

Abstract
Hypoxia plays a crucial role in the angiogenic switch, modulating a large set of genes mainly through the activation of hypoxia-inducible factor (HIF) transcriptional complex. Endothelial cells play a central role in new vessels formation and express placental growth factor (PlGF), a member of vascular endothelial growth factor (VEGF) family, mainly involved in pathological angiogenesis. Despite several observations suggest a hypoxia-mediated positive modulation of PlGF, the molecular mechanism governing this regulation has not been fully elucidated. We decided to investigate if epigenetic modifications are involved in hypoxia-induced PlGF expression. We report that PlGF expression was induced in cultured human and mouse endothelial cells exposed to hypoxia (1% O2), although DNA methylation at the Plgf CpG-island remains unchanged. Remarkably, robust hyperacetylation of histones H3 and H4 was observed in the second intron of Plgf, where hypoxia responsive elements (HREs), never described before, are located. HIF-1α, but not HIF-2α, binds to identified HREs. Noteworthy, only HIF-1α silencing fully inhibited PlGF upregulation. These results formally demonstrate a direct involvement of HIF-1α in the upregulation of PlGF expression in hypoxia through chromatin remodeling of HREs sites. Therefore, PlGF may be considered one of the putative targets of anti-HIF therapeutic applications.
Keywords: hypoxia, hypoxia inducible factor (HIF), chromatin, histone modification, DNA methylation, angiogenesis, VEGF family, placental growth factor (PlGF)
Introduction
Placental growth factor (PlGF), the second member of vascular endothelial growth factor (VEGF) family discovered,1 is mainly involved in pathological angiogenesis,2,3 a complex biological phenomenon associated to many multifactorial diseases, such as cancer, atherosclerosis, arthritis, diabetic retinopathy and age-related macular degeneration.4,5
Low oxygen tension is one of the major stimuli responsible for angiogenic switch, a time-restricted event during tumor progression where the balance between pro- and anti-angiogenic factors tilts toward a pro-angiogenic outcome, resulting in the transition from dormant avascularized hyperplasia to outgrowing vascularized tumor.6 The main response of cells to hypoxia is represented by the activation of hypoxia inducible factor (HIF) transcriptional complex, which modulates the expression of a large set of genes through the binding of the hypoxia responsive element (HRE) located in their promoters7,8 or along the gene body.9,10 Several genes upregulated by hypoxia encode for proteins having as target endothelial cells (ECs), whose proliferation, migration and differentiation is essential for new vessel formation.11 The EC itself produces several factors involved in angiogenesis, such as the pro-angiogenic members of the VEGF family and related receptors, which play relevant autocrine and paracrine functions with a central role in the mechanisms underlying new vessels formation.12-14
Among the pro-angiogenic members of the VEGF family and related receptors, it has long been known that, in hypoxic condition, HIF is directly involved in the increase of transcription of VEGF-A and VEGF Receptor-1 (VEGFR-1).15,16 Despite the strict biochemical and functional relationship between VEGFR-1, PlGF, and VEGF-A,17-20 a direct involvement of HIF in the modulation of PlGF transcription has not been demonstrated. Furthermore, even though the analysis of promoter/enhancer region of mouse and human Plgf showed the presence of putative HREs, their functionality has never been demonstrated.21-23
Nevertheless, some reports indicate a hypoxia-induced positive modulation of Plgf transcription through the involvement of metal responsive transcription factor 1 (MTF-1) in immortalized/H-Ras-transformed mouse embryonic fibroblasts (mEFs),21 and of nuclear factor κB (NF-κB), in human embryonic kidney 293 (HEK-293) cells.24 Surprisingly, no PlGF upregulation by hypoxia has been observed in human aortic and human umbilical vein endothelial cells.24 However, overexpression of HIF-1α in human endothelial cells25 or in mouse primary cardiac and vascular cells26 positively affects PlGF expression. In vivo, PlGF upregulation occurs in cardiomyocytes and neovessels in the model of myocardial infarct.2,27 Recently, its expression has been reported in human colorectal carcinomas28 and in pediatric medulloblastomas.29 Overall, these data suggest a possible involvement of HIF-1α in the modulation of PlGF expression even if a direct functional link between HIF activity and PlGF expression has never been demonstrated.
Due to the central role of ECs in angiogenic switch, we decided to investigate whether PlGF expression is effectively modulated by hypoxia via HIF in human and mouse ECs. Studies previously accomplished excluded a direct role of the HRE located in Plgf promoter. We decided to investigate if this modulation could be mediated by additional intragenic HREs, taking into account that alteration of chromatin structure has an important role in the response to hypoxia.30
In this frame, CpG methylation and histone H3 and H4 acetylation have been examined in order to identify whether epigenetic changes occur for hypoxia-induced Plgf transcriptional regulation. The data here presented highlight for the first time a direct functional link between HIF-1α and PlGF overexpression in hypoxic condition.
Results
Hypoxia increases PlGF expression in HUVEC and H5V cells
To evaluate the impact of hypoxia on PlGF expression, HUVEC or H5V31 cells were exposed to 1% O2. At 3, 6, 12 and 24 h, RNA was extracted to quantify the expression of PlGF and, as control, of VEGF-A by qRT-PCR. In HUVECs, no change of PlGF was detected until 6 h, while the VEGF mRNA was already increased at 3 h, compared with normoxic condition (at 6 h, P < 0.05). At 12 h a significant increase (~6-folds, P < 0.0005) of PlGF mRNA was observed and maintained up to 24 h (~3.5-fold, P < 0.005). The mRNA level of VEGF-A raised until 12 h (~8-fold increase, P < 0.0005), and at 24 h the upregulation is still evident (~5.5-fold, P < 0.005) (Fig. 1A). In mouse endothelial cells, the increase of PlGF showed a trend similar to that of VEGF-A, with a peak of expression at 12 h, as in human endothelial cells (~7.6- and ~9.6-fold increase compared with normoxic condition, respectively, P < 0.0005). These levels of expression were maintained also at 24 h (~8.6- and ~10.5-fold increase, respectively, P < 0.0005) (Fig. 1B).

Figure 1. Overexpression of PlGF mRNA and protein in ECs exposed to low oxygen tension. Time dependent PlGF and VEGF-A mRNA expression determined by qRT-PCR in HUVEC (A) and mouse H5V (B) ECs exposed to 1% O2. PlGF and VEGF mRNA levels in hypoxic conditions were compared with the normoxic levels and normalized against RPL32, in human cells, and Rpl13a, in mouse cells. Data are expressed as fold induction compared with normoxic condition and represent the mean ± SEM of two independent experiments performed in triplicate. *P < 0.0005; §P < 0.005; ¶P < 0.001; #P < 0.05 vs normoxic condition. Determination of PlGF concentration in the culture medium of HUVEC (C) and H5V (D) exposed to 1% O2 for 24 h, as assessed by sandwich ELISA. As control, cells cultured in normoxic condition were used. Data represent the mean ± SEM of two independent experiments performed in triplicate. *P = 0.0013 and §P = 0.0008 vs normoxic condition. Western blot analysis of human HIF-1α and HIF-2α (E) and of mouse HIF-1α (F) performed on HUVEC and H5V protein extracts, after cells exposure to 1% O2 for 24 h. Densitometry analyses are reported as percentage of arbitrary densitometry units (ADU) of the ratio of HIF-1α or HIF-2α and β-Tubulin, assigning the value of 100 to the relative ratio obtained in normoxic condition.
In order to verify whether a protein increase corresponded to the PlGF mRNA overexpression, ELISA assays were performed to quantify human and mouse PlGF secreted in the culture medium. An increase of ~4.6- (P = 0.0013) and ~11.5-fold (P = 0.0008) of PlGF protein was detectable after 24 h of hypoxia in HUVEC and H5V medium culture, respectively, compared with normoxic condition (Fig. 1C and D). The effectiveness of hypoxia condition was confirmed by the increase of HIF-1α in both cell lines, as well as of HIF-2α in HUVECs, as assessed by western blot analysis (Fig. 1E and F).
These data clearly indicate that hypoxic condition induces the upregulation of mRNA and protein of human and mouse PlGF.
Since four isoforms of human PlGF have been described (PlGF 1 to 4),1 differently from mouse in which a single PlGF form corresponding to human PlGF-2 has been identified, qRT-PCR was performed to evaluate whether hypoxia could differentially modulate human PlGF isoforms.32 The low oxygen tension significantly upregulated the two main isoforms PlGF-1 and PlGF-2, with a preference for the soluble PlGF-1 isoform, starting from 12 h of exposure to 1% O2 (Table 1).
Table 1. Time-dependent differential modulation of human PlGF isoforms by hypoxia.
| Hours | PlGF -1 | PlGF-2 | PlGF-3 | PlGF-4 |
| 0 | 1 | 1 | 1 | 1 |
| 3 | 0.96 ± 0.04 | 1.43 ± 0.12 | 0.90 ± 0.02 | 1.18 ± 0.20 |
| 6 | 0.71 ± 0.12 | 1.10 ± 0.13 | 1.14 ± 0.22 | 1.21 ± 0.24 |
| 12 | 3.86 ± 0.09 * | 2.73 ± 0.18 * | 1.18 ± 0.27 | 1.00 ± 0.03 |
| 24 | 1.76 ± 0.28 § | 1.71 ± 0.20 § | 0.97 ± 0.03 | 1.07 ± 0.07 |
Data are expressed as fold induction compared with normoxic condition and represent the mean ± SEM of two independent experiments performed in triplicate. In bold the values indicating the upregulation of PlGF-1 and PlGF-2 after 12 and 24 h of exposure to 1% O2. *P < 0.005 and §P < 0.05.
Hypoxia does not change DNA methylation status at human Plgf promoter
In order to investigate a possible role of DNA methylation in hypoxia-induced PlGF upregulation, we compared the cytosine methylation profile of Plgf promoter region under hypoxic and normoxic conditions. A CpG island overlapping the exon 1 (nucleotides -388/+156) was identified on human Plgf gene (Fig. 2A). Genomic DNA extracted from HUVECs grown in normoxic or in hypoxic conditions was subjected to bisulfite sequencing.33 The analysis of 20 clones for each condition showed general low CpG site methylation in both samples, without any significant change in hypoxia compared with normoxic condition (Fig. 2B).

Figure 2. Methylation status of human Plgf promoter in normoxic and hypoxic conditions. (A) Schematic representation of the CpG island located in human Plgf promoter at position -388/+156 with respect to the transcription start site. (B) Sequence analysis of bisulfite-treated genomic DNA from HUVECs exposed (bottom) or not (top) to hypoxia for 9 h. Twenty clones for each group have been analyzed. Each row indicates a single clone, whereas each column represents a single CpG site. White circles indicate unmethylated cytosines, whereas black circles represent the methylated ones.
Hypoxia determines Plgf histone acetylation changes
To establish whether hypoxia might induce changes in the chromatin structure of the Plgf gene, we measured histone H3 and histone H4 acetylation levels, marks of permissive chromatin. Ten different regions spanning Plgf gene were analyzed by ChIP assay (Fig. 3A; Table 2). Under hypoxic condition, we found an increase of H3 and H4 acetylation in Plgf promoter region 6, as assessed by qRT-PCR (P < 0.05 vs normoxic control) (Fig. 3A and B). No increase in histone acetylation was observed in promoter regions 4 and 5, where putative HREs, already reported by other authors, are present (not shown).21,34 Surprisingly, a strong enrichment of H3 and H4 acetylation was instead observed in region 7, located in the second Plgf intron (P < 0.005 for histone H3 and P < 0.01 for histone H4, vs normoxic control) (Fig. 3B). The sequence analysis of this region evidenced the presence of three previously unknown HRE elements centered in position +2324, +2407 and +2422 (Fig. 3A). The first one is also flanked by an additional consensus sequence frequently associated with functional HRE sites (Table 3).9,35

Figure 3. Histone acetylation enrichment and HIF-1α binding to human Plgf second intron. (A) Schematic representation of part of human Plgf gene. Regions analyzed by ChIP analysis are indicated by numerated black rectangles. Gray boxes represent exons. An arrow indicates the transcription start site. Black ovals indicate the putative HRE located in the promoter and in the second intron, identified by numbers indicating the central bases (G) of HRE core consensus sequence, with respect to the transcription start site. ChIP analysis for H3 (AcH3) and H4 (AcH4) acetylation (B) or for HIF-1α or HIF-2α (C), performed starting from chromatin sample from HUVEC exposed to hypoxia for 9 h. As control, species matched IgG were used. Data obtained by qRT-PCR are expressed as enrichment of chromatin-associated DNA fragments immunoprecipitated by specific antibody compared with input (% Input) and represent the mean ± SEM of two independent experiments performed in triplicate. *P < 0.05; §P < 0.01 and #P < 0.005 vs normoxic controls. Numbers indicate the amplified regions of Plgf gene, as reported in (A). V-PC and V-NC represent Vegf-a positive and negative controls, respectively, containing or not active HRE. V-2-Int, indicates Vegf-a second intron region containing a putative HRE.
Table 2. Regions of Plgf and Vegf-a genes analyzed by ChIP.
| Target | Amplicon # | Region | HRE |
| hPlgf | 1 | -9730 / -9570 | |
| 2 | -4903 / -4747 | ||
| 3 | -3561 / -3419 | ||
| 4 | -1702/ -1554 | putative (-1654) | |
| 5 | -1168 / -1022 | putative (-1047) | |
| 6 | -350 / -176 | ||
| 7 | +2208 / +2282 | putative (+2324, +2407, +2422) | |
| 8 | +8199 / +8305 | ||
| 9 | +13445 / +13578 | ||
| 10 | +15976 / +16108 | ||
| hVegf | V-PC | -1005 / -868 | active (-978) |
| V-NC | -1762 / -1364 | absent | |
| V-2-Int | +5161 / +5391 | ||
| mPlgf | 11 | -1945 / -1812 | putative (-3100) |
| 12 | -1001 / -889 | ||
| 13 | -314 / -202 | ||
| 14 | +1740 / +1841 | putative (+1767, +2168) | |
| 15 | +3892 / +4011 | putative (+4030) | |
| 16 | +6584 / +6708 | putative (+6593) | |
| mVegf | V-PC | -944 / -831 | active (-899) |
| V-NC | -1903 / -2040 | absent |
The numbers in the region column indicated the area of genes analyzed with respect to transcription start site. In bold are indicated regions in which known and discovered active HRE are located. Numbers in HRE column refer to center position of HREs respect to transcription start site. V-PC, VEGF-A positive control; V-NC, VEGF-A negative control, V-2-Int, region of the second intron of Vegf-a gene where a putative HRE is located.
Table 3. HREs located in the second intron of PlGF genes.
| Species | HRE +2324 | HRE +2407 | HRE +2422 |
| Human | aaGACGTGCa aagtggcCAC ACACc | acaCGCGTGa Tag | atcTgCGTGC Tgg |
| Rhesus | aaGACGTGCa gagcggcCAC ACgCc | acaCGCGTGa Tag | atcTgCaTGC Tgg |
| Dog | ggGACGTGCa gcaaagcCAC ACgCc | acaCtCGTGa Tag | ctcCgaaaGC Tgg |
| Mouse | gaGACaTGGa ggatggcCAC AtACc | acaCAgaTaa tag | ttctT—GGCctgg |
The positions of HREs are referred to human gene and indicate the central position of common core of putative HRE consensus, respect to the transcription start site. HRE consensus DNA sequence (G/T/C) A/G CGTG (C/G) (T/G/C) and the additional sequence CACACA G/C often associated with functional HRE sites are in uppercase and underlined (underlined lowercase indicates mismatched bases). In rhesus all three putative HRE are conserved (position +1965, +2048, +2063), in dog only the first two (position +1970, +2054). In mouse only the first one (+2168) is conserved but an additional HRE (agGACGTGaTcg) centered at position +1767 has been observed.
A comparative analysis among different species revealed the conservation of one or more HREs in the same area of the second intron of rhesus, dog and mouse Plgf gene. In all species, at least one HRE resulted flanked by the additional consensus sequence (Table 3). Interestingly, also in the second intron of Vegf-a gene, a HRE was identified centered at position +5184.
HIF-1α, but not HIF-2α, binds to the HREs on the human Plgf second intron
ChIP analyses using anti-HIF-1α and anti-HIF-2α antibodies were performed to establish whether HIFs are involved in the binding of HREs located in the second intron of Plgf. We also investigated two Plgf promoter regions where two known putative HRE elements are located (regions 4 and 5, Fig. 3A; Table 2). As positive control, the canonical Vegf-a promoter area including an active HRE was analyzed (V-CP), whereas a region lacking HRE sites (V-NC) was amplified as negative control (Table 2).15,36 Moreover we also analyzed the Vegf-a second intron region where, as in the Plgf gene, a HRE is located (V-2-Int).
A direct binding of HIF-1α to the H3 and H4 hyperacetylated region 7 of Plgf second intron was observed, as well as to the Vegf-a promoter area (P < 0.005 vs normoxic controls) (Fig. 3C). Conversely, the two putative HREs located in the Plgf promoter regions 4 and 5 not differentially H3 and H4 acetylated, were not recognized by HIF-1α and/or HIF-2α. Interestingly, while both HIF-1α and HIF-2α were able to bind the HRE in V-PC region (P < 0.01 and 0.05 vs normoxic control),37 HIF-2α did not interact with Plgf second intron HRE (Fig. 3C).
Finally, the HRE located in the second intron of Vegf-a was not recognized by HIFs, indicating that the involvement of this intragenic site is a peculiarity of Plgf gene.
Silencing of HIF-1α abrogates human PlGF overexpression in hypoxic condition
To demonstrate a direct functional link between HIF-1α and PlGF overexpression in hypoxic condition, we knocked down HIF-1α by introducing specific siRNA in HUVECs. As control, HUVECs transfected with siRNA for HIF-2α or non-targeting (NT) siRNA, as well as mock transfected cells, were used. After confirming by western blot analysis that HIF-1α or HIF-2α were efficiently silenced (Fig. 4C), we observed the abrogation of hypoxia-mediated PlGF overexpression only in cells transfected with specific siRNA for HIF-1α, whereas silencing of HIF-2α or transfection of NT-siRNA were ineffective (Fig. 4A and B).
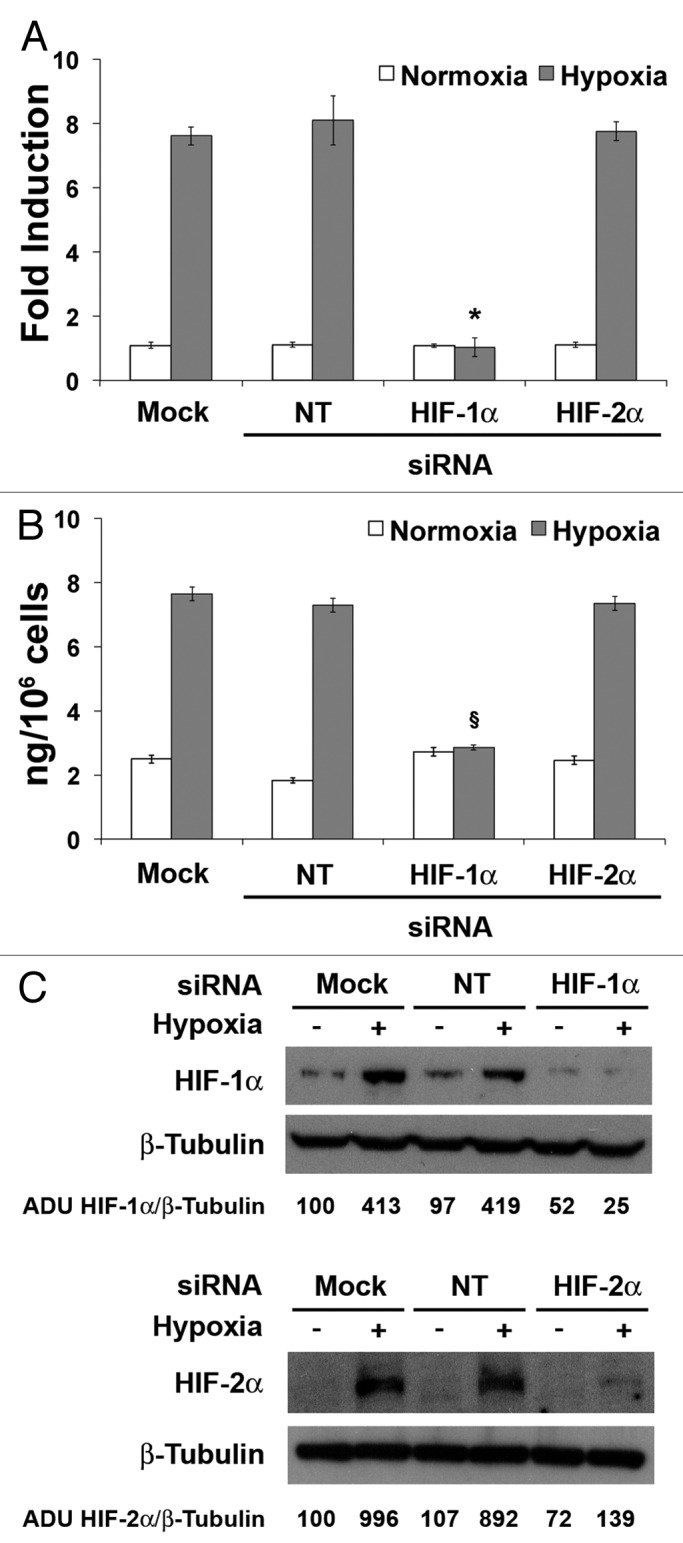
Figure 4. Knock down of HIF-1α, but not that of HIF-2α, inhibits hypoxia-induced human PlGF expression in HUVECs. (A) qRT-PCR analysis of human PlGF mRNA after silencing of HIF-1α or HIF-2α in HUVECs exposed to 1% O2 for 9 h and determination of PlGF concentration by sandwich ELISA (B) in HUVEC culture medium harvested after silencing of HIF-1α or HIF-2α and 24 h of exposure to 1% O2. As control, cells transfected with non-targeting (NT) siRNA and mock-transfected cells were used. Data obtained by qRT-PCR are expressed as fold induction and represent the mean ± SEM of two independent experiments performed in triplicate. Data obtained by ELISAs represent the mean ± SEM of two independent experiments performed in triplicate. *P < 0.0005 and §P < 0.001 vs controls and HIF-2α in hypoxia. (C) Western blot analysis of human HIF-1α (up) and HIF-2α (down) performed on HUVEC protein extracts, after cells exposure to 1% O2 for 24 h. β-Tubulin detection on the same filter was used for normalization. Densitometry analyses are reported as percentage of arbitrary densitometry units (ADU) of the ratio of HIF-1α or HIF-2α and β-Tubulin, assigning the value of 100 to the relative ratio obtained in mock-transfected cells in normoxic condition.
These data clearly demonstrate the direct role of HIF-1α in the modulation of PlGF expression under hypoxic condition.
HIF-1α also modulates the hypoxia-induced overexpression of mouse Plgf gene
To assess whether hypoxia-mediated PlGF upregulation is conserved in mouse, we examined histone acetylation and HIF-1α binding in murine Plgf gene by ChIP analysis. For mouse Plgf gene, a total of six regions were analyzed to monitor the level of H3 and H4 acetylation, covering all putative HRE sequences detected along the entire gene (Fig. 5A; Table 2). The positive (V-PC) and negative (V-NC) controls were represented by regions of mouse Vegf-a promoter containing or not functional HREs.38 An increase in histone acetylation was observed in the promoter region 13, and again only in the region of second intron (region 14) where a HRE, similar to the first one observed in human Plgf second intron, is located (Fig. 5B; Table 3). ChIP analysis performed with anti-HIF-1α confirmed a direct binding exclusively to the HRE in the second intron (Fig. 5C).

Figure 5. HIF-1α is directly involved in the hypoxia-modulation of mouse Plgf expression in H5V cells by HRE binding located on second intron. (A) Schematic representation of part of mouse Plgf gene. Areas analyzed by ChIP analysis are indicated by numbered black rectangles. Gray boxes represent exons. An arrow indicates the transcription start site. Black ovals indicate the putative HRE located along the gene. ChIP analysis for H3 (AcH3) and H4 (AcH4) acetylation (B) or for HIF-1α (C) performed starting from chromatin sample of H5V exposed to hypoxia for 12 h. As control, species matched IgG were used. Data obtained by qRT-PCR are expressed as enrichment of chromatin-associated DNA fragments immunoprecipitated by specific antibody compared with input (% Input) and represent the mean ± SEM of two independent experiments performed in triplicate. §P < 0.05; *P < 0.01 and #P < 0.005 vs normoxic control. Numbers indicate the amplified regions of Plgf gene, as reported in (A). V-PC and V-NC represent Vegf-a positive and negative controls, respectively, containing or not active HRE. (D) qRT-PCR analysis of mouse PlGF mRNA after silencing of HIF-1α in H5V exposed to 1% O2 for 12 h and determination of PlGF concentration by sandwich ELISA (E) in H5V culture medium harvested after silencing of HIF-1α and 24 h of exposure to 1% O2. As control, cells transfected with non-targeting (NT) siRNA and mock-transfected cells were used. Data obtained by qRT-PCR are expressed as fold induction and represent the mean ± SEM of two independent experiments performed in triplicate. Data obtained by ELISAs represent the mean ± SEM of two independent experiments performed in triplicate. *P < 0.0001 and §P < 0.0005 vs NT and mock in hypoxia. (F) Western blot analysis of mouse HIF-1α performed on H5V protein extracts, after cells exposure to 1% O2 for 24 h. β-Tubulin detection on the same filter was used for normalization. Densitometry analyses are reported as percentage of arbitrary densitometry units (ADU) of the ratio of HIF-1α and β-Tubulin, assigning the value of 100 to the relative ratio obtained in mock-transfected cells in normoxic condition.
Finally, silencing of HIF-1α in H5V cells, confirmed by western blot analysis (Fig. 5F), fully prevent the hypoxia-induced upregulation of mouse PlGF mRNA and protein, as assessed by qRT-PCR and ELISA, whereas the NT-siRNA was ineffective (Fig. 5D and E).
Discussion
PlGF is redundant for developmental and physiological processes but plays an important role in different pathological contexts in which angiogenesis and inflammation are involved through endothelial stimulation and bone marrow-derived cells recruitment and activation.39-41 Moreover, since PlGF is able to act on different cell types, thanks to the broad expression of its specific receptor VEGFR-1,19,42 the list of biological processes in which its pleiotropic activities are involved is still growing.1,3
Considering this scenario, the control of PlGF expression levels is of great interest, as confirmed also by many preclinical models that clearly showed how PlGF deregulation resulted directly correlated with pathological conditions. For these reasons, we decided to investigate the molecular mechanism governing the modulation of PlGF by oxygen tension. We focused our interest on ECs because of their central role in new vessels formation.
Despite several reports indicate that hypoxia, as well as the overexpression of HIF-1α, are responsible in vitro and in vivo for a positive modulation of PlGF,2,25-29 the direct involvement of the HIF transcriptional complex has not been evidenced with classical transcriptional studies on Plgf promoter.21,24,34 Considering that it has recently been reported that epigenetic regulation of chromatin structure plays an important role in defining the response to hypoxia,30 we decided to investigate the role of epigenetic modifications, such as DNA methylation and histone acetylation, in the modulation of PlGF expression under hypoxic stimuli.
First, we assessed that exposure to 1% O2 of human and mouse ECs is sufficient to unambiguously induce an increase of PlGF mRNA and protein. Then, we verified whether hypoxia might influence the DNA methylation status of the Plgf promoter. Detailed CpG methylation profile evidenced hypomethylation at Plgf promoter without significant changes between normoxic and hypoxic conditions. Previous reports indicated that low CpG methylation level at the HREs binding sites is required to allow gene transcriptional induction.43,44 However, our findings suggest a role for specific histone modifications rather than DNA methylation changes in the hypoxia-mediated PlGF induction. To date, only one previous study has investigated Plgf promoter methylation.45 Interestingly, this study reported that hypermethylation of the Plgf promoter is associated with PlGF downregulation in human lung and colon carcinoma tissues, as well as in correspondent cancer cell lines in normoxic conditions, suggesting that the methylation of the Plgf promoter may change in different cell and tissue contexts.
We found that alteration of chromatin structure may influence the modulation of PlGF expression under hypoxic condition. Analyzing the H3 and H4 acetylation along the Plgf gene we observed an enrichment of histone acetylation in the second intron, in addition to the expected increase in the promoter. Moreover, in human Plgf second intron, three putative HREs never described before were found close to the region showing histone acetylation increase. The first one, centered at position +2324 with respect to the transcriptional start site, is also followed by a second consensus sequence often associated to the active HRE (Table 3).9,35 We then confirmed the evolutionary conservation of one or more HREs together with the second consensus sequence in the second intron of Plgf among different species, thus indicating a functional role of these regions in PlGF regulation. Consistently, in hypoxic conditions, HIF-1α exclusively interacts with the second intron of human and mouse Plgf genes.
Moreover, we evaluated whether an involvement of HIF-2α occurred because, differently from HIF-1α that is ubiquitously expressed, HIF-2α is expressed in a restricted number of cell types among which ECs are included.46,47 No direct binding of HIF-2α to the second Plgf intron, or other regions of the gene, was detected.
The absence of an active role of HIF-2α was confirmed by silencing experiments. Indeed, the specific silencing of HIF-2α did not affect the hypoxia-mediated upregulation of PlGF. Conversely, HIF-1α silencing fully abrogates upregulation of PlGF mRNA and protein in both EC lines analyzed, demonstrating for the first time its direct role in this biological process, at least in ECs.
The involvement of MTF-1 in the hypoxic modulation of PlGF expression has been demonstrated in H-ras transformed mEFs; however, the same cells transformed with SV40 large T antigen were unresponsive.21 Moreover, the activity of NF-κB on PlGF has been reported in HEK-293 cells overexpressing NF-κB p65.24 It is important to note that both H-ras and NF-κB positively modulate HIF-1α expression.48-50 Therefore, the upregulation of PlGF in these two peculiar cellular contexts was probably due, at least in part, to the direct activation of HIF-1α. Nonetheless, a strict collaboration between these three modulators of gene expression is probably required for a fully modulation of PlGF in hypoxic conditions.
Our data corroborate also the view that increased level of PlGF might contribute to the tumor escape strategy that follows anti-angiogenic therapy targeting VEGF-A or RTKs, included VEGFR-2.51-54 Indeed, it is well known that these therapies induce an increase of the hypoxic status of the tumor that, as confirmed by our data, positively affects PlGF expression. On the other hand, it has been reported that in the peritumor area of human hepatocellular carcinoma, the level of PlGF was significantly increased and correlated with augmented levels of HIF-1α.55 Finally, since PlGF also positively modulates HIF-1α expression in ECs,56 a positive loop may be established to maintain high levels of PlGF expression when required.
In conclusion, our data demonstrate that epigenetic changes, such as histone acetylation, are involved in the modulation of PlGF expression under hypoxic conditions in ECs, possibly by determining the exposition of a HRE located in the second intron of Plgf specifically recognized by HIF-1α. There is growing evidence that formation of chromatin loops mediated by transcription factors allows the interaction between regions far on linear DNA (i.e., promoter and gene body), thus providing an efficient control of gene expression.57 In addition, the transcription process appears to be compartmentalized in factories occupying distinct loci in the nuclear space and genes are thought to be looped out from chromosomes territories toward these loci.58 In line with these findings, it is reasonable to hypothesize that HIF-1α binding mediates a spatial association of the transcriptional start site and the regulatory site in the second intron. Further studies will be necessary to investigate these aspects and to confirm the three-dimensional chromatin structure of Plgf regulatory regions.
Materials and Methods
Cell culture
Human umbilical vein endothelial cells (HUVECs, Clonetics) were cultured in endothelial basal medium (EBM-2) supplemented with endothelial growth factors (EGM-2 bullet kit, Cambrex). HUVECs at passages 4–7 were used for all the experiments. Murine-immortalized heart microvascular endothelial cell line (H5V)31 was cultured in DMEM, supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine and standard concentration of antibiotics (Euroclone). For exposure to hypoxia, sub-confluent cells were placed in an appropriate incubator at 1% oxygen concentration. As control, sub-confluent cells were cultured in normoxic condition.
RNA preparation and quantitative real-time-PCR (qRT-PCR)
RNA was isolated using Trizol reagent (Invitrogen). The first strand of cDNA was obtained by reverse-transcription using Quantitect RT Kit (Qiagen). qRT-PCR was performed using SYBR green quantitative PCR on CFX96TM Real Time PCR Detection Systems (BioRad). The annealing temperatures were 58 °C for human gene and 62 °C for mouse gene. The primers were: hPlGF upper (559) ATGTTCAGCC CATCCTGTGT; lower (759) CTTCATCTTC TCCCGCAGAG - hVEGF-A upper (1130) AGGGCAGAAT CATCACGAAG; lower (1357) ATCCGCATAA TCTGCATGGT - hRPL-32 upper (324) AGTTCCTGGT CCACAACGTC; lower (519) TGCACATGAG CTGCCTACTC – mPlGF upper (325) GCTGGTCATG AAGCTGTTC; lower (454) ACCCCACACT TCGTTGAAAG - mVEGF-A upper (642) CAGGCTGCTG TAACGATGAA; lower (781) GCATTCACAT CTGCTGTGCT - mRpl13a upper (345) CCCTCCACCC TATGACAAGA; lower (565) CTGCCTGTTT CCGTAACCTC. The numbers identify the first nucleotide (5′ position) of upper primers or the last one (3′ position) of lower primers with respect to the transcription start site. The PlGF and VEGF expression levels in hypoxic condition were calculated with respect to the normoxic level and normalized against RPL32, in human cells, and Rpl13a, in mouse cells. Human PlGF isoforms (PlGF 1 to 4) were detected using specific primers as previously described.32
Western blot analysis
Western blot analyses were performed. with antibodies against HIF-1α (1:200, Santa Cruz Biotechnology), HIF-2α (1:500, Novus Biologicals) and β-Tubulin (1:1000, Santa Cruz Biotechnology) using standard protocols. Densitometry analyses were performed using ImageQuant 5.2 software (Molecular Dynamics). Values of arbitrary densitometry units (%) were calculated as ratio of HIF-1α or HIF-2α respect to β-Tubulin, assigning the value of 100 to ratio obtained in normoxic condition.
ELISA assay
All the reagents used in ELISA were from R&D Systems (Minneapolis, MN). The assays were performed as described elsewhere.20,59
Bisulfite analysis
Genomic DNA was isolated from HUVEC exposed (9 h) or not to hypoxia, using a Purelink Genomic DNA kit (Invitrogen). 1µg of genomic DNA was subjected to bisulfite modification using the Epitect Bisulfite kit (Qiagen). Nested PCR strategy was adopted to amplify the target region from bisulfite modified genomic DNA. The conditions for the 1st PCR were: 95 °C for 30s, 55 °C for 45s, and 72 °C, for 1 min - 25 cycles, while for the 2nd PCR: 95 °C for 30s, 55 °C for 45s, and 72 °C for 45s - 30 cycles. The primers used were for the 1st PCR: upper (-309) GATTTTTGGA TGTTTTTATT TTAGGTGAT; lower (+315) AAAAAAAACC ACCATACTCA TCCC and for the 2nd PCR: upper (-264) GTAGGGTTGT GGGTTTTGTG G; lower (+223) CCTCCCTCAC TACTACCCC. The numbers identify the first nucleotide (5′ position) of upper primers or the last one (3′ position) of lower primers with respect to the transcription start site. PCR products were cloned into the pCR2.1 TOPO vectors (Invitrogen). The sequence of 20 clones for each group has been performed using M13 forward and reverse primers.
Chromatin immunoprecipitation (ChIP) assay
ChIP experiments were essentially performed as previously described.60 Briefly, HUVEC and H5V cells were exposed to hypoxia (1% O2) for 9 or 12 h, respectively. 1 × 107 cells were fixed with 1% formaldehyde. After cross-linking, chromatin was isolated and subjected to sonication, resulting in 200–1000 bp DNA fragments. After immunoprecipitation with anti-acetylated histone H3 (Upstate 06-599), and H4 (Upstate 06-866), or anti-HIF-1α (Santa Cruz Biotechnology) and anti-HIF-2α (Novus Biologicals), immunocomplexes were purified by co-precipitation with protein A-Sepharose (GE Healthcare). Species matched IgG were used as negative control. The amount of recovered DNA was determined and the quantification of chromatin-immunoprecipitated DNA fragments was performed by qRT-PCR using the primers listed in Table S1. The enrichment of DNA was calculated in terms of % input = 2-ΔCt × 100, where ΔCt (threshold cycle) is determined by CtIP sample - CtInput and 100 refers to the input being 1% of the chromatin amount exposed to IP.
Silencing experiments
HUVECs and H5V were plated into 6-well plates, at 3 × 105 and 5 × 105 cells/well density, respectively. 24 h later, cells were transfected with 150nM of siRNA for human HIF-1α, human HIF-2α, or mouse HIF-1α and, as control, with non-targeting siRNA 2 (siGENOME SMART pool, Dharmacon), using nucleofection technology (Amaxa). Sixteen hours later, HUVEC and H5V cells were exposed to hypoxia (1% O2) for 9 or 12 h, respectively, or cultured in normoxic condition for the same time. Therefore, RNA was extracted and gene expression was quantified as described before. PlGF concentration in the culture medium was evaluated after 24 h of exposure to hypoxia.
Statistical Analysis
Results are expressed as mean ± standard error of the mean (SEM), with P values < 0.05 considered statistically significant. Differences between groups were compared by Student’s t test and two-tailed P values are reported.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Anna Maria Aliperti for manuscript editing. This work was supported by AIRC (Associazione Italiana Ricerca sul Cancro, grant number IG 11420) and the Italian Ministry of Scientific Research (Grant MERIT RBNE08YFN3_006) to S.D.F. and by UE Initial Training Network Project n. 238242 “DISCHROM” and the Epigenomics Flagship Project Epigen, Italian Ministry of Scientific Research – National Research Council, to M.D.E. and M.R.M.
Glossary
Abbreviations:
- HIF-1α
hypoxia inducible factor-1α
- PlGF
Placental Growth Factor
- VEGF
vascular endothelial growth factor
- HRE
hypoxia responsive element
- EC
endothelial cell
- VEGFR-1
VEGF Receptor-1
- MTF-1
metal responsive transcription factor 1
- NF-κB
nuclear factor κB
- ChIP
chromatin immunoprecipitation
- HUVEC
human umbilical vein endothelial cell
References
- 1.De Falco S. The discovery of placenta growth factor and its biological activity. Exp Mol Med. 2012;44:1–9. doi: 10.3858/emm.2012.44.1.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F, Devy L, Beck H, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7:575–83. doi: 10.1038/87904. [DOI] [PubMed] [Google Scholar]
- 3.Dewerchin M, Carmeliet P. PlGF: a multitasking cytokine with disease-restricted activity. Cold Spring Harb Perspect Med 2012; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–87. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009;19:329–37. doi: 10.1016/j.semcancer.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/S0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 8.Maxwell PH, Ratcliffe PJ. Oxygen sensors and angiogenesis. Semin Cell Dev Biol. 2002;13:29–37. doi: 10.1006/scdb.2001.0287. [DOI] [PubMed] [Google Scholar]
- 9.Simon MP, Tournaire R, Pouyssegur J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J Cell Physiol. 2008;217:809–18. doi: 10.1002/jcp.21558. [DOI] [PubMed] [Google Scholar]
- 10.Eyries M, Siegfried G, Ciumas M, Montagne K, Agrapart M, Lebrin F, Soubrier F. Hypoxia-induced apelin expression regulates endothelial cell proliferation and regenerative angiogenesis. Circ Res. 2008;103:432–40. doi: 10.1161/CIRCRESAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- 11.Michiels C, Arnould T, Remacle J. Endothelial cell responses to hypoxia: initiation of a cascade of cellular interactions. Biochim Biophys Acta. 2000;1497:1–10. doi: 10.1016/S0167-4889(00)00041-0. [DOI] [PubMed] [Google Scholar]
- 12.Nomura M, Yamagishi S, Harada S, Hayashi Y, Yamashima T, Yamashita J, Yamamoto H. Possible participation of autocrine and paracrine vascular endothelial growth factors in hypoxia-induced proliferation of endothelial cells and pericytes. J Biol Chem. 1995;270:28316–24. doi: 10.1074/jbc.270.47.28316. [DOI] [PubMed] [Google Scholar]
- 13.Yonekura H, Sakurai S, Liu X, Migita H, Wang H, Yamagishi S, Nomura M, Abedin MJ, Unoki H, Yamamoto Y, et al. Placenta growth factor and vascular endothelial growth factor B and C expression in microvascular endothelial cells and pericytes. Implication in autocrine and paracrine regulation of angiogenesis. J Biol Chem. 1999;274:35172–8. doi: 10.1074/jbc.274.49.35172. [DOI] [PubMed] [Google Scholar]
- 14.Adini A, Kornaga T, Firoozbakht F, Benjamin LE. Placental growth factor is a survival factor for tumor endothelial cells and macrophages. Cancer Res. 2002;62:2749–52. [PubMed] [Google Scholar]
- 15.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerber HP, Condorelli F, Park J, Ferrara N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem. 1997;272:23659–67. doi: 10.1074/jbc.272.38.23659. [DOI] [PubMed] [Google Scholar]
- 17.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 18.Tugues S, Koch S, Gualandi L, Li X, Claesson-Welsh L. Vascular endothelial growth factors and receptors: anti-angiogenic therapy in the treatment of cancer. Mol Aspects Med. 2011;32:88–111. doi: 10.1016/j.mam.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Cao Y. Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci Signal. 2009;2:re1. doi: 10.1126/scisignal.259re1. [DOI] [PubMed] [Google Scholar]
- 20.Tarallo V, Vesci L, Capasso O, Esposito MT, Riccioni T, Pastore L, Orlandi A, Pisano C, De Falco S. A placental growth factor variant unable to recognize vascular endothelial growth factor (VEGF) receptor-1 inhibits VEGF-dependent tumor angiogenesis via heterodimerization. Cancer Res. 2010;70:1804–13. doi: 10.1158/0008-5472.CAN-09-2609. [DOI] [PubMed] [Google Scholar]
- 21.Green CJ, Lichtlen P, Huynh NT, Yanovsky M, Laderoute KR, Schaffner W, Murphy BJ. Placenta growth factor gene expression is induced by hypoxia in fibroblasts: a central role for metal transcription factor-1. Cancer Res. 2001;61:2696–703. [PubMed] [Google Scholar]
- 22.Oura H, Bertoncini J, Velasco P, Brown LF, Carmeliet P, Detmar M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood. 2003;101:560–7. doi: 10.1182/blood-2002-05-1516. [DOI] [PubMed] [Google Scholar]
- 23.Selvaraj SK, Giri RK, Perelman N, Johnson C, Malik P, Kalra VK. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood. 2003;102:1515–24. doi: 10.1182/blood-2002-11-3423. [DOI] [PubMed] [Google Scholar]
- 24.Cramer M, Nagy I, Murphy BJ, Gassmann M, Hottiger MO, Georgiev O, Schaffner W. NF-kappaB contributes to transcription of placenta growth factor and interacts with metal responsive transcription factor-1 in hypoxic human cells. Biol Chem. 2005;386:865–72. doi: 10.1515/BC.2005.101. [DOI] [PubMed] [Google Scholar]
- 25.Yamakawa M, Liu LX, Date T, Belanger AJ, Vincent KA, Akita GY, Kuriyama T, Cheng SH, Gregory RJ, Jiang C. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res. 2003;93:664–73. doi: 10.1161/01.RES.0000093984.48643.D7. [DOI] [PubMed] [Google Scholar]
- 26.Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, Rowan A, Yan Z, Campochiaro PA, Semenza GL. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–81. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 27.Torry RJ, Tomanek RJ, Zheng W, Miller SJ, Labarrere CA, Torry DS. Hypoxia increases placenta growth factor expression in human myocardium and cultured neonatal rat cardiomyocytes. J Heart Lung Transplant. 2009;28:183–90. doi: 10.1016/j.healun.2008.11.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sung CY, Son MW, Ahn TS, Jung DJ, Lee MS, Baek MJ. Expression of placenta growth factor in colorectal carcinomas. J Korean Soc Coloproctol. 2012;28:315–20. doi: 10.3393/jksc.2012.28.6.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snuderl M, Batista A, Kirkpatrick ND, Ruiz de Almodovar C, Riedemann L, Walsh EC, Anolik R, Huang Y, Martin JD, Kamoun W, et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell. 2013;152:1065–76. doi: 10.1016/j.cell.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117:e207–17. doi: 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garlanda C, Parravicini C, Sironi M, De Rossi M, Wainstok de Calmanovici R, Carozzi F, Bussolino F, Colotta F, Mantovani A, Vecchi A. Progressive growth in immunodeficient mice and host cell recruitment by mouse endothelial cells transformed by polyoma middle-sized T antigen: implications for the pathogenesis of opportunistic vascular tumors. Proc Natl Acad Sci U S A. 1994;91:7291–5. doi: 10.1073/pnas.91.15.7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultze A, Ben Batalla I, Riethdorf S, Bubenheim M, Yekebas E, Erbersdobler A, Reichelt U, Effenberger KE, Schmidt T, Izbicki JR, et al. VEGFR-1 expression levels predict occurrence of disseminated tumor cells in the bone marrow of patients with esophageal carcinoma. Clin Exp Metastasis. 2012;29:879–87. doi: 10.1007/s10585-012-9477-1. [DOI] [PubMed] [Google Scholar]
- 33.Matarazzo MR, De Bonis ML, Gregory RI, Vacca M, Hansen RS, Mercadante G, D’Urso M, Feil R, D’Esposito M. Allelic inactivation of the pseudoautosomal gene SYBL1 is controlled by epigenetic mechanisms common to the X and Y chromosomes. Hum Mol Genet. 2002;11:3191–8. doi: 10.1093/hmg/11.25.3191. [DOI] [PubMed] [Google Scholar]
- 34.Gobble RM, Groesch KA, Chang M, Torry RJ, Torry DS. Differential regulation of human PlGF gene expression in trophoblast and nontrophoblast cells by oxygen tension. Placenta. 2009;30:869–75. doi: 10.1016/j.placenta.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tazuke SI, Mazure NM, Sugawara J, Carland G, Faessen GH, Suen LF, Irwin JC, Powell DR, Giaccia AJ, Giudice LC. Hypoxia stimulates insulin-like growth factor binding protein 1 (IGFBP-1) gene expression in HepG2 cells: a possible model for IGFBP-1 expression in fetal hypoxia. Proc Natl Acad Sci U S A. 1998;95:10188–93. doi: 10.1073/pnas.95.17.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang HJ, Kim HJ, Rih JK, Mattson TL, Kim KW, Cho CH, Isaacs JS, Bae I. BRCA1 plays a role in the hypoxic response by regulating HIF-1alpha stability and by modulating vascular endothelial growth factor expression. J Biol Chem. 2006;281:13047–56. doi: 10.1074/jbc.M513033200. [DOI] [PubMed] [Google Scholar]
- 37.Maemura K, Hsieh CM, Jain MK, Fukumoto S, Layne MD, Liu Y, Kourembanas S, Yet SF, Perrella MA, Lee ME. Generation of a dominant-negative mutant of endothelial PAS domain protein 1 by deletion of a potent C-terminal transactivation domain. J Biol Chem. 1999;274:31565–70. doi: 10.1074/jbc.274.44.31565. [DOI] [PubMed] [Google Scholar]
- 38.Chavez JC, Baranova O, Lin J, Pichiule P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J Neurosci. 2006;26:9471–81. doi: 10.1523/JNEUROSCI.2838-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loges S, Schmidt T, Carmeliet P. “Antimyeloangiogenic” therapy for cancer by inhibiting PlGF. Clin Cancer Res. 2009;15:3648–53. doi: 10.1158/1078-0432.CCR-08-2276. [DOI] [PubMed] [Google Scholar]
- 40.Yoo SA, Yoon HJ, Kim HS, Chae CB, De Falco S, Cho CS, Kim WU. Role of placenta growth factor and its receptor flt-1 in rheumatoid inflammation: a link between angiogenesis and inflammation. Arthritis Rheum. 2009;60:345–54. doi: 10.1002/art.24289. [DOI] [PubMed] [Google Scholar]
- 41.Carnevale D, Lembo G. Placental growth factor and cardiac inflammation. Trends Cardiovasc Med. 2012;22:209–12. doi: 10.1016/j.tcm.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 42.de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–91. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 43.Wenger RH, Kvietikova I, Rolfs A, Camenisch G, Gassmann M. Oxygen-regulated erythropoietin gene expression is dependent on a CpG methylation-free hypoxia-inducible factor-1 DNA-binding site. Eur J Biochem. 1998;253:771–7. doi: 10.1046/j.1432-1327.1998.2530771.x. [DOI] [PubMed] [Google Scholar]
- 44.Rössler J, Stolze I, Frede S, Freitag P, Schweigerer L, Havers W, Fandrey J. Hypoxia-induced erythropoietin expression in human neuroblastoma requires a methylation free HIF-1 binding site. J Cell Biochem. 2004;93:153–61. doi: 10.1002/jcb.20133. [DOI] [PubMed] [Google Scholar]
- 45.Xu L, Jain RK. Down-regulation of placenta growth factor by promoter hypermethylation in human lung and colon carcinoma. Mol Cancer Res. 2007;5:873–80. doi: 10.1158/1541-7786.MCR-06-0141. [DOI] [PubMed] [Google Scholar]
- 46.Flamme I, Fröhlich T, von Reutern M, Kappel A, Damert A, Risau W. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1 alpha and developmentally expressed in blood vessels. Mech Dev. 1997;63:51–60. doi: 10.1016/S0925-4773(97)00674-6. [DOI] [PubMed] [Google Scholar]
- 47.Yamashita T, Ohneda K, Nagano M, Miyoshi C, Kaneko N, Miwa Y, Yamamoto M, Ohneda O, Fujii-Kuriyama Y. Hypoxia-inducible transcription factor-2alpha in endothelial cells regulates tumor neovascularization through activation of ephrin A1. J Biol Chem. 2008;283:18926–36. doi: 10.1074/jbc.M709133200. [DOI] [PubMed] [Google Scholar]
- 48.Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519–25. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- 49.Chun SY, Johnson C, Washburn JG, Cruz-Correa MR, Dang DT, Dang LH. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Mol Cancer. 2010;9:293. doi: 10.1186/1476-4598-9-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bocci G, Man S, Green SK, Francia G, Ebos JM, du Manoir JM, Weinerman A, Emmenegger U, Ma L, Thorpe P, et al. Increased plasma vascular endothelial growth factor (VEGF) as a surrogate marker for optimal therapeutic dosing of VEGF receptor-2 monoclonal antibodies. Cancer Res. 2004;64:6616–25. doi: 10.1158/0008-5472.CAN-04-0401. [DOI] [PubMed] [Google Scholar]
- 52.Bertolini F, Shaked Y, Mancuso P, Kerbel RS. The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat Rev Cancer. 2006;6:835–45. doi: 10.1038/nrc1971. [DOI] [PubMed] [Google Scholar]
- 53.Rini BI, Michaelson MD, Rosenberg JE, Bukowski RM, Sosman JA, Stadler WM, Hutson TE, Margolin K, Harmon CS, DePrimo SE, et al. Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J Clin Oncol. 2008;26:3743–8. doi: 10.1200/JCO.2007.15.5416. [DOI] [PubMed] [Google Scholar]
- 54.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu HX, Zhu XD, Zhuang PY, Zhang JB, Zhang W, Kong LQ, Wang WQ, Liang Y, Wu WZ, Wang L, et al. Expression and prognostic significance of placental growth factor in hepatocellular carcinoma and peritumoral liver tissue. Int J Cancer. 2011;128:1559–69. doi: 10.1002/ijc.25492. [DOI] [PubMed] [Google Scholar]
- 56.Patel N, Kalra VK. Placenta growth factor-induced early growth response 1 (Egr-1) regulates hypoxia-inducible factor-1alpha (HIF-1alpha) in endothelial cells. J Biol Chem. 2010;285:20570–9. doi: 10.1074/jbc.M110.119495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fraser P. Transcriptional control thrown for a loop. Curr Opin Genet Dev. 2006;16:490–5. doi: 10.1016/j.gde.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 58.Matarazzo MR, Boyle S, D’Esposito M, Bickmore WA. Chromosome territory reorganization in a human disease with altered DNA methylation. Proc Natl Acad Sci U S A. 2007;104:16546–51. doi: 10.1073/pnas.0702924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Errico M, Riccioni T, Iyer S, Pisano C, Acharya KR, Persico MG, De Falco S. Identification of placenta growth factor determinants for binding and activation of Flt-1 receptor. J Biol Chem. 2004;279:43929–39. doi: 10.1074/jbc.M401418200. [DOI] [PubMed] [Google Scholar]
- 60.De Bonis ML, Cerase A, Matarazzo MR, Ferraro M, Strazzullo M, Hansen RS, Chiurazzi P, Neri G, D’Esposito M. Maintenance of X- and Y-inactivation of the pseudoautosomal (PAR2) gene SPRY3 is independent from DNA methylation and associated to multiple layers of epigenetic modifications. Hum Mol Genet. 2006;15:1123–32. doi: 10.1093/hmg/ddl027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.