

Abstract
The Na,K-ATPase or sodium pump carries out the coupled extrusion of Na+ and uptake of K+ across the plasma membranes of cells of most higher eukaryotes. We have shown earlier that Na,K-ATPase-β1 (NaK-β) protein levels are highly reduced in poorly differentiated kidney carcinoma cells in culture and in patients' tumor samples. The mechanism(s) regulating the expression of NaK-β in tumor tissues has yet to be explored. We hypothesized that DNA methylation plays a role in silencing the NaK-β gene (ATP1B1) expression in kidney cancers. In this study, to the best of our knowledge we provide the first evidence that ATP1B1 is epigenetically silenced by promoter methylation in both renal cell carcinoma (RCC) patients’ tissues and cell lines. We also show that knockdown of the von Hippel-Lindau (VHL) tumor suppressor gene in RCC cell lines results in enhanced ATP1B1 promoter AT hypermethylation, which is accompanied by reduced expression of NaK-β. Furthermore, treatment with 5-Aza-2′-deoxycytidine rescued the expression of ATP1B1 mRNA as well as NaK-β protein in these cells. These data demonstrate that promoter hypermethylation is associated with reduced NaK-β expression, which might contribute to RCC initiation and/or disease progression.
Keywords: epigenetics; DNA methylation; renal cell carcinoma; Na,K-ATPase; ATP1B1
Introduction
Kidney cancers account for about 2% of all cancers, and more than 200 000 new cases of kidney cancer are diagnosed worldwide each year.1 The most common form of kidney cancer in adult is renal cell carcinoma (RCC). Approximately 75% of the RCC cases are classified as clear cell RCC (ccRCC).2 The most common genetic event in the evolution of sporadic ccRCC is inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene (TSG).3-6 VHL inactivation leads to stabilization of the hypoxia-inducible transcription factor (HIF) and activation of a wide repertoire of hypoxia response genes.7 The frequency of VHL mutations in sporadic ccRCC has been reported to be as high as 80% (although VHL mutations are rare in non-clear-cell forms of RCC).8
TSG inactivation may result from genetic or epigenetic events, and it is well recognized that epigenetic silencing of TSGs has a significant role in the pathogenesis of human cancers. Indeed, epigenetic silencing via promoter hypermethylation of VHL in RCC5 was one of the first examples of this phenomenon. In fact, VHL mutation and methylation have been shown to be mutually exclusive, with methylation-induced silencing of VHL observed in 7% of RCCs.9 From initial reports, approximately 60 genes were suggested to be epigenetically dysregulated in RCC.10 Subsequently, work from The Cancer Genome Atlas (TCGA) that profiled over 400 tumors indicated that 289 genes display evidence of silencing by DNA methylation in at least 5% of tumors.9 Identification of potential tumor suppressors silenced by methylation will facilitate a better understanding of the etiology of the disease and promote novel therapeutic approaches to treat ccRCC.9,11,12
The Na,K-ATPase is an abundantly expressed protein in epithelial cells and plays a crucial role in kidney function. Localized to the basolateral plasma membrane in epithelial cells, the oligomeric Na,K-ATPase catalyzes the ATP-dependent transport of three Na+ out and two K+ into the cell per pump cycle to maintain Na+ and K+ gradients across the plasma membrane. This Na+ and K+ homeostasis in epithelia is necessary regulate the functions of various ion and solute transporters which is essential for the directional transport of solutes across the epithelial cell layer (vectorial transport).13 The Na,K,ATPase is composed of two essential polypeptide subunits, the α-subunit (112 kDa) and the β-subunit (55 kDa),14,15 and an optional regulatory γ-subunit with tissue-specific expression (7 kDa).16 Of the four α- and three β-subunit isoforms known, α1 (NaK-α) and β1 (NaK-β) are predominantly expressed in kidney.14,15
Our laboratory previously demonstrated that NaK-β protein expression is reduced in ccRCC patients’ tumor samples.17 Subsequently, we showed that oncogenic transformation of kidney epithelial cells resulted in the reduced expression of NaK-β and promoted invasive and metastatic behaviors of these cells.18,19 NaK-β levels were also reduced in a wide variety of carcinoma cells that have undergone epithelial to mesenchymal transition (EMT), which is one of the events associated with cancer progression into metastatic disease.20 Ectopic expression of NaK-β in these transformed kidney epithelial cells delayed EMT,21 abolished anchorage independent growth (the ability of tumor cells to grow in soft agar), and suppressed the growth of tumor xenografts in vivo.19 Anchorage-independent growth and the ability to form tumors in immunocompromised mice (tumorigenicity) are primary features of malignant transformation, and TSGs inhibit both of these characteristics.22-25 Since NaK-β repletion suppressed these characteristics, we proposed that NaK-β is a potential tumor suppressor.19
Inactivating mutations of the NaK-β gene (ATP1B1) have not been reported in cancer, yet many tumor suppressors can be silenced by promoter hypermethylation instead of mutation.26 Given the putative tumor suppressor role of NaK-β and its general reduction in ccRCC, we hypothesized that NaK-β is epigenetically repressed in ccRCC. In this study, by analyzing a large number of patients’ data from TCGA in conjunction with our own in vitro experiments we show a strong correlation between ATP1B1 expression and methylation. Using methylation specific PCR (MSP) in ccRCC patients’ tumor samples, the ATP1B1 promoter displays a stage-dependent increase in hypermethylation. Furthermore, we demonstrate that the ATP1B1 promoter is preferentially hypermethylated in RCC cell lines deficient in VHL expression, which correlates with an increase in the expression of DNA methyltransferase (DNMT) 1 and 3A in these cells. Importantly, inhibition of DNMT activity using 5-Aza-2′-deoxycytidine (5-Aza-dC) rescued NaK-β expression in VHL knockdown cell lines.
Results
ATP1B1 promoter is hypermethylated in ccRCC patient tumor samples
Analysis of the ATP1B1 promoter sequences using MethPrimer27 showed two CpG islands located at bases -944 to -1064 and −500 to -649 (termed as CpG regions R1 and R2, respectively) (Fig. 1). This finding suggests that methylation of 5′ regulatory CpG sites might be one of the mechanisms involved in the transcriptional repression of ATP1B1 in ccRCC.
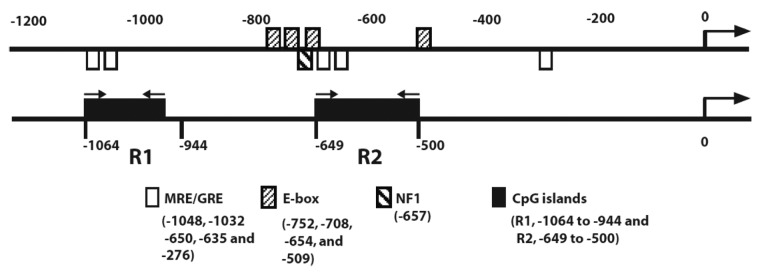
Figure 1. Promoter methylation analysis of ATP1B1. Schematic representation of NaK-β1 subunit promoter elements (upper panel) and CpG islands (lower panel). GRE, glucocorticoid responsive elements.
We then analyzed ATP1B1 promoter methylation in tumor samples obtained from RCC patients. To determine DNA methylation, methylation-specific PCR (MSP) primers were designed using the online tool Methprimer to the R1 and R2 regions (Fig. 1) (Table 1). Analyses of the methylation pattern of ccRCC tumor samples with matched morphologically normal tissues from six patients are shown in Figure 2. Compared with matched normal tissues, tumor tissues showed more intense bands corresponding to methylated promoter regions (compare lanes 8 vs 4). Interestingly, the intensity of methylated promoter regions positively correlated with the stage of the tumor (lane 8). In the R1 region the promoter is largely unmethylated in normal compared with tumor tissues (compare lanes 1 vs 5). In the R2 region of stage 4, adjacent normal tissues showed higher methylation compared with stages 1, 2, and 3 (lane 4). The intensity of the methylated band was maximal in stage 4 tumors (lane 8).
Table 1. PCR primers.
| Na,K,ATPase β1: |
|---|
| 5′-CATCAGAGAGCAACTGATCC-3′ and |
| 5′-ATGCCATTCCGAATCTCAGC-3′ |
| R1 region |
|---|
| Unmethylation |
| 5′-GAA GGG TTG GAA TTG TAA TTG T-3′ |
| 5′-AAAAAA AAT CAC TCC AAA ACA C-3′ |
| Methylation |
| 5′-GAA GGG TCG GAA TTG TAA TC-3′ |
| 5′-AAA AAAATC ACT CCGAAA CG-3′ |
| R2 region: |
|---|
| Unmethylation |
| 5′-TTTTTT TGT GTT GGTTTT GA -3′ |
| 5′-AAAAAA TTAACT ACA CCC ACC-3′ |
| Methylation |
| 5′-GGT TTT TTT GTG TCG GTTTC-3′ |
| 5′-GAAAAA TTAACT ACG CCC GC-3′ |

Figure 2.ATP1B1 promoter methylation, tumor stage, and gene expression in patient RCC tumor samples. (A) Analysis of RCC patient tumor samples. U and M indicate amplicons generated using primers specific for unmethylated and methylated NaK-β1 promoter alleles.
To validate our observation of ATP1B1 promoter methylation and explore the relation of ATP1B1 methylation with gene expression in RCC, we accessed data from The Cancer Genome Atlas (TCGA) of renal cell carcinoma (tumor n = 272, normal n = 21).9 There are 15 CpG sites associated with ATP1B1 on the Illumina 450K methylation array platform; six CpGs 5′ of the TSS, three CpGs in the first exon, and six CpGs in the gene body with nine of the CpGs in a CpG island. Interestingly, the nine CpG island CpGs were relatively unmethylated in RCC, with none of the nine sites having a maximum average β greater than 0.17 (17% methylated). Among the nine CpG island CpG sites, one CpG was mapped to each of the regions measured with MSP (R1 and R2), though not within the primer sequences. The maximum methylation among TCGA RCCs was 0.14 (14% methylated) for the CpG in R1, and 0.09 (9% methylated) for the CpG in R2. The range of median methylation values among the nine CpG island CpG sites was from 0.009 to 0.054 (less than 1% methylated to 5% methylated). However, five of the remaining six CpG sites each demonstrated a wide range of methylation values among RCC tumors, with median methylation ranging from 0.29 to 0.60 (29% methylated to 60% methylated). Methylation of each of these five ATP1B1 CpG sites was significantly negatively associated with ATP1B1 gene expression. For example, methylation of cg13104274 in the South CpG island shore region of ATP1B1 was significantly associated with reduced ATP1B1 gene expression (P = 5.3E-09, Fig. 3A). In addition, methylation of cg13104274 was significantly lower in normal kidney samples (n = 21, mean methylation 0.203) compared with RCC (n = 272, mean methylation 0.46; P = 3.0E-06, Fig. 3B). Concomitantly, ATP1B1 gene expression was significantly lower in RCC compared with normal kidney (P = 3.0E-12, Fig. 3C). Altogether, these results indicate that the ATP1B1 promoter is hypermethylated in ccRCC tumors relative to matched morphologically normal tissues.

Figure 3. TCGA data analysis of ATP1B1 methylation and gene expression in normal kidney and RCC. (A) Normalized gene expression of ATP1B1 is plotted vs. ATP1B1 methylation at cg13104274 in RCC (n = 272) and demonstrates a significant association between DNA methylation and reduced gene expression (P = 5.3E-09). Above the plot is a scheme of CpG sites measured on the Illumina 450K platform in black, the yellow regions represent R1 (left) and R2 regions measured with MSP, the red line is the TSS, and the blue line is the plotted CpG site. (B) Methylation of cg13104274 is significantly higher in RCC (n = 272) compared with normal kidney (n = 21, P = 3.0E-06). Black bars represent mean methylation values: normal tissue mean methylation = 0.20, tumor mean methylation = 0.46 (C) ATP1B1 gene expression is significantly higher in normal kidney (n = 21) compared with RCC (n = 272, P = 3.0E-12). Black bars represent mean expression values: normal tissue mean expression = 6.1E+04, tumor mean methylation = 2.2E+04.
Knockdown of VHL enhances ATP1B1 promoter methylation
To further understand the molecular mechanism of ATP1B1 repression by promoter hypermethylation, we used two well characterized isogenic pairs of human ccRCC cell lines that differ only in VHL expression levels due to lentiviral transfer of VHL-specific shRNA (SN12C and SN12C-VHL; ACHN and ACHN-VHL).28 Methylation-specific PCR of the genomic DNA isolated from these cell lines revealed enhanced methylation of ATP1B1 promoter in the VHL knockdown cells compared with the parental cells (Fig. 4A). In the R1 region, methylation of the promoter was not detected in the parental cell lines (lanes 1), whereas it was considerably methylated in VHL-knockdown cell lines (lanes 5). In the R2 region moderate methylation was observed in VHL-positive cell lines (lane 3) while the promoter was heavily methylated in VHL-knockdown cell lines (lane 7). Taken together, these data suggest that ATP1B1 promoter is hypermethylated in ccRCC cell lines and that VHL deficiency associated with ATP1B1 promoter methylation.

Figure 4.ATP1B1 promoter methylation, NaK-β expression, and DNMT expression in isogenic ccRCC cell lines. (A) Methylation-specific PCR analysis of ATP1B1 promoter methylation in the isogenic ccRCC cells lines. U and M indicate amplicons generated using primers specific for unmethylated and methylated ATP1B1 promoter alleles. (B) Analysis of ATP1B1 transcript expression. Graph depicts mean ± SD of one representative experiment. (C) Analysis of NaK-β1 protein expression. (D) Analysis of DNMT1 and DNMT3A protein expression.
Knockdown of VHL decreases NaK-β expression but increases DNMT1 and 3A expression
Since we observed enhanced ATP1B1 promoter methylation in the VHL knockdown cells, we assessed whether NaK-β expression is reduced in these cells. We found that knockdown of VHL produced a dramatic reduction in both NaK-β transcript (Fig. 4B) and protein (Fig. 4C) expression. There was a 60% reduction in the NaK-β protein levels. These data confirm that enhanced ATP1B1 promoter methylation leads to reduced NaK-β expression in VHL-knockdown cells.
DNA methylation is performed by methyltransferase enzymes such as DNMT1 and DNMT3A.29 We investigated whether expression of DNMT1 and DNMT3A were affected in the isogenic ccRCC cells. Strikingly, the VHL-knockdown cells exhibited an increase in both DNMT1 and DNMT3A protein expression levels compared with the parental cell lines (Fig. 4D). The increase was more pronounced for both DNMT1 and DNMT3A in the SN12C cells (2.5 and 3.0-fold) than the ACHN cells (1.8 and 1.6-fold) indicating that there may be additional cell-type specific effects that affect DNMT enzyme expression.
DNMT inhibitor 5-AZA-dC rescues NaK-β expression in VHL knockdown cell lines
5-Aza-dC has been effectively used to re-express genes that have been silenced by promoter hypermethylation.30 We treated the isogenic ccRCC cell lines with 5-Aza-dC and found that treatment enhanced NaK-β protein expression in the VHL-knockdown cells but not the parental cells (Fig. 5A). In support of this, transcript analysis revealed a 50% increase in the NaK-β mRNA levels in 5-Aza-dC-treated VHL-knockdown cells (Fig. 5B). These results indicate that inhibition of methyltransferase enzymes rescues NaK-β expression in VHL-knockdown cells.

Figure 5. Effect of 5-Aza-dC on the expression of NaK-β in ccRCC cell lines. (A) Analysis of NaK-β protein expression in isogenic SN12C and ACHN cells treated with increasing concentrations of 5-Aza-dC (0, 1, 5 and 10 μM) for 48hr. (B) Analysis of NaK-β transcript expression in VHL-knockdown cells treated with 5-Aza-dC.
To further probe this effect, we performed bisulfite pyrosequencing to quantitatively analyze the ATP1B1 promoter methylation and its loss of methylation following 5-Aza-dC treatment in the isogenic ccRCC cell lines (Fig. S1). SN12C cells showed 51% methylation, and ACHN cells showed 16% promoter methylation (16%). 5-Aza-dC treatment had no significant effect on these cell lines (-4% and +1%, respectively). In contrast, the SN12C and ACHN VHL-knockdown cell lines showed a methylation rate of 72 and 74%, respectively, and 5-Aza-dC treatment reduced promoter methylation in these cell lines (-20% and -9%, respectively). These results further confirm that in VHL-knockdown cells the ATP1B1 promoter is methylated and that 5-Aza-dC treatment results in loss of methylation.
Discussion
Aberrant DNA methylation patterns have been linked to altered gene expression in certain genetic diseases and tumors.31,32 In this study, to our knowledge, we provide the first evidence that the ATP1B1 promoter is hypermethylated in ccRCC compared with paired adjacent non-malignant tissues, a finding that we validated using TCGA tumor methylation data. TCGA RCC tumor data indicated a significant association between ATP1B1 methylation and decreased ATP1B1 gene expression. Furthermore, in the TCGA data, normal kidney tissue samples had both significantly lower methylation and significantly higher gene expression compared with with RCCs. In our tumor samples, we observed that ATP1B1 promoter methylation increased with increasing tumor stage, although we did not observe a consistent trend for increasing methylation with tumor stage in the TCGA data. Using isogenic ccRCC cell lines that differ in only VHL expression, we further showed that ATP1B1 hypermethylation is enhanced in VHL-deficient cell lines and that inhibition of DNMTs by 5-Aza-dC relieves promoter hypermethylation which leads to renewed expression of Na,K-β transcript and protein. We reported earlier that ectopic expression of Na,K-β in kidney carcinoma cells reduces in vivo tumor growth and abolishes anchorage-independent growth which are hallmark functions of TSGs.19 Along with previous observations, the present study further confirms that Na,K-β has tumor suppressor functions in kidney epithelial cells.
Patients’ tumor samples obtained from stage IV tumors showed heavy methylation by methylation-specific PCR (MSP) compared with stage I, II, and III tumors. Interestingly, the matched morphologically normal adjacent tissue from stage IV patients showed higher methylation than the stage II or III patients. This result indicates that ATP1B1 promoter is also hypermethylated in adjacent normal tissues, and suggests that ATP1B1 promoter methylation may be an early event that occurs prior to changes in the morphology of kidney cells. Based upon our MSP results, the R2 region appears to be methylated to a greater degree than the R1 region in tumors, although this relationship was not confirmed in the TCGA data. While these results may seem inconsistent, there are key differences in the methods used. Provided that there is correlation among the methylated CpG sites in the primers, MSP assays can detect relatively low levels of methylated DNA (< 5%).33 Interestingly, despite the canonical relation of CpG island hypermethylation with gene silencing for tumor suppressor genes in cancer, TCGA methylation and expression data indicated a strong relation of methylation at island-adjacent CpG sites with gene expression. The importance of so-called CpG island shore methylation in human cancer has become well recognized in recent years.34,35 We proposed earlier that Na,K-β expression could be used as a biomarker whose expression is reduced in ccRCC.17 Since the morphologically normal tissues adjacent to advanced tumors showed ATP1B1 promoter hypermethylation by MSP, and significant differences between normal and tumor tissue were found from the TCGA data, the methylation pattern of ATP1B1 promoter may have potential as an early biomarker for the diagnosis of ccRCC.
During EMT, the expression of cell-cell adhesion molecules is reduced which facilitates invasion and metastatic dissemination of carcinoma cells. NaK-β is a key cell-cell adhesion molecule in kidney epithelial cells, and its expression is downregulated during EMT21 by the transcriptional suppressor Snail which binds to E-boxes within the ATP1B1 promoter and downregulates its expression.20 Although we cannot rule out that Snail is induced and involved in the suppression of NaK-β in VHL-knockdown cells, the enhanced NaK-β transcript and protein levels following 5-Aza-dC treatment suggests that hypermethylation is one of the mechanisms involved in reducing NaK-β expression in ccRCC. Furthermore, loss of VHL function leads to constitutive expression of hypoxia-inducible factor (HIF) which promotes EMT.36 Therefore, it is possible that HIF might be involved in the downregulation of NaK-β levels in VHL-knockdown cells.
Enhanced expression of both DNMT1 and DNMT3A in VHL-knockdown cells suggest that loss of VHL might be associated with hypermethylation of multiple genes associated with the EMT process. In fact, it has been shown that DNMTs are induced during EMT in several cancers, including pancreatic cancer, non-small cell lung cancer,37 and breast cancer.38 There are no reports available on the influence of HIF-1α on DNMT activity; however multiple reports are available on the interaction between HIF-1α and histone demethylases JMJD1A, (demethylates dimethylated histone H3 lysine 9 (H3K9Me2) and JMJD2B (demethylates trimethylated H3K9).39,40 A recent report suggests that VHL depletion results in the HIF-dependent regulation of H3K4 trimethylation by decreasing histone demethylase JAR1D1C.41 Future investigations are necessary to understand the mechanisms of DNMT induction by loss VHL in ccRCC. We reported earlier that ectopic expression of NaK-β in kidney carcinoma cells reduces in vivo tumor growth and abolishes anchorage-independent growth, which are hallmark functions of TSGs.19 Along with our previous observations, the present study further confirms that NaK-β has tumor suppressor functions in kidney epithelial cells.
Materials and Methods
Reagents
5-Aza-2′-deoxycytidine and β-actin mouse antibody were purchased from Sigma-Aldrich. Antibody against NaK-β (M17-P5-F11) was kindly provided by Dr William Ball, Jr. (University of Cincinnati). Primary antibodies specific for GAPDH, DNMT1, and DNMT3, as well as horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Cell Signaling.
Patient tissues
Fresh-frozen clinical tissue samples were obtained with informed consent from the Helen F Graham Cancer Center Tumor Bank, Christiana Care Hospital with appropriate ethical approval from the Institutional Review Boards of the Christiana Care Hospital and the Nemours/Alfred I. duPont Hospital for Children. Primary RCCs and matched adjacent macroscopically normal renal tissues from six patients undergoing renal surgery were analyzed. Tissues were evaluated by pathologists, and DNA was isolated from fresh frozen samples using the DNeasy Blood and Tissue Kit (Qiagen Inc.) and used for methylation specific PCR as described below.
The Cancer Genome Atlas data and methods
Using The Cancer Genome Atlas (TCGA) data portal, we downloaded level 3 data from 272 RCC tumors and 21 normal tissues with available matched Illumina 450K DNA methylation array and Illumina HiSeq RNASeqV2 data. DNA methylation and RNASeq files were loaded into and parsed using the R software environment (v.3.0.1), matching samples based on their TCGA ID.
Tissue culture and drug treatments
Human clear cell renal cell carcinoma cell lines (SN12C, ACHN) and isogenic VHL-knockdown cell lines (SN12C-VHL, ACHN-VHL)28 were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2mM L-glutamine, 25 U/mL penicillin, and 25 μg/mL streptomycin at 37 °C in 5% CO2. Cell lines were treated with 5-aza-2′-deoxycytidine (5-Aza-dC) (Sigma) for 48 h.
Methylation-specific PCR
DNA methylation within the CpG region of the NaK-β promoter was analyzed using methylation-specific PCR after sodium bisulfite treatment of genomic DNA as described previously.22 CpGenome universally methylated DNA (Chemicon), after bisulfite modification, served as the methylated NaK-β. PCR was performed in 50μl reaction mixtures containing 1.0 μl of modified DNA, 1 × Qiagen buffer, 2.5 mM MgCl2, 0.2 mM of each dNTP, 0.2 μM of each primer and 1.5 U of HotStar Taq (Qiagen Inc.).
Quantitative real-time PCR
Total RNA was harvested using RNAase isolation kit (Qiagen Inc.). CDNA (cDNA) was synthesized from 2 μg of total RNA using iScript cDNA synthesis kit (Bio-Rad laboratories, Hercules, CA, USA). CDNA was amplified in a 25 μL reaction containing 12.5 μL SybrGreen PCR Master Mix (Bio-Rad), 100 pmol primers and 0.25 μg cDNA using a Mini-Opticon Q-RTPCR machine (Bio-Rad) with the following program: 1 cycle at 95 °C for 5 min; 45 cycles at 95 °C for 45 s, 55 °C 45 s, 72 °C for 45 s. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression was used to normalize mRNA expression.
Pyrosequencing
Pyrosequencing analysis was performed using PCR product generated from bisulfite-treated DNA. PCR was performed in 50 μl reaction mixtures containing 1.0 μL of bisulfite-modified DNA, 1 × Qiagen PCR buffer, 2.0 mM MgCl2, 0.2 mM of each dNTP, 0.2 μM of each primer and 1.5 U of Hotstar Taq DNA polymerase. The amplification program consisted of 1 cycle at 95 °C for 15 min, 45 cycles at 95 °C for 20s, 52 °C for 20s and 72 °C for 20s. Pyrosequencing analysis was performed by EpigenDx Inc.
Immunoblot analysis
At the end of treatments with 5-Aza-2′-deoxycytidine and untreated control were analyzed by immunoblot analysis as described before (23). Briefly, cells were washed with cold PBS and lysed in buffer (20 mmol/L TRIS-HCl pH 7.4, 100 mmol/L NaCl, 1% Triton X-100, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L sodium glycerolphosphate, 1 mmol/L sodium orthovanadate) supplemented with protease inhibitors. Cell lysates were centrifuged at 14 000 g for 15 min at 4 °C and protein concentration was determined in the supernatants using the Biorad protein assay reagent (Bio-Rad) using bovine serum albumin as standard. Equal amounts of protein were then resolved by SDS-PAGE and transferred to nitrocellulose. The membranes were blocked for one hour at room temperature in TBST (50 mM TRIS-HCl pH 7.5, 150 mM NaCl, 0.2% Tween-20) containing 5% nonfat milk. Blots were sequentially incubated with the primary and secondary antibodies, washed in TBS-T. Membranes were developed using Amersham ECL Prime (GE Healthcare) and images were acquired using Photoshop (Adobe Systems Inc.).
Statistical analysis
Statistical analyses were performed by one-way ANOVA followed by Dunnett’s Test. Statistical comparisons were considered significant at P < 0.05. Linear regression analysis was used to test the relation of ATP1B1 gene expression with promoter DNA methylation. The Wilcoxon rank sum test was used to test the relation of DNA methylation or ATP1B1 gene expression with tumor status (tumor vs. normal).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Charles Sawyers for providing the VHL-deficient cell lines and Dr. William James Ball, Jr. for the Na,K-ATPase β1 antibody. This research was supported by NIH grants DK56216 (A.K.R.), P20GM103464 (T.S.), P20-GM104416 (B.C.C.), and the Nemours Foundation.
Glossary
Abbreviations:
- Na,K-ATPase
sodium, potassium adenosine triphosphatase
- NaK-β
Na,K-ATPase-β1 subunit
- ATP1B1
NaK-β gene
- RCC
renal cell carcinoma
- ccRCC
clear cell RCC
- VHL
von Hippel-Lindau
- HIF
hypoxia-inducible factor
- TSG
tumor suppressor gene
- TCGA
the cancer genome atlas
- EMT
epithelial-mesenchymal transition
- MSP
methylation-specific polymerase chain reaction
- DNMT
DNA methyltransferase
- 5-Aza-dC
5-Aza-2′-deoxycytidine
References
- 1.Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol. 2007;18:581–92. doi: 10.1093/annonc/mdl498. [DOI] [PubMed] [Google Scholar]
- 2.Mancini V, Battaglia M, Ditonno P, Palazzo S, Lastilla G, Montironi R, Bettocchi C, Cavalcanti E, Ranieri E, Selvaggi FP. Current insights in renal cell cancer pathology. Urol Oncol. 2008;26:225–38. doi: 10.1016/j.urolonc.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 3.Clifford SC, Prowse AH, Affara NA, Buys CH, Maher ER. Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL-independent pathway in clear cell renal tumourigenesis. Genes Chromosomes Cancer. 1998;22:200–9. doi: 10.1002/(SICI)1098-2264(199807)22:3<200::AID-GCC5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 4.Foster K, Crossey PA, Cairns P, Hetherington JW, Richards FM, Jones MH, Bentley E, Affara NA, Ferguson-Smith MA, Maher ER. Molecular genetic investigation of sporadic renal cell carcinoma: analysis of allele loss on chromosomes 3p, 5q, 11p, 17 and 22. Br J Cancer. 1994;69:230–4. doi: 10.1038/bjc.1994.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994;91:9700–4. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 7.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 8.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–34. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris MR, Maher ER. Epigenetics of renal cell carcinoma: the path towards new diagnostics and therapeutics. Genome Med. 2010;2:59. doi: 10.1186/gm180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Avissar-Whiting M, Koestler DC, Houseman EA, Christensen BC, Kelsey KT, Marsit CJ. Polycomb group genes are targets of aberrant DNA methylation in renal cell carcinoma. Epigenetics. 2011;6:703–9. doi: 10.4161/epi.6.6.16158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricketts CJ, Morris MR, Gentle D, Brown M, Wake N, Woodward ER, Clarke N, Latif F, Maher ER. Genome-wide CpG island methylation analysis implicates novel genes in the pathogenesis of renal cell carcinoma. Epigenetics. 2012;7:278–90. doi: 10.4161/epi.7.3.19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajasekaran SA, Barwe SP, Rajasekaran AK. Multiple functions of Na,K-ATPase in epithelial cells. Semin Nephrol. 2005;25:328–34. doi: 10.1016/j.semnephrol.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 14.Rajasekaran SA, Rajasekaran AK. Na,K-ATPase and epithelial tight junctions. Front Biosci (Landmark Ed) 2009;14:2130–48. doi: 10.2741/3367. [DOI] [PubMed] [Google Scholar]
- 15.Vagin O, Dada LA, Tokhtaeva E, Sachs G. The Na-K-ATPase α₁β₁ heterodimer as a cell adhesion molecule in epithelia. Am J Physiol Cell Physiol. 2012;302:C1271–81. doi: 10.1152/ajpcell.00456.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–66. doi: 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- 17.Rajasekaran SA, Ball WJ, Jr., Bander NH, Liu H, Pardee JD, Rajasekaran AK. Reduced expression of beta-subunit of Na,K-ATPase in human clear-cell renal cell carcinoma. J Urol. 1999;162:574–80. doi: 10.1016/S0022-5347(05)68629-6. [DOI] [PubMed] [Google Scholar]
- 18.Rajasekaran SA, Palmer LG, Quan K, Harper JF, Ball WJ, Jr., Bander NH, Peralta Soler A, Rajasekaran AK. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol Biol Cell. 2001;12:279–95. doi: 10.1091/mbc.12.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inge LJ, Rajasekaran SA, Yoshimoto K, Mischel PS, McBride W, Landaw E, Rajasekaran AK. Evidence for a potential tumor suppressor role for the Na,K-ATPase beta1-subunit. Histol Histopathol. 2008;23:459–67. doi: 10.14670/hh-23.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Espineda CE, Chang JH, Twiss J, Rajasekaran SA, Rajasekaran AK. Repression of Na,K-ATPase beta1-subunit by the transcription factor snail in carcinoma. Mol Biol Cell. 2004;15:1364–73. doi: 10.1091/mbc.E03-09-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajasekaran SA, Huynh TP, Wolle DG, Espineda CE, Inge LJ, Skay A, Lassman C, Nicholas SB, Harper JF, Reeves AE, et al. Na,K-ATPase subunits as markers for epithelial-mesenchymal transition in cancer and fibrosis. Mol Cancer Ther. 2010;9:1515–24. doi: 10.1158/1535-7163.MCT-09-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodrigues M, Balicki D, Newrock KM, Mukherjee BB. Lack of correlation between loss of anchorage-independent growth and levels of transformation-specific p53 protein in retinoic acid-treated F9 embryonal carcinoma cells. Exp Cell Res. 1985;156:22–30. doi: 10.1016/0014-4827(85)90258-7. [DOI] [PubMed] [Google Scholar]
- 23.Leone A, Flatow U, King CR, Sandeen MA, Margulies IM, Liotta LA, Steeg PS. Reduced tumor incidence, metastatic potential, and cytokine responsiveness of nm23-transfected melanoma cells. Cell. 1991;65:25–35. doi: 10.1016/0092-8674(91)90404-M. [DOI] [PubMed] [Google Scholar]
- 24.Weinberg RA. Tumor suppressor genes. Science. 1991;254:1138–46. doi: 10.1126/science.1659741. [DOI] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 26.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–31. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 28.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–7. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 29.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–93. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 30.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 31.Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–92. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- 32.Balch C, Nephew KP. Epigenetic targeting therapies to overcome chemotherapy resistance. Adv Exp Med Biol. 2013;754:285–311. doi: 10.1007/978-1-4419-9967-2_14. [DOI] [PubMed] [Google Scholar]
- 33.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanderkraats ND, Hiken JF, Decker KF, Edwards JR. Discovering high-resolution patterns of differential DNA methylation that correlate with gene expression changes. Nucleic Acids Res. 2013;41:6816–27. doi: 10.1093/nar/gkt482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27:157–69. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mateen S, Raina K, Agarwal C, Chan D, Agarwal R. Silibinin synergizes with histone deacetylase and DNA methyltransferase inhibitors in upregulating E-cadherin expression together with inhibition of migration and invasion of human non-small cell lung cancer cells. J Pharmacol Exp Ther. 2013;345:206–14. doi: 10.1124/jpet.113.203471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chik F, Szyf M. Effects of specific DNMT gene depletion on cancer cell transformation and breast cancer cell invasion; toward selective DNMT inhibitors. Carcinogenesis. 2011;32:224–32. doi: 10.1093/carcin/bgq221. [DOI] [PubMed] [Google Scholar]
- 39.Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem. 2008;283:36542–52. doi: 10.1074/jbc.M804578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A. 2009;106:4260–5. doi: 10.1073/pnas.0810067106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niu X, Zhang T, Liao L, Zhou L, Lindner DJ, Zhou M, Rini B, Yan Q, Yang H. The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene. 2012;31:776–86. doi: 10.1038/onc.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.