

Abstract
Alterations in DNA methylation have been proposed to create a field cancerization state in the colon, where molecular alterations that predispose cells to transformation occur in histologically normal tissue. However, our understanding of the role of DNA methylation in field cancerization is limited by an incomplete characterization of the methylation state of the normal colon. In order to determine the colon’s normal methylation state, we extracted DNA from normal colon biopsies from the rectum, sigmoid, transverse, and ascending colon and assessed the methylation status of the DNA by pyrosequencing candidate loci as well as with HumanMethylation450 arrays. We found that methylation levels of repetitive elements LINE-1 and SAT-α showed minimal variability throughout the colon in contrast to other loci. Promoter methylation of EVL was highest in the rectum and progressively lower in the proximal segments, whereas ESR1 methylation was higher in older individuals. Genome-wide methylation analysis of normal DNA revealed 8388, 82, and 93 differentially methylated loci that distinguished right from left colon, males from females, and older vs. younger individuals, respectively. Although variability in methylation between biopsies and among different colon segments was minimal for repetitive elements, analyses of specific cancer-related genes as well as a genome-wide methylation analysis demonstrated differential methylation based on colon location, individual age, and gender. These studies advance our knowledge regarding the variation of DNA methylation in the normal colon, a prerequisite for future studies aimed at understanding methylation differences indicative of a colon field effect.
Keywords: DNA methylation, methylation microarray, normal colon methylation, colorectal cancer, field effect
Introduction
Modifications in DNA methylation are of particular interest in cancer formation because they may arise as part of a field cancerization process, or “field effect,” which predisposes tissue to neoplastic transformation.1-3 Field carcinogenesis, which has been proposed to occur in various organs, including the colon, lung, pancreas, oral cavity, esophagus, and prostate, was originally recognized by an increased risk of cancer formation in normal tissue adjacent to a cancer.4 In the current era, it is recognized by the detection of cancer-related molecular alterations in the histologically normal mucosa of an organ.5 With respect to the colon, this suggests that normal-appearing mucosa within the colon could be assessed for molecular alterations as a way to gauge the risk of harboring concurrent colon adenomas or cancer or developing metachronous adenomas or cancer.6 In fact, previous studies evaluating the morphologically-normal colon mucosa (MNCM) have demonstrated an association between DNA methylation of certain cancer-related genes and neoplastic lesions located elsewhere in the colon.2,3,7-11 A direct correlation between the aberrant methylation of APC, DKK1, CDKN2A/p16, and SFRP4 in the apparently normal colon mucosa and the presence of adjacent colon cancer has been reported.12 Another group showed that a panel of methylated genes isolated from normal rectal biopsies of 113 subjects was able to discriminate between those with and without an adenoma present at the time of biopsy.13 The results of these and other studies demonstrate the potential for aberrant DNA methylation in the normal colon to serve as a marker of field cancerization in the colon, but the results remain to be validated in independent populations in more thoroughly characterized tissue sample sets.
A fundamental issue related to the validation and confirmation of the studies previously described is an accurate assessment of genome-wide DNA methylation in the normal colon mucosa of individuals without colon neoplasia. Indeed, it is already known that there are numerous functional and molecular differences between the various segments of the colon. The proximal, or right colon (cecum, ascending colon, and proximal two thirds of the transverse colon) is derived from the embryonic midgut, whereas the distal, or left colon (distal one third of the transverse colon, descending colon, sigmoid colon, and rectum) is derived from the embryonic hindgut.14,15 Consistent with the differences in the embryological sites of origin of the different segments of the colon, the DNA methylation status of certain genes in the normal colon has been shown to vary by anatomical location, but whether or to what extent this variability in DNA methylation is related to differences in age, embryology, or some environmental exposure is not known.16 Other loci have been shown to be methylated in histologically normal colon in an age-dependent fashion, and half of the genes involved in this age-related methylation are the same as those involved in colon carcinogenesis.1,17 There is also evidence that the degree of methylation of certain genes in the normal colon varies between males and females.18,19 A better understanding of variations in genome-wide methylation in the normal colon related to age, gender, and anatomical location is needed in order to identify methylated genes that are specifically associated with the development of neoplasia.
The relative lack of characterization of the DNA methylation state of the normal colon led us to conduct epigenetic studies of the normal colon using a genome-wide approach combined with analysis of specific cancer-related genes and repetitive elements that are indicative of global methylation. We focused on repetitive elements that have been shown to be susceptible to aberrant methylation in colon cancer (LINE-1; SAT-α) and genes previously shown to be indicative of a colon field effect (EVL; MGMT), a gene known to be aberrantly methylated in colon cancer but not adjacent normal tissue (CDKN2A) and a gene shown to be associated with age-related methylation (ESR1). On a genome-wide level, we hypothesized that in the normal colon, methylation is likely highly variable, with differences occurring as a function of age, anatomical location, and gender. We assessed the variability in DNA methylation in immediately adjacent regions of the colon from single individuals as well as between individuals. We correlated these findings with the anatomic location of the colon and with the age and gender of the individual.
Results
Methylation levels in loci containing LINE-1 and SAT-α repetitive elements from adjacent biopsy sites and from different colon segments show minimal variability
Global DNA hypomethylation and regional hypermethylation are commonly observed in a variety of cancers, including colorectal cancer.20,21 The regions that are hypomethylated often involve repetitive sequence elements, such as LINE-1, ALU, and SAT-α. A number of studies have demonstrated a loss of methylation in these regions in normal tissue that is at risk of developing cancer.22-24 This led us to initially assess the methylation status of loci containing LINE-1 and SAT- α repetitive elements in normal colon mucosa. DNA methylation in these loci was compared between two normal colon biopsy specimens obtained from within two centimeters of each other in order to determine the degree of variability between adjacent regions in the colon. In general, for both LINE-1 and SAT-α loci, there was very little difference between the adjacent sites regardless of the anatomic region of the colon (P > 0.18 for both loci in all colon sites using t test and equivalent test). Additionally, LINE-1 and SAT-α methylation levels demonstrated minimal variation between the eight subjects examined (P values for LINE-1 were > 0.05 except for the sigmoid colon, where P = 0.028; P values for SAT-α were not significant; P values were generated using ANOVA) (Fig. 1). When the methylation level of the LINE-1 and SAT-α loci in DNA isolated from different segments of the colon was directly compared, we found no significant differences between methylation levels in the rectum, sigmoid, transverse, and ascending colon for LINE-1 (P = not significant using ANOVA test to compare means). Although methylation differences between colonic segments were statistically different for SAT-α, the mean difference was less than one percent between segments and it is unlikely that this is biologically meaningful (P ≤ 0.001 using ANOVA test to compare means) (Fig. 2). There were no differences in the methylation levels of these repetitive element-containing loci with respect to age or gender.

Figure 1. DNA methylation in repetitive elements in the various anatomical colon segments from eight patients. Each panel shows the degree of methylation in two adjacent biopsies for each patient in LINE-1 (A–D) and SAT-α (E–H). There was minimal variability in methylation between adjacent biopsies from each patient, minimal patient-to-patient variability, and low variability between colon segments.
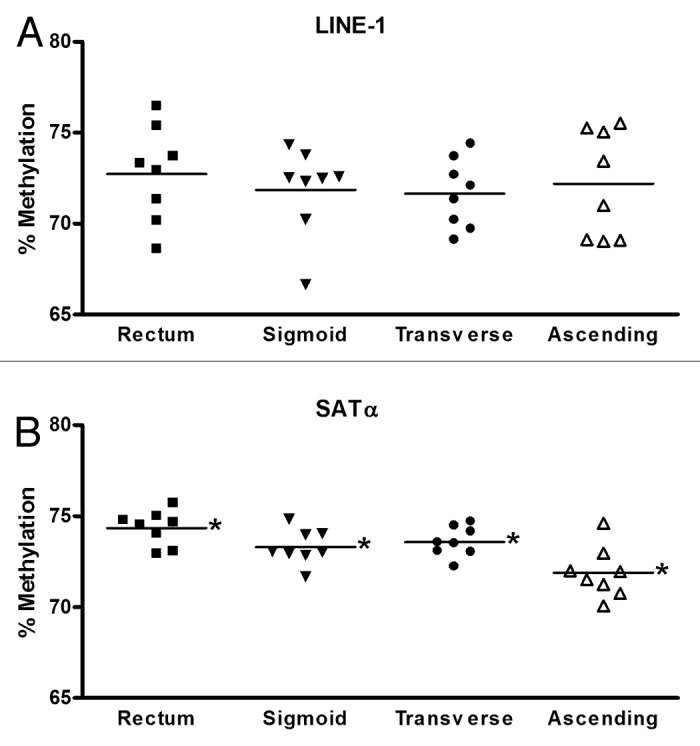
Figure 2. A single colon biopsy from each colon site from each individual was used to compare methylation of LINE-1 (A) and SAT-α (B). Each data point represents the percent methylation for one individual. The average methylation of LINE-1 did not differ between rectum, sigmoid, transverse, and ascending colon. The differences in methylation of SAT-α between colon sites did meet criteria for statistical significance but overall the variability between colon segments was minimal. *P < 0.001 for mean methylation level between each site.
Promoter methylation of ESR1 and EVL differs between individuals and between regions of the normal colon
In light of prior studies that provided evidence that there are different classes of loci that are either susceptible to cancer-associated aberrant hypermethylation (“C” genes) or to age-associated hypermethylation (“A” genes),25 we next chose to study representative loci from these two classes of genes. EVL and MGMT are susceptible to cancer-specific methylation and also have been found to be frequently methylated in the normal colon mucosa of colon cancer patients suggesting they may be epigenetic cancerization markers. In contrast, CDKN2A has been found to be methylated in colon neoplasms but not in normal colon mucosa from colon cancer patients. ESR1 has been shown to be more highly methylated in the normal colon of older compared with younger individuals and thus appears to undergo age-associated methylation.1,17 Based on these prior findings, we chose to assess the DNA methylation status of the promoter regions of the genes EVL, MGMT, CDKN2A, and ESR1 in the normal colon using pyrosequencing (Fig. 3). EVL methylation varied according to the location of the colon biopsy. The highest level of methylation was detected in the rectum, and there was a gradual, step-wise decrease in EVL methylation moving proximally to the sigmoid, transverse, and eventually ascending colon (P < 0.001 for mean methylation level for each site using ANOVA). We also examined the promoter methylation status of EVL in the ascending colon (A) and rectal (R) DNA samples that were assayed with the HumanMethylation450 (HM450) BeadChip. There were three probes targeting the EVL promoter on the HM450 arrays, and although two of the three probes showed increased methylation in the A compared with R samples, these results were not statistically significant. We suspect this discrepancy between the pyrosequencing results and HumanMethylation450 array results is due to the fact that these assays assess different regions of the CpG island in the EVL promoter.

Figure 3. Methylation status of cancer-related genes EVL, CDKN2A, MGMT, and ESR1 is shown by colon location for each of eight individuals. Each data point represents the methylation level for a single individual, and the average methylation level for each colon site is shown. EVL showed a statistically significant decrease in average methylation between rectum, sigmoid, transverse, and ascending colon samples (P < 0.01 using ANOVA test to compare means of four groups).
The other three genes demonstrated no significant differences in their levels of methylation between the various colon sites. As would be expected from previously published studies, in the normal colon the MGMT and CDKN2A promoters demonstrated minimal methylation in all regions among all individuals. Although promoter methylation of the ESR1 gene did not differ by colon site, there appeared to be a greater range of methylation values in the normal colon among the eight individuals examined compared with the other three genes, with normal methylation ranging from approximately 10–32% in the sigmoid colon, for example (Fig. 3D). This finding, which appears to correlate with the age of an individual, is described in more detail below.
Methylation of ESR1 in the normal colon increases with increasing age
Promoter methylation of ESR1, which has previously been noted to be increased in the colon mucosa of older individuals compared with younger individuals,1,26 correlated with age in the eight normal individuals in this study. A weak positive correlation between age and promoter methylation of ESR1 was seen in all four segments of the colon; this correlation was strongest in the rectum (r2 = 0.45) (Fig. 4). There was no correlation between age and methylation level in EVL, MGMT, and CDKN2A.

Figure 4. Relationship of ESR1 methylation to the age of each individual. In DNA isolated from all colon sites (A–D), there was a weak positive correlation (r2) between ESR1 methylation and age.
Specific CpG dinucleotides in the EVL gene promoter demonstrate variable levels of methylation
We used pyrosequencing to assess the methylation status of a number of CpGs located in the EVL promoter, which allowed us to precisely quantitate the proportion of methylated CpGs in this promoter (Fig. S1). An examination of the degree of methylation of eight sequential CpG dinucleotides revealed a gradual decrease in mean methylation from CpG 1 to CpG 8. This pattern was seen in DNA isolated from the rectum, sigmoid, transverse, and ascending colon. The difference in mean methylation was greatest in the rectum, ranging from approximately 24% in CpG 1 to approximately 5% in CpG 8, and the difference in methylation between the first four CpGs was statistically different than the last four CpGs (P < 1 × 10−5) With respect to MGMT, CDKN2A, and ESR1, there were not substantial differences in methylation between sequential CpG dinucleotides in their respective targeted promoter region.
Genome-wide methylation analysis of ascending colon and rectal DNA reveals multiple differentially methylated genes
Our studies of the methylation status of specific loci in the normal colon revealed substantial variability in their methylation status, which led us to conduct an assessment of loci throughout the genome of normal colon epithelial cells. Assessment of genome-wide methylation of DNA isolated from the colon of the eight individuals described above plus an additional four normal individuals (i.e., with no neoplasia present anywhere in the colon) was conducted using the HumanMethylation450 BeadChip arrays (Illumina). Mean methylation levels of the matched ascending colon DNA and rectal DNA was compared at more than 430 000 CpG sites located throughout the genome. We found 8388 differentially methylated loci (DML) when comparing ascending colon (A) and rectal (R) samples (FDR q < 1 × 10−4). The top 50 DML are illustrated in Figure 5A and the top 20 annotated loci based on lowest FDR q value are listed in Table 1. Of the 8388 DML, 5456 (64.8%) showed higher methylation in rectal DNA vs. ascending colon DNA, whereas the entire top 20 annotated DML demonstrated higher methylation in the ascending colon samples compared with the rectal samples.
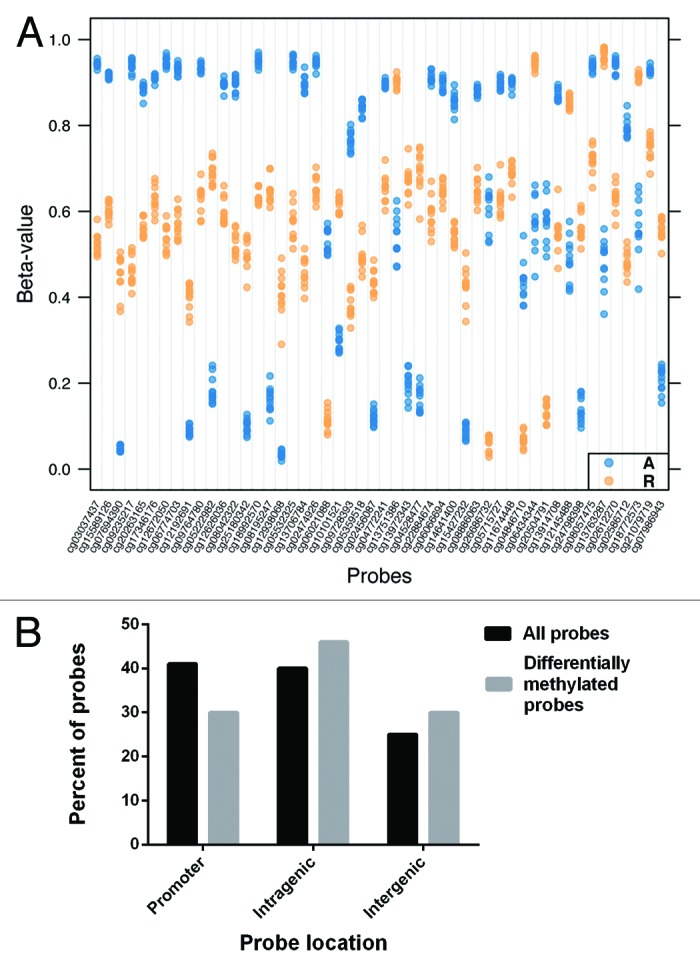
Figure 5. (A) Strip plot showing the top 50 (smallest FDR q value) differentially methylated loci between the ascending colon and rectal DNA samples. Each ascending colon sample is depicted as a blue dot, and each rectal sample as an orange dot. The magnitude of methylation difference (difference in β value) can be seen for each group of samples at each locus. The corresponding genes and probe locations of the top 20 loci are shown in Table 1. (B) The overall percentage of HM450 array probe locations (black bars) compared with the probe locations of the 8388 differentially methylated loci (gray bars) is shown. A greater proportion of the differentially methylated loci (DML) were located in intragenic and intergenic regions as opposed to promoter regions. The total percent of probe types is greater than 100% because a few probes were classified as belonging in more than one type of CpG class. (P < 0.001 using hypergeometric testing of expected vs. actual number of DML for promoter, intragenic, and intergenic probes.)
Table 1. Top 20 annotated differentially methylated loci comparing ascending colon and rectum.
| Probe ID | Gene | Probe location/CGI? | Avg β value: Ascending (A) | Avg β value: Rectum (R) | Avg M value: Ascending (A) | Avg M value: Rectum (R) |
|---|---|---|---|---|---|---|
| cg03037437 | B3GNT7 | Body/Y | 0.94 | 0.53 | 4.06 | 0.15 |
| cg15589126 | B3GNT7 | Body/N | 0.92 | 0.60 | 3.45 | 0.61 |
| cg09235217 | B3GNT7 | Body/Y | 0.94 | 0.46 | 4.02 | −0.22 |
| cg20263165 | FLJ12825 | Body/N | 0.88 | 0.57 | 2.95 | 0.38 |
| cg17346176 | MCM9 | Body/N | 0.91 | 0.63 | 3.35 | 0.76 |
| cg12672050 | ATG4B | Body/Y | 0.95 | 0.54 | 4.24 | 0.23 |
| cg06774703 | KRT36 | TSS1500/N | 0.93 | 0.57 | 3.78 | 0.42 |
| cg09764780 | EIF4G3 | 5′UTR/N | 0.93 | 0.63 | 3.83 | 0.77 |
| cg08042322 | NT5DC3 | Body/N | 0.90 | 0.52 | 3.14 | 0.14 |
| cg05532325 | B3GNT7 | Body/Y | 0.95 | 0.57 | 4.26 | 0.43 |
| cg13706784 | LMF1 | Body/Y | 0.90 | 0.46 | 3.14 | −0.23 |
| cg02474926 | PARP11 | Body/N | 0.95 | 0.65 | 4.18 | 0.92 |
| cg06021088 | BIN1 | Body/N | 0.53 | 0.11 | 0.18 | −2.98 |
| cg09728393 | KIF13A | Body/N | 0.77 | 0.38 | 1.71 | −0.71 |
| cg05359518 | TCF12 | Body/N | 0.84 | 0.50 | 2.42 | −0.02 |
| cg04772241 | ZCCHC14 | Body/Y | 0.90 | 0.65 | 3.13. | 0.92 |
| cg22884674 | PPP2R5E | Body/N | 0.91 | 0.59 | 3.37 | 0.54 |
| cg06066694 | DPEP1 | 5′UTR/Y | 0.90 | 0.65 | 3.13 | 0.87 |
| cg08886063 | VPS53 | Body/Y | 0.88 | 0.65 | 2.87 | 0.89 |
| cg26686732 | FOXA2 | Body/Y | 0.61 | 0.06 | 0.62 | −4.00 |
The top 20 annotated differentially methylated loci (DML) comparing ascending colon and rectal samples are shown. For each DML, the associated gene, location, whether or not the probe is located in a CpG island (CGI: Y, yes; N, no), and β and M values are listed. TSS1500 = CpG located 1500 base pairs downstream from transcription start site; 5′UTR = CpG located in 5′ untranslated region.
Many of the DML between the ascending colon and rectal samples were present in genes with more than one DML, either in the same or different genomic locations within a given gene. Thus, the 8388 DML corresponded to 2953 unique genes, and 1053 (35.3%) of these genes contained more than one DML. In most of the genes with multiple DML, these loci were located in similar genomic elements (both in the gene promoter, for example), but in 454 genes (15.4%), the DML were located in distinct genomic regions (promoter and gene body, for example). In general, when a gene had multiple DML in the same region, these DML demonstrated similar levels of methylation, which is consistent with previous publications that have demonstrated strong positive correlations between methylation levels of CpGs in proximity to each other.27,28 DML that were located in different genomic regions could either show similar or dissimilar degrees of methylation. An example of a gene (B3GNT7) with multiple DML in different genomic locations in rectum vs. ascending colon samples is shown in Figure S2.
A greater proportion of loci that demonstrated differential methylation between ascending colon (A) and rectal (R) samples were located in intergenic and non-promoter regions (as opposed to promoter regions) than was expected based on the total number of each probe types on the array. For example, while 41% of the annotated probes on the HM450 BeadChip target CpGs in promoter regions, only 30% of the differentially methylated probes were located in gene promoters. Similarly, 25% of the annotated HM450 probes target CpGs in intergenic regions, but 30% of the differentially methylated probes were located in intergenic regions (P < 0.001 using hypergeometric testing of expected vs. actual number of DML for promoter, intragenic, and intergenic probes) (Fig. 5B).
There are differences in DNA methylation of multiple loci when comparing females to males and comparing individuals of various ages
When genome-wide methylation patterns of females (n = 6) and males (n = 6) were compared, we found a total of 82 DML after excluding loci located on the X and Y chromosomes (FDR q < 1 × 10−3). The annotated 20 DML with the lowest FDR q values between females and are shown in Table 2. Among these 20 DML, 12/20 (60.0%) were more highly methylated in females, and among the 82 DML (FDR q < 1 × 10−3), 57/82 (69.5%) were more highly methylated in females. There were no differences in methylation between males and females when we considered the right and left colon separately.
Table 2. Top 20 annotated differentially methylated loci comparing females and males.
| Probe ID | Gene | Probe location/CGI? | Avg β value: Females | Avg β value: Males | Avg M value: Females | Avg M value: Males |
|---|---|---|---|---|---|---|
| cg25294185 | RNASEH2C | Body/Y | 0.03 | 0.14 | −4.93 | −2.57 |
| cg17307919 | CSMD1 | Body/Y | 0.96 | 0.86 | 4.71 | 2.68 |
| cg03911306 | DAZL | TSS1500/N | 0.93 | 0.75 | 3.83 | 1.58 |
| cg16218221 | CCNYL1 | 1st Exon/Y | 0.23 | 0.08 | –1.77 | –3.50 |
| cg22149355 | MYO5A | 5′UTR/Y | 0.34 | 0.13 | –0.97 | –2.80 |
| cg00804338 | TFDP1 | 5′UTR/Y | 0.18 | 0.05 | –2.21 | –4.13 |
| cg02675179 | ACSM1 | Body/N | 0.87 | 0.77 | 2.68 | 1.76 |
| cg12019814 | RAD21 | Body/N | 0.62 | 0.75 | 0.71 | 1.60 |
| cg24016844 | LRIF1 | TSS200/Y | 0.05 | 0.09 | –4.33 | –3.36 |
| cg08656326 | TLE1 | TSS1500/Y | 0.31 | 0.13 | –1.13 | –2.80 |
| cg22345911 | CSNK1D | 5′UTR/Y | 0.06 | 0.02 | –4.10 | –5.43 |
| cg02556954 | ETF1 | Body/N | 0.58 | 0.69 | 0.44 | 1.16 |
| cg16383222 | CYHR1 | Body/Y | 0.12 | 0.07 | –2.93 | –3.66 |
| cg20926353 | TLE1 | 5′UTR/Y | 0.50 | 0.23 | –0.01 | –1.83 |
| cg06644124 | ZNF281 | 1st Exon/Y | 0.33 | 0.23 | –1.03 | –1.73 |
| cg13150977 | UBE2Q2P1 | TSS200/Y | 0.11 | 0.26 | –3.06 | –1.47 |
| cg17226602 | KIF4B | 1st Exon/Y | 0.49 | 0.36 | –0.07 | –0.80 |
| cg02325951 | FOXN3 | Body/N | 0.61 | 0.72 | 0.66 | 1.34 |
| cg02798874 | TRPV1 | 3′UTR/N | 0.76 | 0.85 | 1.69 | 2.56 |
| cg17403731 | HCN2 | Body/Y | 0.73 | 0.91 | 1.44 | 3.36 |
The top 20 annotated differentially methylated loci comparing females and males, along with the gene name, location, whether or not the probe is located in a CpG island (CGI; Y = yes, n = no), and β/M values are shown. TSS200/1500 = CpG located 200 base pairs upstream or 1500 base pairs downstream from transcription start site; 5′UTR;3′UTR = CpG located in 5′ or 3′ untranslated region, respectively.
Next, we compared DNA methylation in individuals aged less than 60 (n = 6) to those aged greater than 60 (n = 6). There were a total of 93 DML between these two groups (FDR q < 0.10). A strip plot showing the top 12 DML (FDR q < 0.01) is shown in Figure 6 and the top 12 annotated loci (FDR q < 0.05) are listed in Table 3. In general, the level of methylation tended to increase in conjunction with increasing age of the individuals examined, with 71 out of the 93 DML (76.3%) and 9 of the top 12 DML (75.0%) demonstrating higher methylation in the older age group. When we investigated the relationship of DNA methylation to age as a continuous variable in the 12 study subjects, we found a direct correlation between increasing methylation and age in the top 12 DML (Fig. 6B).

Figure 6. (A) Strip plot depicting the top 12 DML (FDR q value < 0.01) when comparing individuals aged >60 to those aged <60. Each sample is shown as a blue dot (age >60) or red dot (age <60). In 11 of 12 top DML, methylation was higher in the older age group. The corresponding genes and probe locations of the loci are listed in Table 3. (B) Regression curves for the same top 12 DML illustrating the linear relationship between methylation and age. For the same 11 out of 12 loci, the level of methylation increased along with increasing age; methylation decreased with age at one locus.
Table 3. Top 12 annotated differentially methylated loci comparing individuals age >60 vs. <60.
| Probe ID | Gene | Probe location/CGI? | Avg β value: Age >60 | Avg β value: Age <60 | Avg M value: Age >60 | Avg M value: Age <60 |
|---|---|---|---|---|---|---|
| cg19731612 | NSD1 | TSS1500/Y | 0.84 | 0.75 | 2.41 | 1.57 |
| cg21823080 | CNTNAP2 | Body/N | 0.72 | 0.47 | 1.37 | –0.17 |
| cg09381737 | C9orf167 | Body/Y | 0.80 | 0.72 | 1.99 | 1.34 |
| cg10635895 | CACNA1C | Body/Y | 0.84 | 0.74 | 2.42 | 1.52 |
| cg23390118 | CACNA1C | Body/Y | 0.79 | 0.69 | 1.92 | 1.16 |
| cg07063463 | MATN4 | Body/Y | 0.71 | 0.57 | 1.30 | 0.44 |
| cg14581491 | ATF6B | Body/Y | 0.01 | 0.01 | –6.25 | –6.88 |
| cg07223935 | RPH3AL | Body/Y | 0.84 | 0.91 | 2.38 | 3.40 |
| cg21156776 | HCG26 | Body/N | 0.68 | 0.80 | 1.12 | 2.00 |
| cg20459037 | WDR16 | 3′UTR/N | 0.33 | 0.90 | –2.26 | 4.12 |
| cg21169611 | SMC2 | TSS1500/Y | 0.02 | 0.01 | –5.34 | –6.15 |
| cg07175007 | UHMK1 | TSS1500/N | 0.89 | 0.83 | 3.10 | 2.29 |
The top 12 annotated differentially methylated loci (DML) comparing individuals age >60 vs. <60 are shown. Along with the DML, the associated gene name, location, whether or not the probe is located in a CpG island (CGI: Y, yes; N, no), and β/M values are listed. TSS1500 = CpG located 1500 base pairs downstream from transcription start site; 3′UTR = CpG located in 3′untranslated region.
Gene set enrichment analysis (GSEA) demonstrated over- and under-represented processes and pathways
The differentially methylated CpG sites between rectum and ascending colon samples were next used to identify biological processes that were either over-represented or under-represented in these samples. We utilized both the Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for the GSEA. Using a cut-off P value < 0.01, there were 281 over-represented GO terms in the rectum compared with ascending colon samples, many of which were associated with morphogenesis or cell differentiation, including gland morphogenesis, regulation of epithelial cell differentiation, and morphogenesis of an epithelial fold. There were nine under-represented GO terms in the rectum vs. ascending colon samples, including RNA processing and translational termination. When we examined the KEGG pathways using the differentially methylated CpGs (P value < 0.01), we found 15 over-represented pathways (including adherens junctions and Wnt signaling pathway) and four under-represented pathways (including olfactory transduction and oxidative phosphorylation) in rectum compared with ascending colon samples.
Discussion
In the present study, we describe patterns in DNA methylation in biopsy specimens obtained from the normal colon. In the first series of studies we focused on the methylation status of the repetitive genomic elements LINE-1 and SAT-α, and within the promoter regions of the cancer-related and age-related genes EVL, MGMT, CDKN2A, and ESR1. In colorectal cancer, it is known that genome-wide hypomethylation, which is particularly evident in repetitive DNA sequences, is associated with aneuploidy.29,30 EVL and MGMT have previously been shown to be aberrantly methylated in the normal colon mucosa of individuals with sporadic colon cancer, and prior studies of these loci have provided evidence for an epigenetic ‘field effect’ involving tumor suppressor genes in the colon.2,31 Aberrant methylation of CDKN2A, meanwhile, is frequently seen in neoplastic lesions of the colon but not in uninvolved normal colon mucosa.32 The estrogen receptor gene, ESR1, has been shown to exhibit increasing promoter methylation that is associated with colon neoplasia as well as with increasing age.1,32,33
We confirmed that LINE-1 and SAT-α show a relatively high level of DNA methylation as expected in normal colon mucosa, ranging from approximately 70–80% in the loci we examined. The degree of methylation did not differ substantially between various colon segments or between the eight normal study subjects. Additionally, the variability of methylation between biopsies taken at the same site from the same individual was minimal, providing further characterization of the methylation state in the normal colon.
With respect to the four cancer- or age-related genes we examined, we found very low methylation levels in the promoters of the tumor-suppressor genes CDKN2A and MGMT with almost no variation in methylation between individuals or by anatomical location. Of these four genes, EVL was the only one that demonstrated significant methylation differences by colon site, with average promoter methylation decreasing from the rectum, where it was approximately 15%, to the sigmoid colon, transverse colon and, finally ascending colon, where it was approximately 5%. This finding, along with our genome-wide methylation array results, which demonstrated multiple epigenetic differences between DNA isolated from the right and left colon, highlight the importance of the biopsy location when interpreting the results of DNA methylation studies of colonic tissue. ESR1 demonstrated somewhat higher promoter methylation (average approximately 15–20%) that varied minimally by colon site but did show greater inter-individual variability than EVL, CDKN2A, and MGMT. It is likely that this inter-individual variability is related to subject age as we noted a direct correlation between ESR1 promoter methylation and age, consistent with prior reports.1,32,33 These results have obvious implications regarding the development of biomarker assays based on methylated DNA.
In addition to studies of specific CpGs, we assessed the genome-wide methylation status of the colon epithelium using HumanMethylation450 BeadChips. We compared methylation patterns between matched normal ascending and rectal DNA samples from 12 individuals without colorectal cancer or adenomas. The results of these methylation arrays, which allowed us to simultaneously compare the methylation status of more than 430 000 CpG dinucleotides distributed throughout the genome, highlight some of the differences in methylation in the normal human colon between individuals and between regions of the colon. We found more than 8000 DML when comparing the average methylation in ascending colon and rectal biopsies. Interestingly, more of the DML than would be predicted based on the overall frequency of HM450 probe locations were found in intragenic (within a gene body) and intergenic (between gene) locations as opposed to promoter regions. One explanation for this finding is that the right and left colon arise from different embryological sources (midgut vs. hindgut) and it has been shown previously that alterations in methylation of the CpG islands located remotely from promoters and transcription start sites is frequently associated with particular tissue types and cell lineages.34-36
We anticipate that the results of these studies will help inform the development of epigenetic biomarkers from the normal colon that are reflective of a “field cancerization process.” An understanding of epigenetic variability in the normal colon, where no neoplasia exists, is a prerequisite to determining whether particular alterations in DNA methylation are genuine markers of a field effect and not merely reflective of a predictable degree of variability related to colon site, patient age, environmental factors, and so forth. In addition to these demographic and biological variables, there are numerous additional factors that were not evaluated here that might affect methylation in the normal colon, such as tobacco, alcohol, certain medications such as oral contraceptives, and dietary factors. These factors will need to be considered in future studies concerned with alterations of DNA methylation in the normal colon that might be related to the presence or risk of developing colorectal cancer.
Material and Methods
Sample acquisition and preparation
Colon biopsy specimens were obtained from the rectum, sigmoid colon, transverse colon, and ascending colon from healthy subjects who underwent screening colonoscopy at the University of Pittsburgh Medical Center. Colon biopsies were snap frozen in liquid nitrogen and transferred to a −80 °C freezer for long-term storage. Two biopsies were taken with standard biopsy forceps from each of the four colon sites totaling eight biopsies per subject. The two biopsies from a particular segment were taken immediately adjacent to each other (no more than 2 cm apart). Ascending colon biopsies were taken no more than 20 cm distal to the cecum/ileocecal valve; the triangular-shaped mucosal folds were used to identify biopsy sites for the transverse colon; the sigmoid colon biopsies were taken from an area between 20 and 40 cm from the anal verge, and the rectal biopsies taken from an area 5–10 cm from the anal verge. Normal specimens from a total of eight subjects with no neoplasia present were included in pyrosequencing-based methylation studies of specific genes (EVL, MGMT, CDKN2A, ESR1) or loci with repetitive elements (LINE-1, SAT-α). DNA from these same eight subjects plus an additional four normal subjects (also without colon neoplasia) was assessed using the HumanMethylation450 arrays. DNA was extracted using the Qiagen Core Kit A and quantified using the Quant-iT PicoGreen DS DNA assay kit following the manufacturers’ instructions (Qiagen #1042601 and Invitrogen #P7589). Five hundred nanograms of DNA from each sample was bisulfite converted using the EZ DNA Methylation Kit (ZymoResearch #D5002) and eluted in either an 8 µL volume for Illumina methylation arrays or a 40 µL volume for pyrosequencing. Bisulfite converted DNA was submitted to the Genomics Core at the Fred Hutchinson Cancer Research Center (FHCRC) for processing, application, and scanning on the HumanMethylation450 (HM450) BeadChip following the manufacturer’s instructions (http://www.Illumina.com).
Pyrosequencing
Assays to assess the methylation status of CpG dinucleotides in the loci of interest were designed using the PyroMark Assay Design 2.0 software (Qiagen, Inc.). For LINE-1 and SAT-α, primers were designed to target consensus sequences based on previously published reports.37 For EVL, MGMT, CDKN2A, and ESR1, primers were designed to assess sequential CpG dinucleotides located in a CpG islands near the transcription start site of each respective gene. Following PCR amplification using the Qiagen PyroMark PCR kit (Qiagen #978703), the final biotin-labeled PCR product was purified, sequenced, and analyzed using the PyroMark Q24 MDx machine. The accuracy of the assays was confirmed using a series of known standard DNA samples before assessing the test samples. Methylated, unmethylated, and 50% methylated control DNA was included with each reaction. Pyrosequencing primer sequences and annealing temperatures for each gene assay are shown in Table S1.
Genome-wide methylation arrays
HumanMethylation450 (HM450) BeadChips (Illumina #WG-314-1003) were used to compare patterns of DNA methylation in normal colon epithelial samples from 12 normal study subjects. For each subject, DNA from both ascending colon and rectum was included on the array. Data from the BeadChips was exported into the GenomeStudio Methylation Module software (Illumina), which contains information on probe location, CpG island location, and methylation values. Methylation values from the HM450 arrays are reported as “β values” (β = M/M+U+100), where 0.0 is equivalent to 0% methylation and 1.0 is equivalent to 100% methylation at a given CpG dinucleotide. We also converted β values to “M values” (M = log2[M/U]), which are logarithmic scores similar to those used in gene expression microarrays. M values were used for statistical analyses as they have been shown to be more reliable than β values for many statistical calculations and more accurate when evaluating methylation data at the lowest and highest extremes.38 Probes with detection P values > 0.05 (poor starting DNA quality) were excluded from downstream analyses. Bioconductor minfi package software was used for background level correction, color adjustment, and Subset-quantile Within-Array Normalization (SWAN).39 Non-specific filtering was next used to remove probes that were located on or within 10 base pairs from single-nucleotide polymorphisms (SNPs) and probes that targeted non-CpG sites. Finally, probes targeting CpG sites on the X and Y chromosomes were removed in order to reduce confounding due to gender bias.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge the outstanding service of the Genomics Core Shared Resource (FHCRC) and the study participants at the UPMC who kindly provided tissues for these studies.
Glossary
Abbreviations:
- CRC
colorectal cancer
- MNCM
morphologically normal colon mucosa
- LINE-1
long interspersed nucleotide element
- HM450
HumanMethylation450 array
- DML
differentially methylated loci
References
- 1.Ahuja N, Li Q, Mohan AL, Baylin SB, Issa J-P. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–94. [PubMed] [Google Scholar]
- 2.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330–8. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 3.Rashid A, Shen L, Morris JS, Issa JP, Hamilton SR. CpG island methylation in colorectal adenomas. Am J Pathol. 2001;159:1129–35. doi: 10.1016/S0002-9440(10)61789-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–8. doi: 10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 5.Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727–30. [PubMed] [Google Scholar]
- 6.Backman V, Roy HK. Light-scattering technologies for field carcinogenesis detection: a modality for endoscopic prescreening. Gastroenterology. 2011;140:35–41. doi: 10.1053/j.gastro.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belshaw NJ, Pal N, Tapp HS, Dainty JR, Lewis MP, Williams MR, Lund EK, Johnson IT. Patterns of DNA methylation in individual colonic crypts reveal aging and cancer-related field defects in the morphologically normal mucosa. Carcinogenesis. 2010;31:1158–63. doi: 10.1093/carcin/bgq077. [DOI] [PubMed] [Google Scholar]
- 8.Hiraoka S, Kato J, Horii J, Saito S, Harada K, Fujita H, Kuriyama M, Takemoto K, Uraoka T, Yamamoto K. Methylation status of normal background mucosa is correlated with occurrence and development of neoplasia in the distal colon. Hum Pathol. 2010;41:38–47. doi: 10.1016/j.humpath.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Tominaga K, Fujii S, Mukawa K, Fujita M, Ichikawa K, Tomita S, Imai Y, Kanke K, Ono Y, Terano A, et al. Prediction of colorectal neoplasia by quantitative methylation analysis of estrogen receptor gene in nonneoplastic epithelium from patients with ulcerative colitis. Clin Cancer Res. 2005;11:8880–5. doi: 10.1158/1078-0432.CCR-05-1309. [DOI] [PubMed] [Google Scholar]
- 10.Ally MS, Al-Ghnaniem R, Pufulete M. The relationship between gene-specific DNA methylation in leukocytes and normal colorectal mucosa in subjects with and without colorectal tumors. Cancer Epidemiol Biomarkers Prev. 2009;18:922–8. doi: 10.1158/1055-9965.EPI-08-0703. [DOI] [PubMed] [Google Scholar]
- 11.Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Grieu F, Watanabe G, Iacopetta B. DNA hypermethylation in the normal colonic mucosa of patients with colorectal cancer. Br J Cancer. 2006;94:593–8. doi: 10.1038/sj.bjc.6602940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belshaw NJ, Elliott GO, Foxall RJ, Dainty JR, Pal N, Coupe A, Garg D, Bradburn DM, Mathers JC, Johnson IT. Profiling CpG island field methylation in both morphologically normal and neoplastic human colonic mucosa. Br J Cancer. 2008;99:136–42. doi: 10.1038/sj.bjc.6604432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paun BC, Kukuruga D, Jin Z, Mori Y, Cheng Y, Duncan M, Stass SA, Montgomery E, Hutcheon D, Meltzer SJ. Relation between normal rectal methylation, smoking status, and the presence or absence of colorectal adenomas. Cancer. 2010;116:4495–501. doi: 10.1002/cncr.25348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iacopetta B. Are there two sides to colorectal cancer? Int J Cancer. 2002;101:403–8. doi: 10.1002/ijc.10635. [DOI] [PubMed] [Google Scholar]
- 15.Glebov OK, Rodriguez LM, Nakahara K, Jenkins J, Cliatt J, Humbyrd CJ, DeNobile J, Soballe P, Simon R, Wright G, et al. Distinguishing right from left colon by the pattern of gene expression. Cancer Epidemiol Biomarkers Prev. 2003;12:755–62. [PubMed] [Google Scholar]
- 16.Worthley DL, Whitehall VL, Le Leu RK, Irahara N, Buttenshaw RL, Mallitt KA, Greco SA, Ramsnes I, Winter J, Hu Y, et al. DNA methylation in the rectal mucosa is associated with crypt proliferation and fecal short-chain fatty acids. Dig Dis Sci. 2011;56:387–96. doi: 10.1007/s10620-010-1312-4. [DOI] [PubMed] [Google Scholar]
- 17.Ahuja N, Issa JP. Aging, methylation and cancer. Histol Histopathol. 2000;15:835–42. doi: 10.14670/HH-15.835. [DOI] [PubMed] [Google Scholar]
- 18.Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011;6:828–37. doi: 10.4161/epi.6.7.16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tapp HS, Commane DM, Bradburn DM, Arasaradnam R, Mathers JC, Johnson IT, Belshaw NJ. Nutritional factors and gender influence age-related DNA methylation in the human rectal mucosa. Aging Cell. 2013;12:148–55. doi: 10.1111/acel.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida T, Yamashita S, Takamura-Enya T, Niwa T, Ando T, Enomoto S, Maekita T, Nakazawa K, Tatematsu M, Ichinose M, et al. Alu and Satα hypomethylation in Helicobacter pylori-infected gastric mucosae. Int J Cancer. 2011;128:33–9. doi: 10.1002/ijc.25534. [DOI] [PubMed] [Google Scholar]
- 23.Kamiyama H, Suzuki K, Maeda T, Koizumi K, Miyaki Y, Okada S, Kawamura YJ, Samuelsson JK, Alonso S, Konishi F, et al. DNA demethylation in normal colon tissue predicts predisposition to multiple cancers. Oncogene. 2012;31:5029–37. doi: 10.1038/onc.2011.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol. 2007;40:142–50. doi: 10.5483/BMBRep.2007.40.2.142. [DOI] [PubMed] [Google Scholar]
- 25.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–93. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 26.Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–7. [PubMed] [Google Scholar]
- 27.Kaz AM, Wong CJ, Luo Y, Virgin JB, Washington MK, Willis JE, Leidner RS, Chak A, Grady WM. DNA methylation profiling in Barrett’s esophagus and esophageal adenocarcinoma reveals unique methylation signatures and molecular subclasses. Epigenetics. 2011;6:1403–12. doi: 10.4161/epi.6.12.18199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du PB. R. methyAnalysis: an R package for DNA methylation data analysis and visualization. In: Genentech I, ed., 2012:1-11. [Google Scholar]
- 29.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 30.Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 31.Grady WM, Parkin RK, Mitchell PS, Lee JH, Kim YH, Tsuchiya KD, Washington MK, Paraskeva C, Willson JK, Kaz AM, et al. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene. 2008;27:3880–8. doi: 10.1038/onc.2008.10. [DOI] [PubMed] [Google Scholar]
- 32.Worthley DL, Whitehall VL, Buttenshaw RL, Irahara N, Greco SA, Ramsnes I, Mallitt KA, Le Leu RK, Winter J, Hu Y, et al. DNA methylation within the normal colorectal mucosa is associated with pathway-specific predisposition to cancer. Oncogene. 2010;29:1653–62. doi: 10.1038/onc.2009.449. [DOI] [PubMed] [Google Scholar]
- 33.Toyota M, Issa JP. CpG island methylator phenotypes in aging and cancer. Semin Cancer Biol. 1999;9:349–57. doi: 10.1006/scbi.1999.0135. [DOI] [PubMed] [Google Scholar]
- 34.Illingworth R, Kerr A, Desousa D, Jørgensen H, Ellis P, Stalker J, Jackson D, Clee C, Plumb R, Rogers J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, Rehli M. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009;19:1165–74. doi: 10.1101/gr.091470.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–80. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 38.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.