. W. Artz. 1983. Effects of the hisT mutation of Salmonella typhimurium on translation elongation rate. J. Bacteriol. 153:357-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perret, V., A. Garcia, J. Puglisi, H. Grosjean, J. P. Ebel, C. Florentz, and R. Giegé. 1990. Conformation in solution of yeast tRNA(Asp) transcripts deprived of modified nucleotides. Biochimie 72:735-743. [DOI] [PubMed] [Google Scholar]
- 30.Persson, B. C., G. O. Bylund, D. E. Berg, and P. M. Wikström. 1995. Functional analysis of the ffh-trmD region of the Escherichia coli chromosome by using reverse genetics. J. Bacteriol. 177:5554-5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Persson, B. C., C. Gustafsson, D. E. Berg, and G. R. Björk. 1992. The gene for a tRNA modifying enzyme, m5U54-methyltransferase, is essential for viability in Escherichia coli. Proc. Natl. Acad. Sci. USA 89:3995-3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qian, Q., and G. R. Björk. 1997. Structural alterations far from the anticodon of the tRNAProGGG of Salmonella typhimurium induce +1 frameshifting at the peptidyl-site. J. Mol. Biol. 273:978-992. [DOI] [PubMed] [Google Scholar]
- 33.Qian, Q., and G. R. Björk. 1997. Structural requirements for the formation of 1-methylguanosine in vivo in tRNA(Pro)GGG of Salmonella typhimurium. J. Mol. Biol. 266:283-296. [DOI] [PubMed] [Google Scholar]
- 34.Redlak, M., C. Andraos-Selim, R. Giegé, C. Florentz, and W. M. Holmes. 1997. Interaction of tRNA with tRNA (guanosine-1)methyltransferase: binding specificity determinants involve the dinucleotide G36pG37 and tertiary structure. Biochemistry 36:8699-8709. [DOI] [PubMed] [Google Scholar]
- 35.Setzer, D. R. 1999. Measuring equilibrium and kinetic constants using gel retardation assays. Methods Mol. Biol. 118:115-128. [DOI] [PubMed] [Google Scholar]
- 36.Thanassi, J. A., S. L. Hartman-Neumann, T. J. Dougherty, B. A. Dougherty, and M. J. Pucci. 2002. Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res. 30:3152-3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vidgren, J. 1998. X-ray crystallography of catechol O-methyltransferase: perspectives for target-based drug development. Adv. Pharmacol. 42:328-331. [DOI] [PubMed] [Google Scholar]
- 39.Wikström, P. M., and G. R. Björk. 1989. A regulatory element within a gene of a ribosomal protein operon of Escherichia coli negatively controls expression by decreasing the translational efficiency. Mol. Gen. Genet. 219:381-389. [DOI] [PubMed] [Google Scholar]
- 40.Wikström, P. M., L. K. Lind, D. E. Berg, and G. R. Björk. 1992. Importance of mRNA folding and start codon accessibility in the expression of genes in a ribosomal protein operon of Escherichia coli. J. Mol. Biol. 224:949-966. [DOI] [PubMed] [Google Scholar]
- 41.Yebra, M. J., H. Sanchez, C. G. Martin, C. Hardisson, and C. Barbes. 1991. The effect of sinefungin and synthetic analogues on RNA and DNA methyltransferases from Streptomyces. J. Antibiot. 44:1141-1147. [DOI] [PubMed] [Google Scholar]
- 42.Zalacain, M., S. Biswas, K. A. Ingraham, J. Ambrad, A. Bryant, A. F. Chalker, S. Iordanescu, J. Fan, F. Fan, R. D. Lunsford, K. O'Dwyer, L. M. Palmer, C. So, D. Sylvester, C. Volker, P. Warren, D. McDevitt, J. R. Brown, D. J. Holmes, and M. K. R. Burnham. 2004. A global approach to identify novel broad spectrum antibacterial targets among proteins of unknown function. J. Mol. Microbiol. Biotechnol. 6:109-126. [DOI] [PubMed]